REVIEW: Statin Drugs and Dietary Isoprenoids Downregulate Protein Prenylation in Signal Transduction and Are Antithrombotic and Prothrombolytic Agents
J. W. Fenton, II1,2*, W. P. Jeske3, J. L. Catalfamo4, D. V. Brezniak1, D. G. Moon5, and G. X. Shen6
1New York State Department of Health, Wadsworth Center, Albany, NY 12201-0509 USA; fax: 518-473-7082 or 2900; E-mail: brezniak@wadsworth.org2Department of Physiology and Cell Biology and Department of Biochemistry and Molecular Biology, Albany Medical College, Albany, NY 12208 USA
3Cardiovascular Institute, Loyola University Medical Center, Maywood, IL 60153 USA
4College of Veterinary Medicine, Cornell University, Ithaca, NY 14852 USA
5Department of Biology, Albany College of Pharmacy, Albany, NY 12208 USA
6Departments of Internal Medicine and Physiology, University of Manitoba, Winnipeg, MB, R3E 3I7 Canada
* To whom correspondence should be addressed.
Received May 22, 2001; Revision received June 20, 2001
Statins and various isoprenoids of dietary origins inhibit L-mevalonic acid synthesis, which in turn downregulates cholesterol and various other dependent substances, including farnesyl- and geranylgeranyl-conjugated proteins involved in cell signaling processes. Such signaling processes are stimulated by protease-activated receptor-1 (PAR-1), which upon activation, causes the expression of various substances including tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1). Tissue factor promotes thrombin generation, where thrombin stimulates a variety of cellular processes, as well as activating PAR-1 to produce more thrombin. Statins downregulate TF mitigating thrombin generation and also downregulate PAI-1, which normally consumes tissue plasminogen activator (tPA). In the absence of PAI-1, tPA activates plasminogen to generate plasmin. Thus, statins behave as antithrombotic agents and prothrombolytic agents.
KEY WORDS: statins, dietary isoprenoids, protein prenylation inhibition, protease-activated receptor mitigation, thrombin generation downregulation, plasmin generation upregulation
Abbreviations: PAR-1) proteinase activated receptor-1; PAI-1) plasminogen activator inhibitor-1; TF) tissue factor; HMG-CoA) 3-hydroxy-3-methylglutaryl coenzyme A.
While attending plenary lectures at the XI International Symposium on
Atherosclerosis in 1997 at Paris, France, the first and senior authors
envisioned that thrombin somehow participated in the development of
atherosclerotic and prothrombotic diseases. We further envisioned that
statin drugs and other 3-hydroxy-3-methylglutaryl CoA (HMG-CoA)
reductase inhibitors mitigated such diseases by indirectly
downregulating thrombin generation at the cellular level [1-5]. As implied from clinical
trials [6-8], we agreed with
others [9-12] that cholesterol
was a distant product of the isoprenoid pathways and was not a direct
regulant of beneficial pleotropic effects of statin drugs [13]. Rather, these drugs appeared to downregulate
prenylation of proteins involved in signal transduction in cells.
Our model (Fig. 1) for explaining the antithrombotic and prothrombolytic properties of statins, dietary isoprenoids, and other HMG-CoA reductase inhibitors [14-17] couples the downregulation of prenylated proteins with inhibition of chronic thrombin generation at the cellular level. This model involves thrombin activation of protease-activated receptor-1 (PAR-1) and the subsequent upregulation of tissue factor (TF) and plasminogen activator-1 (PAI-1). Thus, TF promotes thrombin generation leading to thrombin-induced processes (e.g., inflammation, cell growth), whereas PAI-1 inactivates tissue plasminogen activator (tPA) and other activators and inhibits thrombolytic processes [18, 19]. Conversely, statins, dietary isoprenoids, or other HMG-CoA reductase inhibitors downregulate TF and PAI-1 with the consequence that less thrombin is generated [20] and plasmin is formed because PAI-1 is insufficient to neutralize tPA, thus promoting thrombolysis [21-24].
