REVIEW: Plasminogen Activators in Vascular Remodeling and Angiogenesis
Ye. V. Parfyonova*, O. S. Plekhanova, and V. A. Tkachuk
Russian Cardiology Research Center, Ministry of Health of the Russian Federation, 3-ya Cherepkovskaya ul. 15a, Moscow, 121552 Russia; fax: (095) 414-6712; E-mail: Yeparfyon@cardio.ru* To whom correspondence should be addressed.
Received August 1, 2001; Revision received October 15, 2001
This review considers cellular and molecular mechanisms of the involvement of plasminogen activators in extracellular proteolysis and cell migration and proliferation. The role of plasminogen activators in vascular remodeling in atherosclerosis, restenosis, and angiogenesis is discussed.
KEY WORDS: urokinase, tissue plasminogen activator, cell migration, vascular remodeling, restenosis, angiogenesis
Abbreviations: uPA) urokinase-type plasminogen activator; tPA) tissue-type plasminogen activator; PAI) plasminogen activator inhibitors; uPAR) urokinase receptor; GFD) growth factor domain, N-terminal domain of urokinase similar in the structure to the epidermal growth factor; GPI-receptor) glycosyl phosphatidylinositol-anchored receptor; bFGF) basic fibroblast growth factor; VEGF) vascular endothelium growth factor; HGF) hepatocyte growth factor; TGF-beta) transforming growth factor-beta; LDL) low density lipoproteins; PDGF) platelet derived growth factor; MAP-kinases) mitogen-activated protein kinases.
PLASMINOGEN ACTIVATORS IN EXTRACELLULAR PROTEOLYSIS AND CELL
MIGRATION AND PROLIFERATION
Extracellular proteolysis is one of many ways the cell influences its environment, this being responsible for structural and functional plasticity of tissues, remodeling of the extracellular matrix and of intercellular spaces, lysis of fibrin clots, and development of vascular bed, ducts, and cavities of the body. Virtually all cells of the body at some stage in their life use extracellular proteolysis to modify environmental structures [1, 2].
Disorders in extracellular proteolysis resulting in its excessive or insufficient activation can cause serious disorders in tissue structure and functions and be primary etiological factors or important links in the genesis of many diseases. The regulation of extracellular proteolysis includes a great number of various mechanisms that should be known for both the understanding of features in the course of many diseases and the elaboration of new therapeutic approaches. The achievements of thrombolytic therapy are a promising example of the successful use of findings on the control mechanisms in the activity and functions of extracellular proteases in the development of a new direction in therapy [2, 3].
The enzyme systems that can degrade extracellular matrix components or activate latent forms of growth factors play the main role in many events during early embryogenesis (fertilization and the subsequent reactions, determination of the developmental axes, mammalian embryo implantation, blood vessel formation, cell migration during organogenesis) and also in the adult organism (inflammation, wound healing, angiogenesis, tissue remodeling, tumor growth, metastasis, etc.) [1, 2].
The plasminogen activator-plasmin system seems to deserve the greatest attention among the active proteolytic cascades of the cell environment. Initially, this system was considered as the main system responsible for fibrinolysis. However, the increasing amount of data on a wide spectrum of physiological and pathological conditions associated with the expression of this system contributed to development of the concept on its involvement in the regulation of turnover of a wide spectrum of extracellular matrix components [2, 4]. The cascade of proteolytic reactions triggered by the plasminogen activators is started by the conversion of the inactive proenzyme plasminogen into the active enzyme plasmin, a protease of wide specificity, which can directly cleave fibrin/fibrinogen, blood coagulation factors V/Va and VIII/VIIIa, some growth factors, and extracellular matrix components, and can also catalyze the degradation of plasmin-resistant matrix proteins, such as natural collagens, due to activation of inactive zymogens of collagenases (matrix metalloproteinases); thus, it can play the dominant role in extracellular proteolysis throughout the body [2]. The dominant role of plasmin was confirmed only within the last 15 years, when plasminogen synthesis and plasmin activation were reported to occur everywhere in the body [5-7].
In addition to the widely expressed plasminogen activator-plasmin system in the body, suggesting its involvement in many biological processes, the organization of this system is an example of a complex extracellular proteolytic cascade. The conversion of the inactive plasminogen into plasmin is controlled by two plasminogen activators found in virtually all mammals: the tissue plasminogen activator (tPA) and the urokinase plasminogen activator (uPA) [1, 4]. These activators of the serine protease family are products of two different genes, but they have in common the same substrate, plasminogen, and two specific inhibitors: the first and the second type inhibitors of the plasminogen activators (PAI-1 and PAI-2, respectively) which are of the family of serine protease inhibitors, serpins [8, 9].
The existence of two plasminogen activators in mammals inevitably suggests the question whether these enzymes have the same or different functions. Some cells are known to synthesize only one plasminogen activator, tPA or uPA, whereas other cells can synthesize both enzymes [10-12]. tPA is most usually believed to be synthesized when fibrinolysis is needed, whereas uPA synthesis is associated with cell migration [1, 3]. Note, that in the granulosa cells of rat ovary tPA is produced, whereas in their close relatives, mice, similar cells produce uPA [13]; this shows that the two enzymes can execute the same function and replace each other. This is indirectly supported by the finding that birds have only one plasminogen activator similar to uPA in structure [14].
The plasminogen activators are synthesized and secreted by various cells: smooth muscle cells (SMC) [10], endothelial cells [11], monocytes/macrophages [12], epithelial cells [15], fibroblasts, and by malignant tumor cells [16]. tPA synthesis by vascular endothelial cells is responsible for adequate fibrinolysis required to maintain vessel patency. uPA, which is synthesized, secreted, and bound on the apical surface of epithelial cells, is important for provision of an adequate proteolysis required to maintain tubular patency [15, 16]. In these cases, the role of urokinase is similar to the role of tPA in the vascular bed. Moreover, urokinase is actively expressed under circumstances associated with cell migration (reparation, inflammation, angiogenesis, metastasizing) [1, 3, 10, 17-19] providing efficient and spatially restricted extracellular proteolysis that is necessary for the cell to move through anatomical boundaries.
Similarly to the majority of serine proteases, the plasminogen activators are secreted as glycosylated single-chain proteins consisting of several domains. uPA consists of three domains: the N-terminal domain similar in the structure to the epidermal growth factor, the growth factor domain (GFD) (amino acids 9-45), the kringle-domain (amino acids 46-143), and the C-terminal proteolytic domain (amino acids 144-411) [20]. The function of the GFD is high affinity interaction with the specific urokinase receptor (uPAR) on the cell surface [21]. The proteolytic domain converts the plasminogen into plasmin and activates some growth factors and matrix metalloproteinases [1, 3]. It contains an active site that includes a catalytic triad of amino acids (His204, Asp255, and Ser356) specific for all serine proteases. The functions of the kringle domain are far from being fully understood. It mediates the binding to heparin through three sequential amino acid residues, Arg108-Arg109-Arg110 [22]. According to the data obtained in our laboratory, it is involved in the urokinase-induced stimulation of cell migration [23].