Within the past few years considerable information has been published strengthening claims for prenylated-protein regulatory mechanisms independent of cholesterol. The product of HMG-CoA reductase, L-mevalonic acid compensates for statins and other HMG-CoA reductase inhibitors [25], whereas cholesterol is noncompensatory, indicating that cholesterol does not participate in the regulatory mechanisms of statins [26]. This is further in agreement with the delayed and proportionally lower response of plasma cholesterol levels to statin drugs than, for example, monocyte TF [20]. Furthermore, in experimental animal models, statins reduce atherosclerotic development independent of cholesterol administration [27]. Despite cholesterol accumulates in foam cells of atherosclerotic lesions [28], the above observations collectively imply cholesterol levels are at best an indirect indicator of statin effectiveness and that more meaningful diagnostic markers are needed.Fig. 1. A composite of isoprenoid-conjugated proteins and cellular thrombin generating pathways. The statin-induced shift is shown from favoring thrombotic to thrombolytic processes (figure courtesy of John W. Fenton, II).
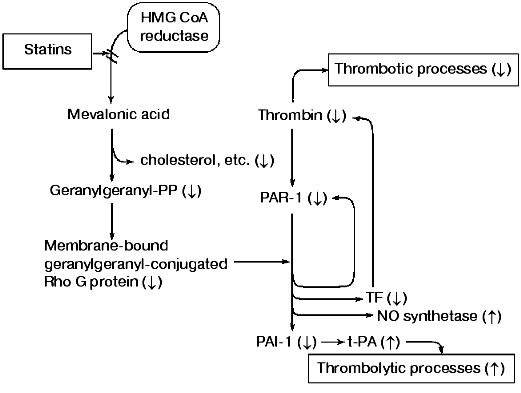
Several investigators have suggested that statins block prenylation of proteins involved in signal transduction at the cellular level. Some investigators have proposed the farnesyl pathway [29], while the majority have implicated the geranylgeranyl route [23, 24, 30-33]. The latter is no surprise, since the majority of prenylated proteins are geranylgeranyl conjugates [34, 35].
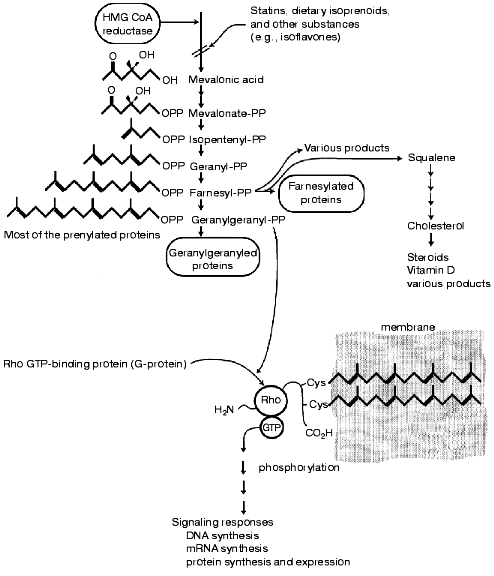
However, both farnesyl- and geranylgeranyl-conjugated proteins may be involved in thrombin-stimulated events [4, 5]. Their most likely gating target(s) may well be selective activation of certain protein kinase C isoforms [36-39]. These are prenylated proteins, and the activation of other prenylated proteins, such as Rho proteins may be subsequent. Both farnesyl- and geranylgeranyl-conjugating groups are L-mevalonic acid dependent and conjugate to proteins via cystine sulfhydryl groups located near the carboxy termini of such proteins [34, 35]. The farnesylated proteins have a single prenylated side chain (15 carbons), whereas the geranylgeranyled proteins have two prenylated attachments (20 carbons each). Such attachments membrane associate (Fig. 2) and the larger geranylgeranyl group should contribute more membrane stability than the smaller farnesyl group.
Because of their sulfhydryl linkages, prenylated conjugates are subject to methylation, oxidation, and cleavage making them susceptible to various turnover processes [25, 34]. In order for prenylated proteins to function as regulatory proteins, they must not only be synthesized but must be turned over by degradation processes. The effect of protein prenylation is to make otherwise “free swimming” proteins membrane-associated, where they may more efficiently carry out processes (e.g., enzyme catalyzed reactions). Decreasing the availability of prenylated moieties for protein conjugation should make such processes less efficient. Still, these processes cannot be completely stopped, since they may inefficiently occur in free solution. Likewise, various degradation processes could limit the availability of prenylated proteins. This may explain the low toxicity of statin drugs and other HMG-CoA reductase inhibitors. This should allow statins to be employed as chronically administered drugs for the treatment of not only cardiovascular problems but also for inflammation and a variety of diseases associated with aging processes (table).Fig. 2. A generalized scheme depicting isoprenoid metabolism to a geranylgeranyl-conjugated Rho GTP-binding in cellular signal transduction. Both mevalonic acid and geranylgeranyl-PP are reported to compensate for statin effects on cells, whereas cholesterol is noncompensatory (see text). Statins should inhibit prenylation of Rho proteins (and other substrates) preventing membrane association. This should not completely block dependent downstream pathways but rather blunt them. This is in accord with the very low toxicity of statins and dietary isoprenoids (figure courtesy of John W. Fenton, II).
Some non-lipid-lowering pleiotropic activities and effects of various
statins

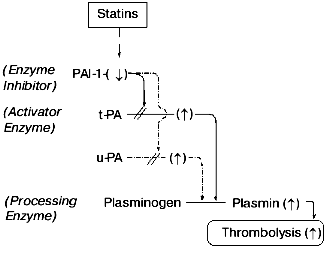
Most investigators have gone as far as to recognize the likely participation of prenylated proteins in accounting for the pleiotropic effects of statins (see above). However, none to our knowledge have gone beyond this point to propose how statins regulate disease processes. In 1998, we proposed that statins downregulated thrombin generation through decreasing TF expression [1-5]. Our proposal (Fig. 1) has essentially remained intact over the past three to four years other than to recognize that if statins disproportionally downregulate PAI-1 relative to tPA, then tPA levels should rise causing plasmin generation and thrombolysis (Fig. 3). Such a mechanism should also enable statins to upregulate urokinase plasminogen activator (uPA) depending on the relative concentrations of PAI-1 and uPA and should cause plasmin generation and thrombolysis. Among the various effects of statins, nitroxide (NO) synthesis is markedly upregulated while most other effects are downregulated (table). It is possible that an activator enzyme mechanism is operative like with PAI-1 and tPA or uPA (Fig. 3), or perhaps a novel mechanism functions in the NO system to invert the more prevalent downregulation caused by statins. Thus, statins depress thrombin generation (by downregulating TF) while promoting plasmin generation (by downregulating PAI-1, allowing tPA to increase) [20-24]. They also upregulate NO synthesis enhancing vascular function and improve circulation. These remarkable systems permit an isoprenoid (or mimic) with very low toxicity to shift the mode from prothrombotic to prothrombolytic and to promote more quiescent states. Moreover, statin drugs should mitigate a variety of thrombin-mediated events [18] and particularly those that occur at the cellular level [1, 2].
In our proposal scheme (Fig. 1), we hypothesized that thrombin initiated its cellular amplification system by activating PAR-1. In addition to thrombin, human platelet PAR-1 is activated by SFLLRN (and related peptides), which correspond to the new amino-terminus generated by thrombin cleavage of PAR-1 and similar receptors [40, 41]. Employing such agonist peptides, we found that two entirely different statins had no immediate effects on SFLLRNP peptide-induced by aggregation of human platelets. Upon incubation for up to 2 h, both statins reduced platelet aggregation by SFLLRNP while neither statin had any effect on ADP-induced aggregation (Jeske, Catalfamo, and Fenton, unpublished data). These data imply that the prenylated protein(s) inhibited by statins do indeed turn over (although moderately slowly in platelets) and the statin blockage involves the PAR-1 pathway, as predicted by our scheme (Fig. 1).Fig. 3. Thrombolysis inversion mechanism. Statins and other HMG-CoA reductase inhibitors (e.g., dietary isoprenoids) downregulate the enzyme inhibitor, PAI-1, which inhibits the activator enzymes, tPA and/or uPA, to a lesser extent and increases activator enzymes. The increased availability of tPA and/or uPA activates plasminogen to the processing enzyme, plasmin, which initiates thrombolysis. Thus, statins and related inhibitors not only downregulate thrombin generation and thrombotic processes but also upregulate plasmin and thrombolytic processes (figure courtesy of John W. Fenton, II).