The two plasminogen activators are mainly different in the structural determinants located in the noncatalytic regions of the proteins that are responsible for the enzyme binding to various components of the cell surface and of the cell environment. Thus, in the noncatalytic N-terminal region of the tPA molecule there are two additional domains as compared to the uPA molecule: a finger-domain and the second kringle-domain, which are responsible for specific binding to fibrin [24].
Precise timing and location of adequate extracellular proteolysis is provided by multiple mechanisms among which the mechanisms regulating the expression of plasminogen activators and of their inhibitors are especially important. A wide spectrum of hormones, growth factors, and cell environment factors are involved in the regulation of expression of the genes of plasminogen activators and of their inhibitors. The expression of uPA is activated by various inflammatory factors: cytokines [25], growth factors [26], and tumor promotors, such as phorbol-12-myristate-13-acetate [27], whereas antiinflammatory agents, such as glucocorticoids, inhibit the expression of uPA [28]. tPA expression is regulated by growth factors, thrombin, and also by angiotensin II [29, 30]. The regulation usually occurs on the level of gene transcription and mRNA stability [26, 27], but it can also occur on the level of translation and protein secretion [31].
The evolutionary advantage of mammals provided by the existence of two plasminogen activators which can “replace” each other is most likely associated with different regulatory sequences of the genes encoding uPA and tPA [31]. This provides a significant enrichment in the potential for the transcriptional and post-transcriptional regulation of their expression. Moreover, the different structure of the noncatalytic regions of the tPA and uPA molecules and different binding sites on the cell surface or on the extracellular matrix components provide different locations of extracellular proteolysis depending on what plasminogen activator is activated under the specific situation.
The single-chain urokinase (scuPA) secreted by cells has a low enzymatic activity which increases ~300-fold after the proteolytic cleavage of the Lys158-Ile159 bond [32] that results in uPA conversion into the two-chain form consisting of two polypeptide chains joined via the Cys148-Cys279 disulfide bond. The N-terminal A-chain includes the GFD and kringle domain; the B-chain contains the proteolytic domain. Plasmin is the most efficient activator of scuPA, although the other enzymes (kallikrein, trypsin, blood coagulation factor XIIa, and cathepsin in tumor cells [32]) could potentially compensate for any loss of plasmin function.
Of the two plasminogen activators, only urokinase has a specific high affinity receptor that has been found in various cells (monocytes, neutrophils, endothelial and smooth muscle cells of blood vessels, keratinocytes, and also various tumor cell lines), and this determines its key role in plasminogen activation on the cell surface [1]. The urokinase receptor is a glycosyl phosphatidylinositol-anchored receptor (GPI-receptor) and lacks both cytoplasmic and transmembrane domains [33]. Therefore, it has great lateral mobility in the membrane, its location and the location of urokinase bound to it are very movable and closely associated with the functional state of the cell. Thus, in a migrating cell the uPAR is clustered on the leading edge and focuses the proteolytic potential on the cell-substrate and cell-cell contact sites that is necessary for directed cell movement [34].
In addition to spatial control of extracellular proteolysis, the urokinase receptor is involved in the activation and inactivation of urokinase. The binding of the secreted single-chain urokinase to the uPAR makes it more sensitive to proteolytic activation by plasmin than the free urokinase [35]. The plasminogen activator activity is regulated by serpins (the serine protease inhibitors) and the most important of them is the first type inhibitor of plasminogen activators (PAI-1), which is a 45-50 kD glycoprotein interacting with both free and bound plasminogen activators [8, 9]. On binding to the two-chain uPA, this inhibitor inactivates it by generating a 1 : 1 covalent complex. The binding of PAI-1 to uPA bound to uPAR results in fast endocytosis of the uPA-uPAR-PAI-1 complex. This endocytosis is provided by several proteins: the alpha2-macroglobulin receptor (LRP), which is a multiligand receptor homologous to the receptor of low density lipoproteins and is involved in the internalization of many proteins [36], megalin, or the glycoprotein 330 [37], and also the receptor of very low density lipoproteins [38]. In the acidic medium of endosomes, urokinase and PAI-1 dissociate and are degraded. However, the urokinase receptor comes back to the cell surface and continues to function.
Pericellular proteolysis can also be regulated by changes in the ratio of the plasminogen activator inhibitors to urokinase and its receptor. Thus, the basic fibroblast growth factor (bFGF) increases the ratio of uPA mRNA to PAI-1 mRNA, and thus increases cell-induced proteolysis [39].
Although tPA binding sites of high and low affinity have been found on vascular SMC and endothelial cells [40], no specific receptor for this protease similar to uPAR has been identified on the cell surface yet. tPA specifically binds to fibrin [2] and activates plasminogen preferentially at the fibrin surface [41]. Unlike tPA, two-chain uPA activates similarly both circulating and fibrin-bound plasminogen, this causing extensive systemic activation of fibrinolysis and possibly resulting in bleeding. Therefore, tPA is more widely used in clinical practice than urokinase [41].
The main role of uPA in extracellular proteolysis is a generation of cell surface associated plasmin cleaving the main components of the basement membrane and of the extracellular matrix and activating matrix metalloproteinases, which degrade the components resistant to plasmin [1, 3, 42]. Essential for the proteolytic function of uPA is its ability to directly or via plasmin production activate latent or release matrix-bound growth factors. Thus, uPA itself proteolytically activates proforms of hepatocyte growth factor (HGF), of macrophage-stimulating protein (MSP), and of an isoform of vascular endothelial growth factor (VEGF189) [43, 44]. Urokinase-generated plasmin activates latent transforming growth factor-beta (TGF-beta) and other VEGF isoforms [45] and promotes the release of basic fibroblast growth factor (bFGF) inactivated by binding to matrix proteins sequestered by the extracellular matrix [42]. Moreover, uPA can directly cleave certain matrix proteins (fibronectin) and activate certain matrix metalloproteinases [46] and, thus, independently of plasmin, affect extracellular matrix remodeling.
However, the functions of uPA are not limited to the proteolytic activation of plasmin and of the growth factors on the cell surface. Recently accumulated data suggest that binding of uPA to membrane receptors on certain cells also directly initiates signaling cascades involved in the regulation of cell migration and proliferation independently of the ability of uPA to activate the growth factors. Thus, uPA-induced cell migration is associated with the activation of Janus kinases and of nonreceptor Src-kinases in certain cells [47-49]. In several cell lines uPA activates signal pathways involved in cytoskeleton reorganization: Hsk-kinases, kinases of focal contacts, paxillin, the p130CAS protein, MAP-kinases, the transcription activators STAT1 and STAT2, protein kinase Cepsilon responsible for cytokeratin phosphorylation [48, 50-53]. In addition to the influence on the protein kinase system, uPA can regulate gene expression. The binding of uPA to uPAR has been shown to stimulate downstream up-regulation of various transcription factors: AP1 in the HT-1080 cell line [54] and c-Jun, c-Myc, and c-fos factors in SaOS2 cells [55]. Recently, the possibility of signal transduction into the cell by the receptor systems associated with urokinase endocytosis was shown [56].