Since activation of PAR-1 stimulated PAR-1 expression [42], statins should inhibit PAR-1 expression and desensitize cells to thrombin stimulation. To our knowledge such experiments have not been performed. Just how statins block the PAR-1 signaling pathways is also unknown. Since PAR-1 activation signals the activation of selective protein kinase C isoforms, where the heterotrimer chain has a prenyl side chain [36-39], this could be an important site for statin blockage. On the other hand, considerable evidence indicates geranylgeranyl conjugation to the Rho GTP-binding protein may be the major rate-limiting step in statin inhibition of thrombin stimulation of cellular mechanisms [30-33, 43, 44]. Beyond this point, where statins act is largely conjecture. Nevertheless, the fact remains that statins are multifaceted pleiotropic drugs where, for the most part, they are well tolerated and clinically approved for chronic administration in reducing blood cholesterol levels [13]. Despite this employment, the beneficial functions of statins appear to be reducing thrombin generation and promoting thrombolytic conditions of more restful, quiescent, or youthful states. That many isoprenoid substances found in foodstuffs are HMG-CoA reductase inhibitors suggests that statins are helping to regulate an important natural pathway in normal development, disease, and longevity. Of particular note is the reduction of cardiovascular diseases and cancers among persons consuming Mediterranean and Eastern diets in contrast to those on northern European diets [14]. Besides cholesterol, eggs and dairy products are rich in dietary isoprenoids [14] suggesting that isoprenoid downregulation may be advantageous in the neonate. In this regard, we believe that protein prenylation is a major physiological regulatory process with perhaps several pleiotropic side paths independent of cholesterol, cholesterol-derived products (e.g., steroids, vitamin D), and other substances. We have proposed a scheme by which statins and other HMG-CoA reductase inhibitors downregulate protein prenylation and by which protein prenylation deprivation downgrades TF and PAI-1 statement (Fig. 1). Diminished TF reduces thrombin generation (antithrombotic) and depressed PAI-1 allows tPA to generate plasmin (prothrombolytic). Furthermore, we believe that thrombin is an important mediator not only of thrombotic processes but also cell regulatory processes [18], which may include cellular excretions (e.g., growth factors), adhesive proteins expression (e.g., P-selectin), protein synthesis (e.g., TF, PAI-1), and cellular proliferation (e.g., smooth muscle cells, atherosclerotic lesions, growth and spreading of thrombin-mediated cancers). Moreover, our scheme has built in it a mechanism where thrombin generates more thrombin, and hence, the “thrombin cycle” at the cellular level, analogous to thrombin amplification by activation of coagulation factors V and VII in the fluid phase of blood. This cellular cycle for thrombin generation may be a formerly unappreciated mechanism for chronic thrombin generation, which is downregulated by statin drugs.
REFERENCES
1.Fenton, J. W., II, and Shen, G. X. (1998) in
Lectures. 15th Int. Congr. on Thrombosis (Ulutin, O. N., ed.)
Thrombosis Society of Turkey, Antalya, Turkey, pp. 112-114.
2.Fenton, J. W., II, and Shen, G. X. (1999)
Haemostasis, 29, 166-169.
3.Fenton, J. W., II, Shen, G. X., Walenga, J. M., and
Ofosu, F. A. (1999) Thromb. Haemost., 82, 191 (Abs.
602).
4.Fenton, J. W., II, Shen, G. X., Minnear, F. L.,
Brezniak, D. V., Walenga, J. M., Bognacki, J. J., and Ofosu, F. A.
(2000) Appl. Thromb. Hemost., 6, 18-21.
5.Fenton, J. W., II, Shen, G. X., Minnear, F. L.,
Brezniak, D. V., Jeske, W. P., Walenga, J. M., Bognacki, J. J., Ofosu,
F. A., and Hassouna, H. I. (2000) Hematol. Oncol. Clin. North
Am., 14, 483-490.
6.The Scandinavian Simvastatin Study Survival Group
(4S) (1994) Lancet, 344, 1383-1389.
7.Shepard, J., Cobbe, S. M., Ford, I., Isles, C. G.,
Lorimer, A. R., MacFarlane, P. W., McKillop, J. H., and Paoletti, P.
(1995) N. Engl. J. Med., 333,1301-1307.
8.Sacks, F. M., Pfeffer, M. A., Moye, L. A., Rouleau,
J. L., Rutherford, J. D., Cole, T. G., Brown, L., Warnica, J. W.,
Arnold, J. A. O., Wun, C. C., Davis, B. R., and Braunwald, E. (1996)
N. Engl. J. Med., 335,1001-1009.
9.Massy, Z. A. (1996) Lancet, 347,
102-103.
10.Vaughan, C. J., Murphy, M. B., and Buckley, B. M.
(1996) Lancet, 348, 1079-1082.
11.Rosenson, R. S., and Tangney, C. C. (1998) J.
Am. Med. Assoc., 279, 1643-1650.
12.Dangas, G., Smith, D. A., Unger, A. H., Shao, J.,
Meraj, P., Fier, C., Cohen, A. M., Fallon, J. T., Badimon, J. J., and
Andersen, H. (2000) Thromb. Haemost., 83, 688-692.
13.Davignon, J. (1998) in Atherosclerosis
(Jacotot, B., Mathe, D., Fruchart, J.-C., eds.) Elsevier, Amsterdam,
pp. 63-77.
14.Elson, C. E. (1995) J. Nutr., 125,
16665-16725.
15.Blanco-Colio, L. M., Valderrama, M.,
Alvarez-Sala, L. A., Bustos, C., Ortego, M., Hernandez-Presa, M. A.,
Cancelas, P., Gomez-Gerique, J., Millan, J., and Eligini, S. (2000)
Circulation, 102, 1020-1026.
16.De La Cruz, J. P., Auxilidora, M., Carmona, J.
A., Martin-Romero, M., Smith-Ageda, J. M., and de la Cuesta, F. S.
(2000) Thromb. Res., 100, 305-315.
17.Kimura, K., Yokoi, K., Matsushita, N., and Okuda,
E. (1997) J. Pharm. Pharmacol., 49, 816-822.
18.Fenton, J. W. (1995) Thromb. Haemost.,
74, 493-498.
19.Shen, G. X. (1998) Intl. J. Mol. Med.,
1, 399-408.
20.Ferro, D., Basili, C., Alessandri, C., Mantovani,
B., Cordova, C., and Violi, F. (1997) Lancet, 350,
1222.
21.Bevilacqua, M., Bettica, P., Milani, M., Vago,
T., Rogolino, A., Riglini, V., Santoli, E., and Norbiato, G. (1997)
Am. J. Cardiol., 79, 84-87.
22.Emeis, J. J., and Cohen, L. H. (1997) Am. J.
Cardiol., 80, 977-978.
23.Essig, M., Nguyen, G., Prie, D., Escoubet, B.,
Sraer, J.-D., and Friedlander, G. (1998) Circ. Res.,
83,683-690.
24.Bourcier, T., and Libby, P. (2000)
Arterioscler. Thromb. Vasc. Biol., 20, 556-562.
25.Raiteri, M., Arnaboldi, L., McGrady, P., Gelb,
H., Verri, D., Tagliabue, C., Quarato, P., Ferraboschi, P.,
Santaniello, E., Paoletti, P., Fumagalli, R., and Corsini, A. (1997)
Am. Soc. Pharmacol. Expl. Therapeutics,
281,1144-1153.
26.Ando, H., Takamura, T., Ota, T., Nagai, Y., and
Kobayashi, K.-I. (2000) J. Pharmacol. Expl. Therapeutics,
294, 1043-1046.
27.Alfon, H., Pueyo Palazon, C., Royo, T., and
Badimon, L. (1999) Thromb. Haemost., 81, 822-827.
28.Ross, R. (1999) N. Engl. J. Med.,
340, 115-126.
29.Goldstein, J. L., and Brown, M. S. (1990)
Nature, 343, 430.
30.Colli, S., Eligini, S., Lalli, M., Camera, M.,
Paoletti, R., and Tremoli, E. (1997) Arterioscler. Thromb. Vas.
Biol., 17, 265-272.
31.Essler, M., Amano, M., Kruse, H.-J., Kaibuchi,
K., Webert, P. C., and Aepfelbacher, M. (1998) J. Biol. Chem.,
273, 21867-21874.
32.Muniyappa, R., Xu, R., Ram, J. L., and Sowers, J.
R. (2000) Am. J. Physiol. Heart Circ. Physiol., 278,
H1762-H1768.
33.Van Nieuw Amerongen, G. P., van Delft, S.,
Vermeer, M. A., Collard, J. G., and van Hinsberg, V. W. M. (2000)
Circ. Res., 87,335-340.
34.Clarke, S. (1992) Ann. Rev. Biochem.,
61, 355-386.
35.Zhang, F. L., and Casey, P. J. (1996) Ann.
Rev. Biochem., 65, 241-269.
36.Shen, G. X., Ren, S., and Fenton, J. W., II
(1998) Sem. Thromb. Hemost., 24, 151-156.
37.Lidington, E. A., Haskard, D. O., and Mason, J.
C. (2000) Blood, 96, 2784-2792.
38.Ludwicka-Bradley, A., Tourkina, E., Suzuki, S.,
Tyson, E., Bonner, M., Fenton, J. W., II, Hoffman, S., and Silver, R.
M. (2000) Am. J. Respir. Cell Mol. Biol., 22,
235-243.