Because uPAR is a GPI-anchored protein, the signal transduction from the uPA-uPAR onto the intracellular effector systems needs a transmembrane adaptor. Integrins and also the transmembrane glycoprotein gp130 are considered as candidates for this role [47, 57].
In addition to urokinase, uPAR interacts with the extracellular matrix protein vitronectin, in this case in the role of a high affinity adhesion molecule [58, 59]. Urokinase stabilizes the “vitronectin-binding” conformation of the receptor and thus stimulates a “vitronectin-dependent” adhesion of some cells. PAI-1 and uPAR have a common binding site on vitronectin and the competition of PAI-1 with uPAR for the binding to this matrix protein prevents the cell adhesion [58-60]. uPAR can also interact with integrins, such as the leukocyte beta2-integrin Mac-1 (CD11b/CD18) [61] and also beta1-, beta3-integrins and with the vitronectin alpha(v)beta5 receptor [61, 63]. Urokinase modifies the interaction of uPAR with integrins [53, 62].
Both the uPA proteolytic function and signaling pathways activation are closely associated with the stimulation by uPA of cell migration and proliferation--the key processes in tissue remodeling.
uPA and uPAR are involved in the regulation of cell migration during various physiological and pathological processes, such as angiogenesis, the implantation of the embryo into the uterus wall, embryonic development, monocyte invasion in tissues, inflammatory reactions, wound healing, and metastasis [1, 18, 19, 64].
The pivotal role of uPA and uPAR in cell migration is shown by both in vitro migration assays [11, 65-71] and by in vivo studies [10, 72, 73]. In migrating cells there is a coordinated expression of uPA and uPAR accumulated at cell-substrate and cell-cell contact sites [11, 65-69, 74, 75] where they concentrate the plasmin production providing extracellular matrix proteolysis, cell-cell contact weakening, activation of growth factors and matrix metalloproteinases, and thus promoting cell motility. Plasmin inhibitors can suppress cell migration both in vitro [75, 76] and in vivo [77, 78], suggesting an important role of plasmin-induced proteolysis in this process.
However, there is also much evidence suggesting the existence of plasmin-independent mechanisms of uPA-induced cell migration. In some cases, to stimulate migration only occupation of uPAR is required, and uPA proteolytic activity is not needed. Thus, urokinase forms that can bind with high affinity to uPAR but are proteolytically inactive or lacking the proteolytic domain stimulated in vitro migration of certain cells [75, 79, 80]. Additionally, antibodies to uPAR or selective blockade of uPAR expression by use of antisense oligonucleotides also completely abrogates monocyte chemotaxis [75, 81]. uPA-induced monocyte migration was shown to require the interaction of uPAR with integrins [61, 76, 81].
Precise temporal and spatial coordination of proteolytic and nonproteolytic mechanisms is responsible for the succession of events during cell migration. With a non-proteolytic mechanism, uPA would stimulate cell migration by enhancing adhesion at the leading edge, through stimulation of binding of uPAR to vitronectin, modulation of uPAR/integrin interaction and/or by initiation of signal-transduction cascade that results in the cytoskeleton reorganization and in the cell “dragging up” to the leading edge. The proteolytic mechanisms include uPA catalyzed plasmin generation at focal adhesion sites that results in extracellular matrix degradation and thus facilitate detachment of the trailing edge. Both mechanisms could be operating simultaneously in the individual migrating cell. The role of PAI-1 in migration depends on its pericellular localization. The anti-migrational effect of PAI-1 seems to be associated with suppression of plasmin generation on the ventral surface of the cell, preventing its unfastening, with an inhibition of vitronectin binding to integrins and to uPAR on the leading edge. However, in an environment with high uPA activity, PAI-1 can protect vitronectin against destruction by urokinase and thus promote the adhesion on the leading edge that is necessary for the “dragging up” of the cell in the migration direction. The pro-migrational effect of PAI-1 can also be caused by stimulation of uPA and uPAR endocytosis and by elimination from the surface of the “dragged up” cell part of adhesion signals induced by the uPA and uPAR binding. Moreover, the endocytosis ensures uPAR recovery on the leading edge, which accelerates a new cycle of adhesion and of cytoskeleton reorganization that are required for cell movement along the substrate. The relative significance of the proteolytic and nonproteolytic mechanisms of the urokinase effect on cell migration and also the effect of PAI-1 depend on the expression in the migrating cells of uPA, uPAR, integrins, and of the endocytosis receptors, on the extracellular matrix composition, and on the plasminogen concentration.
The involvement of urokinase in tissue morphogenesis is also due to its ability to stimulate the cell proliferation. A mitogenic effect of uPA has been shown for various cells; however, its mechanism remains unclear in detail and seems to vary depending on the cell type. It was originally conceived that uPA can affect mitogenesis by a direct proteolytic- or plasmin-mediated activation of latent growth factors, such as HGF, bFGF, and TGF-beta [42-45]. However, the mitogenic effect of urokinase on certain cells such as prostate cancer cells (PC3), human glomerular epithelial cells, osteoblasts was found to be mediated simply via uPAR occupancy and does not require its catalytic activity [82, 83]. To stimulate proliferation of human fibroblasts, human kidney cells, and mesothelial cells, the interaction of uPA with uPAR and its proteolytic activity is necessary, although the effect of urokinase in these cases is not mediated by plasminogen activation [84-86]. In our laboratory, it was shown that a catalytically active uPA and plasmin generation were required for the stimulation of DNA synthesis in vascular SMC [87]. In some cases, the mitogenic effect of urokinase was seen only upon the fucosylation of its GFD at Thr18 [88]. To maintain the certain tumor cell in vivo proliferation, uPAR complexing with fibronectin and the integrin receptor of fibronectin is required [89]. Recently it was demonstrated that uPA stimulates the proliferation of fibrosarcoma HT 1080 cells by interaction with a signal complex that includes nucleolin and casein kinase 2 and by a subsequent activation of casein kinase 2 [90].
Thus, urokinase and its receptor are multifunctional proteins involved in the regulation of cell proliferation, migration, and adhesion on multiple molecular levels. At present, the available data conclusively show that many pericellular molecular and functional interactions between urokinase, its receptor, the plasminogen activator inhibitor, matrix proteins, integrins, endocytosis receptors, and also the activation of intracellular signal pathways provide strict temporal and spatial reorganization of the uPA system during cell migration. A detailed knowledge of these processes is necessary for development of the approaches for intervention in the action of this system in various diseases.