39.Tiruppathi, C., Yan, W., Sandoval, R., Naqvi, T.,
Pronin, A. N., Benovic, J. L., and Malik, A. B. (2000) Proc. Natl.
Acad. Sci. USA, 97, 7440-7445.
40.Vu, T. H., Hung, D. T., Wheaton, V. I., and
Coughlin, S. R. (1991) Cell, 64, 1057-1068.
41.Xu, W.-F., Andersen, H., Whitmore, T. E.,
Presnell, S. R., Yee, D. P., Ching, A., Gilbert, T., Davie, E. W., and
Foster, D. C. (1998) Proc. Natl. Acad. Sci. USA, 95,
6642-6646.
42.Ellis, C. A., Malik, A. B., Gilchrist, A., Hamm,
H., Sandoval, R., Voyno-Yasenetskaya, T., and Tiruppathi, C. (1999)
J. Biol. Chem., 274, 13718-13727.
43.Essig, M., Vrtovsnik, F., Nguyen, G., Spaer,
J.-D., and Friedlander, G. (1998) J. Am. Soc. Nephrol.,
9, 1377-1388.
44.Laufs, U., Marra, D., Node, K., and Liao, J. K.
(1999) J. Biol. Chem., 274, 21926-21931.
45.Laufs, U., La Fata, V., Plutzky, J., and Liao, J.
K. (1998) Circulation, 97, 1129-1135.
46.Kaesemeyer, W. H., Caldwell, R. B., Huang, J.,
and Caldwell, R. W. (1999) J. Am. Coll. Cardiol., 23,
234-241.
47.Laufs, U., and Liao, J. K. (1998) J. Biol.
Chem., 273, 24266-24271.
48.Feron, O., Dessy, C., Desager, J.-P., and
Balligand, J.-L. (2001) Circulation, 103, 113-118.
49.Wu, J.-R., and Gilbert, T. (2000) FEBS
Lett., 484, 108-112.
50.Tomas, M., Senli, M., Garcia-Faria, F., Vila, J.,
Torrento, A., Covas, M., and Martin-Romero, M. (2000) Arterioscler.
Thromb. Vasc. Biol., 20, 2113-2119.
51.Kureishi, Y., Luo, Z., Shiojima, I., Bialik, A.,
Fulton, D., Lefer, D. J., Sessa, W. C., and Walsh, K. (2000) Nature
Med., 6, 1004-1010.
52.Mundy, G., Garrett, R., Harris, S., Chan, J.,
Chen, D., Rossini, G., Boyce, B., Zhao, M., and Gutierrez, G. (1999)
Science, 286, 1946-1949.
53.Chan, K. A., Andrade, S. E., Boles, M., Buist, D.
S. M., Chase, G. A., Donahue, J. G., and Goodman, M. J. (2000)
Lancet, 355, 2185-2188.
54.Edwards, C. J., Hart, D. J., and Spector, T. D.
(2000) Lancet, 355, 2218-2219.
55.Meier, C. R., Schlienger, R. G., Kraenzlin, M.
E., Schlegel, B., and Jick, H. (2000) J. Am. Med. Assoc.,
283, 3205-3210.
56.Aikawa, M., Rabkin, E., Sugiyama, S., Voglic, S.
J., Fukumoto, Y., Furukawa, Y., Shiomi, M., Schoen, F. J., and Libby,
P. (2001) Circulation, 103, 276-283.
57.Sparrow, C. P., Burton, C. A., Hernandez, M.,
Mundt, S., Hassing, H., Patel, S., Rosa, R., Hermanoski-Vosatka, A.,
Wang, P.-R., Zhang, D., Peterson, L., Detmers, P. A., Chao, Y.-S., and
Wright, S. D. (2001) Arterioscler. Thromb. Vasc. Biol.,
21, 115-121.
58.Freeman, D. J., Norrie, J., Sattar, N., Neely, R.
D. G., Cobbe, S. M., Ford, I., Isles, C., Lorimer, A. R., MacFarlane,
P. W., McKillop, J. H., Packard, C. J., Shepard, J., and Gaw, A. (2001)
Circulation, 103, 357-362.
59.Byington, R. P., Davis, B. R., Plehn, J. G.,
White, H. D., Baker, J., Cobbe, S. M., and Shepard, J. (2001)
Circulation, 103, 387-392.
60.Jick, H., Zornberg, G. L., Jick, S. S., Seshadri,
S., and Drachman, D. A. (2000) Lancet, 356,
1627-1631.