THE ROLE OF PLASMINOGEN ACTIVATORS IN VASCULAR REMODELING
Atherosclerosis. Structural remodeling of the vascular wall, both general and local, underlies the development of many cardiovascular disorders: atherosclerosis, arterial hypertension, and post-angioplasty restenoses [91]. The key processes resulting in vascular remodeling are as follows: monocyte and platelet adhesion to damaged vascular wall, invasion of the subintima by monocytes (which later differentiate into macrophages and foam cells), proliferation of vascular SMC, their migration from the media into the intima, phenotypic modulation from a contractile to the secreting state, apoptosis of vascular cells, and excessive accumulation and remodeling of extracellular matrix [92, 93]. All these processes result in a local thickening of the artery internal layer and, finally, in development of an atherosclerotic plaque or of neointima in restenosis. The plasminogen activators and their inhibitors can promote the development of all phases of atherosclerotic damage because extracellular proteolysis is involved in many aspects of atherogenesis and in the development of such complications as stroke and myocardial infarction. Extracellular proteolysis is involved in the migration of macrophages and of SMC into the intima [94, 96]; in the activation of bFGF and TGF-beta, which play important roles in atherogenesis [95, 97]; in the destruction of matrix proteins that weakens the fibrous cap of the atherosclerotic plaque [98]; in the lysis of intravascular thrombi [94, 95].
Considerable evidence supports the idea that disorders in the fibrinolytic system promote the progression of atherosclerosis [2]. Coronary artery disease is, as a rule, associated with a decrease in the blood plasma fibrinolytic capacity [2, 99], which is determined by the ratio between the plasminogen activators and their inhibitors that regulates plasmin generation. Epidemiological studies have revealed a direct correlation between PAI-1 activity and the incidence of repeated myocardial infarctions and also the severity of atherosclerotic lesions of coronary arteries [100]. A disbalance between tPA and PAI-1 seems to be associated with development of coronary artery stenosis and an increase in PAI-1 activity seems to characterize the “activity” of atherosclerotic lesions [101]. Moreover, risk factors for the development of atherosclerosis, such as obesity, insulin-independent diabetes mellitus, hyperlipidemia, and arterial hypertension are found to correlate with an increased level of PAI-1 [102]. The genetic analysis of patients with myocardial infarction has revealed a polymorphism in the PAI-1 gene promotor that can influence the frequency of thrombotic complications in these patients [100]. Our studies indicate that plasma PAI-1 activity in patients with coronary artery disease increased compared to healthy subjects of the same sex and age [103].
PAI-1 expression is also increased in atherosclerotic plaques. It is expressed by smooth muscle cells of the intima, macrophages, and foam cells concurrently with an increased expression of tissue factor, thrombin, and vitronectin [94, 104, 105]. And the level of PAI-1 mRNA correlates with the severity of atherosclerotic lesion [104]. Using immunohistochemistry, we have demonstrated the intense expression of PAI-1 by the endothelium and by the intercellular matrix in a lipid stripe [106]. The more severe lesions, such as lipofibrous plaques also display a high level of the PAI-1 expression by the endothelium that covers the plaque, and also by SMC and foam cells [94, 106]. This expression seems to be stimulated by oxidized LDL [107], native LDL, and Lp(a) [108], and also by growth factors and cytokines which are present in atherosclerotic plaques [100]. The increased PAI-1 expression by endothelial cells in atherosclerotic lesions can locally suppress fibrinolysis and thus promote thrombosis-associated complications [109], and the increased PAI-1 content inside atherosclerotic plaques seems to promote collagen deposition [95].
Thus, the suppression of fibrinolytic activity due to increased levels of PAI-1 in atherosclerotic plaques and in the blood plasma can stimulate the development and/or progression of atherosclerosis promoting thrombogenesis.
However, there is much evidence confirming an increase in plasmin-dependent proteolysis in atherosclerotic plaques. Even at the early reversible stage of the lipid stripe a sufficient number of monocyte/macrophages and of T-lymphocytes are detected in the intima [110]. These cells enter the intima by adhesion to the endothelium and migration through it into the intima. The intensity of these processes depends on the uPA system activity in these cells [111]. By our data and by data of other authors, expression of uPA and its receptor in atherosclerotic plaques is located on monocytes/macrophages and SMC of the intima and is markedly higher than their expression in a vessel region free of atherosclerotic lesions [96, 106]. Modified LDL, such as acetylated LDL, can stimulate uPA secretion by monocytes/macrophages [112]. Foam cells generated from macrophages are also characterized by high expression of uPA mRNA and by high levels of membrane-bound and of intracellular uPA [113]. These cells generate an increased “proteolytic potential” in atherosclerotic lesions. This results in releasing of matrix-bound growth factors and the activation of latent growth factors (interleukin-3, TGF-beta, granulocyte-macrophage-colony-stimulating factor (GM-CSP) [42-45]) and in the activation of zymogens of matrix metalloproteinases [46]. As a chemotactic agent, TGF-beta can stimulate the monocyte invasion into atherosclerotic lesion, whereas GM-CSP can maintain the viability of these cells. Moreover, activated TGF-beta can increase uPAR expression in monocyte/macrophages and in SMC [97].
Because binding of uPA to uPAR stimulates SMC chemotaxis [80, 87, 111], increased expression of these proteins in early atherosclerotic lesions seems to be an important factor promoting the remodeling of the vascular wall by accelerating of SMC migration. uPA mRNA expression is significantly increased in SMC of growing plaques [96].
tPA expression (mRNA and the protein) is also increased in macrophages and SMC in atherosclerotic lesions, its intensity correlating with the severity of the atherosclerotic lesions [96, 114]. The mechanism of tPA expression by intimal macrophages is unclear. Unstimulated macrophages do not produce tPA but, under certain conditions (for instance, on stimulation by lipopolysaccharide or interleukins) [94-96], they began to express tPA. The significance of this increased tPA expression remains unclear. As tPA has features of a mitogen for SMC [115], it is suggested that tPA expression should provide plaque growth. However, in tPA deficient transgenic mice with overexpression of apolipoprotein the lipid stripe development induced by a cholesterol-rich diet was the same as in the wild type animals [64].
Plaque rupture with subsequent thrombosis is the main mechanism of development of acute complications of atherosclerosis, such as myocardial infarction and unstable angina [116]. The ruptured plaques are characterized by an increased concentration of lipids and by macrophagal infiltration compared to “stable” plaques (without ruptures) [117]. Macrophages produce large amounts of tPA and uPA that may result in increased proteolytic activity on these cells and in the activation of matrix metalloproteinases with subsequent degradation of collagens and other matrix proteins. The expression of these proteases is increased in the regions of atherosclerotic plaques that are the most prone for rupture [118].
High contents of urokinase, its receptors, and of matrix metalloproteinases in macrophage-enriched “unstable” plaques suggest the pivotal role of these proteases in the proteolytic mechanisms of plaque “destabilization”. Proteolytic degradation of the extracellular matrix proteins (collagens, elastin, proteoglycans) can weaken the fibrous caps of the plaques, which can rupture under certain conditions that will cause the development of occlusive thrombosis and of its consequences.
Moreover, activation of plasmin-dependent proteolysis in an atherosclerotic plaque can promote its growth also due to stimulation of the plaque neovascularization. The neointima of atherosclerotic and restenotic lesions is shown to be provided by many microvessels, and this increased blood supply promotes the plaque growth [119]. In an atherosclerotic lesion the main angiogenic growth factors VEGF and bFGF are expressed [120]. The increased content and activity of the plasminogen activators in an atherosclerotic plaque can be favorable for angiogenesis via activation of the main angiogenic growth factors and stimulation of SMC and endothelial cell migration and proliferation.
Thus, increased expression of the plasminogen activators in early atherosclerotic lesions can promote their growth and in the advanced plaques it promotes their “destabilization” and rupture and is promising as a new target for treatment. Moreover, both the activation and the suppression of plasmin-dependent proteolysis can be involved in various mechanisms and on various stages of atherosclerotic lesion development.
Restenosis. Vascular reconstruction including coronary angioplasty, endarterectomy, bypass surgery and vascular stents have become succcessful and widely used treatments for patients with atherothrombotic disease. However, chronic restenosis in 30 to 50% of patients remains one of the major limitations of these procedures [121, 122].
The key processes in the development of restenosis are blood monocyte adhesion and migration into the damaged vascular wall, proliferation and migration of vascular smooth muscle cells and of fibroblasts from the media and adventitia into the intima, and remodeling of the extracellular matrix. These processes result in the development of a neointima narrowing the vascular lumen [93, 123]. In addition to the growth factors, these processes are regulated by protease systems, with the plasminogen activator-plasmin system playing the leading role [123, 124].
In uninjured arteries the plasminogen activating system components are expressed very weakly. The mRNA of uPA, its receptor, tPA, PAI-1, and PAI-2 are not detected in SMC or in endothelial cells of an uninjured rat artery [10, 72, 78]. But after balloon-catheter injury associated with overstretching and deendothelization of an artery, the expression of plasminogen activators is rapidly increased, with the maximum coincident with the most intense migration of SMC from the media into the neointima (4-7 days after the injury) [72, 77, 94, 95]. Both plasminogen activators and uPAR were found to be expressed also by the endothelial cells proliferating and migrating at the lesion edge. This expression ended when the migration and proliferation terminated [72].
Using immunohistochemistry we have found that the expression of uPA and uPAR in the media around SMC was very low in uninjured rat arteries [125-127]. But even 6 h after injury, the expression of these proteins increased in both the media and adventitia up to its maximum at the first proliferation wave (on the second day) and the migration of SMC (on the fourth day). It then gradually decreased to the initial level by the fourteenth day, when the proliferation and migration of vascular cells terminated and the neointima was formed. The most intense expression of uPA and uPAR by the neointimal cells was observed during the initial stage of neointima formation (4-7 days).
In an injured blood vessel, various growth factors (bFGF, PDGF, EGF, and TGF-beta) are released from damaged endothelial cells and SMC, activated monocytes/macrophages, and also from platelets adhered to the subendothelial surface [93, 123]. These growth factors can stimulate uPA expression, which is increased in the media even on the first day after the injury. uPA also can activate TGF-beta via plasmin formation and promote bFGF release from the extracellular matrix. These two factors are essential for proliferation and migration of vascular SMC and for the synthesis of matrix proteins required for reconstruction of the damaged vascular wall [97]. Furthermore the chemotactic effect of bFGF seems to be mediated by uPA because uPA-neutralizing antibodies suppress bFGF-induced migration of SMC in vitro [128].
Understanding of the role of the plasminogen system components in the neointima formation has been significantly improved by studies on uPA, tPA, PAI-1, uPAR and plasminogen knockout mice [46, 73, 129-133]. These studies have demonstrated that the rate of neointima formation after vessel injury is significantly reduced in uPA deficient mice [73, 129, 130]. The migration ability of SMC in these animals was also significantly reduced, whereas the proliferative response was unchanged. These findings suggest that the role of urokinase in the neointima formation should be mainly associated with its influence on the migration of vascular cells. Interestingly, in uPA knockout mice the expression of matrix metalloproteinases of types 2, 3, 9, 12, and 13 was less pronounced in all vessel wall layers after the injury to the artery, suggesting the involvement of urokinase in the regulation of expression of the matrix metalloproteinases, which can also influence cell migration .46, 131.. uPAR deficiency failed to affect neointima formation after vessel injury [73, 129]. Moreover, these animals were not different from animals of the wild genotype in proliferation, migration, and fibrinolytic properties of the vascular wall cells and also by the reendothelization rate. These findings show that binding uPA to uPAR is not essential for the stimulation of neointima formation.
Plasminogen-deficient mice display impaired vascular wound healing and reduced arterial neointima formation after arterial injury, suggesting that inhibition of plasmin generation might reduce arterial neointima formation. However alpha2-antiplasmin deficiency had no influence on neointima development .133.. Interestingly, that PAI-1 deficiency stimulated neointima growth after injury while not affecting cell proliferation and the reendothelization rate [132]. Concurrently the migration of smooth muscle cells was increased, and this confirms the great significance of migration in neointima formation. Surprisingly, tPA deficiency had no influence on neointima formation [130], suggesting the different roles of the two plasminogen activators in vascular wound healing.
We obtained similar results using another approach for studies on the role of plasminogen activators in vessel remodeling after intravascular trauma. In an attempt to understand the contribution of uPA proteolytic property and uPA receptor binding property to it's ability to augment early neointima formation we used perivascular application to the balloon injured rat carotid artery of pluronic gel with proteolytically active and proteolytically inactive forms of recombinant uPA (r-uPA). These forms were produced in the Laboratory of Gene Engineering of the Cardiology Center by expression in E. coli and then purified and characterized in our laboratory [23, 87, 125-127]. The following forms of r-uPA were used: wild-type r-uPA, catalytically inactive r-uPA with Glu instead of His in the active center (r-uPA H/Q), proteolytically active r-uPA containing multiple mutations in its growth factor domain, which functionally does not induce cell migration via receptor-dependent mechanisms. The effects of other proteins such as tPA, alpha2-antiplasmin, anti-uPA antibodies were also studied. Perivascular application of wild-type r-uPA or r-uPA containing multiple mutations in its growth factor domain doubled the size of the neointima, induced a thickening of the media and adventitia, and caused greater reductions in vessel lumen size than injury alone (figure). In contrast to these two proteolytically active forms, the proteolytically inactive r-uPA did not stimulate the neointima and neoadventitia formation and prevented the media thickening and the decrease in the artery lumen in response to the injury. The effect of this proteolytically inactive r-uPA resembled the effect of the uPA neutralizing antibodies that suppressed neointima growth and prevented the narrowing of the artery lumen after ballooning. The stimulation of the neointima development by uPA seems to be due to its effect on vascular cell migration because the cell number in the neointima was doubled during the most intense migration from the media. The importance of endogenous uPA for the SMC migration from the media into the neointima is confirmed by the effect of uPA-neutralizing antibodies, which caused twofold decrease in the neointimal cell number without affecting the proliferation index [125-127]. Thus, our findings show in vivo that the proteolytic property of uPA is the major contributor to its ability to augment neointima formation early after balloon catheter injury to the artery.
The development of restenosis after angioplasty is a result not only of an excessive neointima growth but also of changes in the artery diameter, i.e., of the geometric remodeling of the artery, which can vary at the same size of the neointima [93, 134]. Clinical and experimental studies have shown that the remodeling of the artery mainly determines the development of restenosis after angioplasty [93]. The compensatory vessel enlargement (positive remodeling) limits the effect of neointima growth on lumen narrowing. Whereas failure of the artery to enlarge adequately after angioplasty or reduction in the artery diameter (“late recoil”, negative or constrictive remodeling) is sufficient to produce restenosis .134.. The mechanisms of the geometric remodeling of injured artery are far from being fully understood. Changes in the vascular adventitia and in the structure and composition of the extracellular matrix probably play an important role in this remodeling [135, 136].Changes in the structure of rat carotid artery after balloon catheter injury and application of recombinant forms of uPA (wild-type uPA (uPA) and inactive uPA (uPA H/Q)), of uPA-neutralizing antibodies, and of recombinant tissue plasminogen activator (tPA) (in the control the pure gel was applied): a) effects of the substances on changes in the neointimal area; b) on changes in the artery lumen area; c) on changes in the area encircled by the external elastic lamina (EEL); * p < 0.05 (difference from control)
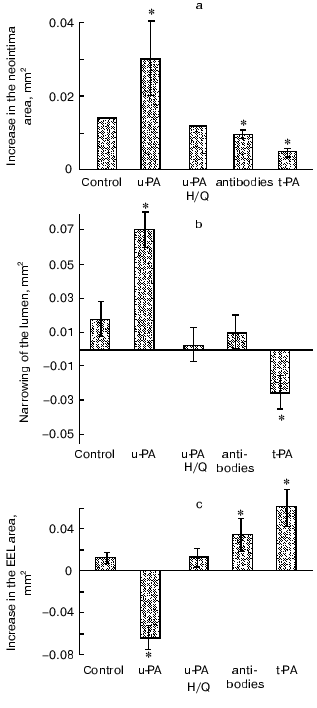
We have demonstrated that uPA induces early negative remodeling of injured vessel additionally to its stimulatory effect on early neointima formation. This is shown by the decrease in the area encircled by the external elastic lamina (figure). A putative mechanisms of this unfavorable remodeling may be represented by the adventitial response to the injury and to the uPA application, namely by generation of a rigid fibrous neoadventitia that squeezes the vessel, preventing its compensatory enlargement. We have found that r-uPA application stimulates the accumulation of alpha-actin positive cells in injured adventitia and increases the size of the adventitia .137., whereas the uPA-neutralizing antibodies decreased the number of cells with contractile phenotype and suppressed the adventitia growth in response to the injury. In fibroblast culture, r-uPA stimulates alpha-actin expression in dose-dependent manner.
All these findings show that uPA that is expressed in the vessel after injury plays an important and possibly the key role in the development of artery stenosis due to its influence on both the formation of excessive neointima and the formation of neoadventitia enriched with contractile phenotype cells.
How specific are the effects of uPA in vascular wound healing? Studies with tPA knockout mice suggest that tPA is not essential for neointima development [130]. Using our experimental approach we have found that uPA and tPA had the opposite effect on early neointima formation and vessel remodeling. In contrast to uPA effects, tPA application reduced neointimal growth, attenuated lumen stenosis, and caused positive vessel remodeling .138. (figure). The mechanism underlying this difference is not clear and needs to be studied. However, together with data obtained on transgenic mice, these findings support the specific significance of uPA for the formation of artery stenoses after injury and suggest that uPA and its proteolytic activity should be a promising new target for measures to prevent restenoses. Our clinical data [139] and data of other authors [140] are in good agreement with these experimental results. According to these data, an increased plasma level of uPA in patients with coronary artery disease is an independent predictor of the restenosis development after coronary angioplasty and stenting.
It has been recently reported that the administration of replication-defective adenovirus expressing chimeric protein consisting of the amino terminal fragment of urokinase and of bovine inhibitor of trypsin markedly suppressed neointima development in mice [141]. The resulting protein bound to the uPAR and inhibited the activation of plasminogen on the cell surface and also prevented the binding of endogenous uPA to the receptor. These findings seem promising for the development of a genetic approach for restenosis prevention using cDNA of the mutant urokinase forms, e.g., of the proteolytically inactive urokinase.
Angiogenesis. Tissue neovascularization provided by generation of new vessels is important in various physiological processes and characterizes various diseases: malignant tumors, hemangiomas, arthritis, retinopathy [142-144]. Tissue neovascularization includes vasculogenesis, i.e., vessel formation by differentiation of cell precursors, angioblasts and stem cells into endothelial cells, angiogenesis, i.e., the sprouting of new capillaries from the preexisting vasculature, and arteriogenesis, i.e., the remodeling of capillaries or of pre-existing collateral vessels into muscle arteries. Until recently, neovascularization by differentiation of cell precusors was thought to occur only during embryogenesis, but it has now been shown that all three processes may be involved in tissue revascularization in the adult organism [145]. To initiate angiogenesis, vessel destabilization is necessary that includes the weakening of intercellular contacts between the endothelial cells, basement membrane destruction, and also local proteolysis of the matrix proteins to allow the endothelial cells to migrate and produce new capillaries.
Under natural conditions, ischemia (hypoxia) and inflammation [146] are the major triggers of angiogenesis, and the angiogenic factors, especially VEGF, bFGF, HGF, TGF-beta, and PDGF inducing endothelial cell proliferation and migration, play the key role in the process [145-147]. Angiogenesis is impossible without activation of extracellular proteolysis that occurs due to uPA-catalyzed generation of plasmin on the cell surface. Plasmin and uPA located in certain regions of the endothelial cell surface initiate a directed degradation of the basement membrane proteins such as fibronectin and laminin. Both uPA itself and uPA-generated plasmin can activate the latent matrix metalloproteinases and elastase [46, 148], which are responsible for the subsequent degradation of the extracellular matrix, during the endothelial cell migration and invasion. Moreover these two proteases activate virtually all growth factors involved in angiogenesis [42-45, 147, 149]. Hypoxia has been shown to increase the expression of angiogenic growth factors, but uPA and plasmin are required to provide proteolytic activation of the latent forms or release of matrix-bound growth factors.
The infiltration of tissue by monocytes/macrophages secreting growth factors and proteases is known to stimulate angiogenesis [150, 151]. Macrophage invasion is also regulated by urokinase and its receptor [61, 76, 81].
The role of uPA in angiogenic processes was initially observed in 1982 in models of corneal revascularization [152]. During the last decade, the involvement of uPA and uPAR in angiogenesis was shown both clinically and experimentally. Quiescent endothelial cells did not normally express uPAR, but being “activated” in vitro by dispersing and plating them onto plastic they started the expression of various genes including the uPAR gene [149]. Endothelial cells engineered to overexpress uPA are characterized by higher in vitro invasiveness than intact cells [153].
Urokinase receptor expression by endothelial cells localizing uPA on cell surface is an important moment in the initiation of angiogenesis. Hypoxia, which is important for the in vivo stimulation of angiogenesis, has been shown to induce both the expression VEGF receptors and of urokinase receptor in umbilical vein endothelial cell culture (HUVEC) [154, 155]. The pro-angiogenic growth factors bFGF and VEGF induce in vitro migration of endothelial cells and up-regulate uPAR [155-157]. To induce angiogenesis, growth factors need the proteolytically active uPA bound to uPAR [158, 159]. Urokinase can mediate the angiogenic effect of some growth factors not only by the localization of cell surface proteolysis but also by non-proteolytic mechanisms [160]. A fusion protein consisting of the amino terminal fragment of urokinase and of human serum albumin inhibits endothelial cell in vitro migration induced by bFGF and by VEGF [161], whereas a fusion protein consisting of the mouse uPA GFD and of the Fc-fragment of IgG inhibits bFGF and VEGF-stimulated branching morphogenesis in an endothelial cell tube formation assay on a fibrin matrix [162].
Retinoids stimulate endothelial cell tube formation also on fibrin matrices and addition of uPA to this system enhances this stimulation, whereas an antibody that blocked urokinase binding to the receptor inhibits the effects of uPA. In the same system, steroid hormones (testosterone, dexamethasone) inhibit tube formation and addition of uPA to the system prevents the effect of steroids [163].
Hormone-dependent tumors are known to become hormone-independent after hormone-ablation therapy and, as a rule, more aggressive, and this is associated with increased expression of uPA and uPAR [164]. Thus, the viability of Shionogi carcinoma cells grown in a dorsal skinfold chamber in immunodeficient mice is supported by androgens. Castration of the animals results in apoptosis of the tumor cells and in a diminution of the tumor vascularization. However, some weeks later the tumor begins to actively grow again, and urokinase and its receptor expression in it increase concurrently with the activation of angiogenesis [165].
It was shown that the soluble uPAR or uPA-neutralizing antibody nearly completely suppressed the formation of capillary-like tubes in a fibrin matrix [166]. Moreover, an antibody that blocked the binding of uPA to uPAR suppressed bFGF- and VEGF-induced tube formation in fibrin matrix, despite the fact that sufficient uPA was present in the conditioned medium around the cells. The amino terminal fragment of urokinase, which binds to uPAR with high affinity but lacks proteolytic activity, also suppressed tube formation in fibrin matrix, possibly due to competition with the endogenous uPA for the binding to uPAR [167].
Taken together, these findings have demonstrated that the uPAR localized cell surface proteolysis plays an important role in tubular angiogenesis. Targeting the uPA-uPAR system may be useful to affect these early events in angiogenesis. This viewpoint is in accordance with the finding that angiogenesis occurring in certain tumors in vivo can be reduced by administration of a catalytically inactive uPA, which retains receptor binding and thus competes for binding of native uPA [168].
The expression of urokinase and its receptor have been shown immunohistochemically in endothelial cells involved in formation of tubules in fibrin matrices [167]. The fact that a few cells in the unstimulated monolayer are positive for uPAR may reflect the existence of a subpopulation of cells that are more able to invade the fibrin matrix if adequate stimuli, which induce uPA expression are provided [169].
Beside a function in controling of spatially focalized degradation of the basement membrane uPA bound to uPAR influences angiogenesis also by the activation of intracellular signal systems in endothelial cells. Thus, it activates MAP-kinases and the phosphorylation of focal adhesion proteins [170]. Recently it was shown that urokinase-dependent angiogenesis in vitro and diacylglycerol production are blocked by antisense oligonucleotides which target uPAR expression [171]. In this study, the down-regulation of the uPAR expression seemed to inhibit signaling pathways depending on the uPA proteolytic activity and also on the binding to the uPAR. The uPA catalytic activity seemed to activate protein kinase C through the matrix destruction and the disruption of integrin ligation, whereas the uPAR occupation activated the MAP-kinase pathway. The two pathways can be simultaneously involved during the migration and invasion of endothelial cells. Therefore, an antagonist capable of inhibiting these two pathways is suggested to display an anti-angiogenic activity.
A complicated and multistep formation of new vessels requires the balance between a protease and its inhibitor to be strictly regulated in time and space because excessive proteolytic activity in endothelial cell would more likely lead to hemangioma rather than capillary formation [172]. In some studies, the pro-angiogenic role of the plasminogen activator inhibitor (PAI-1) is shown. It is also expressed by endothelial cells placed into the matrigel (a reconstituted basement membrane) and is required for proper tube formation. [172]. The pro-angiogenic role of PAI-1 is mainly based on the promotion of fibrin deposition by inhibiting plasminogen activation. Fibrin production and deposition in tumors and during inflammation is associated with angiogenesis providing a matrix for the migration of endothelial cells [149, 173]. PAI-1 is actively expressed in fibroblasts contacting endothelial cells [174]. Such contact during angiogenesis can only occur after the degradation of the basement membrane in the process of endothelial cell migration. Tumor growth and angiogenesis are also suppressed in vivo in PAI-1 deficient mice, suggesting the potential importance of PAI-1 in tumor angiogenesis [173]. In various malignant tumors including breast cancer, the combination of high levels of uPA and PAI-1 indicates the poorest prognosis [175]. This suggests that synergic effects of PAI-1 and uPA can promote tumor angiogenesis and metastasis. It seems that the tumor growth and invasion need both the profibrinolytic and procoagulant activities triggered simultaneously in different regions of the tumor or in different cells. PAI-1 expression in tumor cells is stimulated by TGF-beta which is itself activated by uPA-generated plasmin. The TGF-beta inhibits in vitro proliferation of endothelial cells inducing their apoptosis but stimulates in vivo angiogenesis [176]. TGF-beta also stimulates VEGF expression in fibroblasts, especially under conditions of hypoxia that often occurs in tumors or in tissue ischemia caused by disorders in the main blood flow [177]. The combination of VEGF with fibrin generated by PAI-1-induced suppression of uPA-dependent fibrinolysis can also stimulate migration and proliferation of endothelial cells and angiogenesis.
Recanalization of a mural thrombus associated with an atherosclerotic plaque is a pathological conditions in which neovascularization occurs upon fibrin degradation [178]. Indeed, the uPAR and uPA are expressed in the neovessels supplying an organized fibrin-enriched neointima.
During physiological non-inflammatory angiogenesis in women's reproductive tract the concurrent expression of the uPA and PAI-1 promotes the growth of new vessels and the protection of neovascularized tissues against excessive proteolysis [179]. This also suggests that during the new vessel formation procoagulant and fibrinolytic activities can concurrently exist in different regions of a tissue or of a cell or cyclically replace each other.
In uPA deficient mice, wound healing is impaired at least partly due to suppressed migration of leukocytes, fibroblasts, and smooth muscle cells. However, the denuded vessel re-endothelization in mice deficient in urokinase, its receptor, PAI-1 and plasminogen is unchanged [129-133], suggesting that a lack of fibrinolytic components in transgenic mice probably results in fibrin deposition that stimulates angiogenesis [180]. This finding is often used as an argument in favor of limited importance of the uPA system as a target for treatment pointed to angiogenesis. However, this is not equivalent to effects of inhibition of the same protease activity or receptor in an adult organist because the compensatory mechanisms can be activated during animal development and providing animal's survival. Moreover, the importance of uPA for myocardial angiogenesis has been recently shown. Thus, the peri-infarction angiogenesis was significantly impared in uPA-deficient mice and failed to be stimulated by VEGF, whereas in the wild type mice VEGF displayed a stimulating effect [181].
However, the key significance of the uPA system has been best established for tumor angiogenesis. Recently, adenovirus-mediated delivery of murine ATF directly into the human tumor transplanted into mice suppressed the neovascularization of this tumor and arrested its growth [182]. Because the murine ATF fails to bind to the uPAR of human tumor cells but can interact only with endothelial cells and leukocytes of the host, the tumor growth suppression seems to be mediated only by suppression of angiogenesis of the host (mouse). Immunohistochemical studies have also shown that the uPAR expression may be associated with the most “aggressive” cells within a tumor and detected mostly at the invasive edges of a tumor and, might therefore be a marker of invasive disease, tumor angiogenesis, and metastasis [183, 184].
However, uPA can be also involved in the processes supressing angiogenesis. Thus, the proteolytic cleavage of urokinase between Lys135-Lys136 by plasmin or by tc-uPA or the cleavage of the Glu143-Leu144 bond by some metalloproteinases results in the generation of a so-called connecting peptide consisting of residues 136-143 [185]. This peptide inhibits the interaction of uPA with uPAR, suppresses endothelial cell migration stimulated by bFGF, and markedly suppresses in vivo angiogenesis. This peptide also inhibits tumor cell invasion and, as a result, markedly attenuates the growth and metastasizing of tumors [186]. This peptide is especially effective when used together with cytotoxic agents inhibiting endothelial cell proliferation [187].
The involvement of uPA in plasmin proteolytic degradation with the generation of an angiogenesis inhibitor angiostatin was recently shown in human pancreatic cancer cells [188]. Angiostatin (plasminogen kringles 1-4) is produced in vivo in primary tumors and, entering the blood circulation, it is believed to suppress metastasizing [189]. The induction of apoptosis and the inhibition of endothelial cells migration by angiostatin has recently been described [190]. However, molecular mechanisms of its effect remain unclear. It is well known that the primary tumor ablation often results in increasing metastasis growth. This is probably due to disappearance of angiostatin produced by the primary tumor from the circulation. Quite recently, it has been shown that the fifth kringle domain of plasminogen inhibits the proliferation and migration of endothelial cells even more potently than angiostatin. This fragment of plasminogen is generated under the influence of macrophages elastase [191, 192].
The findings presented suggest the involvement of uPA, uPAR, and PAI-1 in the regulation of various events that determine the new vessel formation in inflammation, tissue ischemia, and in malignant tumors. Intervention in the functions of this system seems promising for suppression of angiogenesis in tumors and in atherosclerotic plaques and also for stimulation of angiogenesis in an ischemia region. But for such intervention the complicated interactions of profibrinolytic and procoagulation mechanisms must be taken into account.
Conclusion. The uPA-system is a key system responsible for a strict temporal and spatial extracellular proteolysis necessary for tissue remodeling under various physiological and pathological conditions. It regulates the migration and proliferation of vascular smooth muscle and endothelial cells, the adhesion and migration of blood monocytes, and the remodeling of the extracellular matrix. It is involved in vascular remodeling during the development of atherosclerosis and restenosis and also in the regulation of angiogenesis. An excessive local activation of this system in the damaged region of a vessel after angioplasty can promote the development of restenosis. Its activation in advanced atherosclerotic plaques enriched with lipids can promote the rupture of such plaques and the development of thrombotic complications, such as myocardial infarction and unstable angina. An excessive expression of this system in tumors can stimulate tumor neoangiogenesis, which promotes the tumor growth and metastasis. In all these cases the uPA system seems to be a promising target for therapeutic interventions designed to locally suppress its activity in a vascular wall or in tumors. On the other hand, the role of urokinase in angiogenesis suggests that a transgenic overexpression of uPA probably can be used together with growth factors to stimulate the development of new vessels in ischemic regions. Significant recent progress in the comprehension of the molecular mechanisms of the involvement of plasminogen activators-plasmin system in tissues remodeling is promising for therapeutic interventions of the activity of this system in various diseases.
REFERENCES
1.Vassalli, J.-D., Sappino, A.-P., and Belin, D.-J.
(1991) Clin. Invest., 88, 1067-1072.
2.Saksela, O. (1985) Biochim. Biophys. Acta,
823, 35-65.
3.Panchenko, E. P., and Dobrovolsky, A. B. (1999)
Sport and Culture [in Russian], Moscow.
4.Vassalli, J.-D. (1994) Fibrinolysis,
8, 172-181.
5.Saksela, O., and Vihko, K. K. (1986) FEBS
Lett., 204, 193-197.
6.Nakajima, K., Tsuzaki, N., Nagata, K., Takemoto,
N., and Kohsaka, S. (1992) FEBS Lett., 308, 179-182.
7.Sappino, A.-P., Madani, R., Huarte, J., Belin, D.,
Kiss, J. Z., Wohlwend, A., and Vassalli, J.-D. (1993) J. Clin.
Invest., 92, 679-685.
8.Manchanda, N., and Schwartz, B. S. (1995) J.
Biol. Chem., 270, 20032-20035.
9.Reinartz, J., Schaefer, B., Bechtel, M. J., and
Kramer, M. D. (1996) Exp. Cell. Res., 223, 91-101.
10.Clowes, A. W., Clowes, M. M., Au, Y. P., Reidy,
M. A., and Belin, D. (1990) Circ. Res., 67, 61-67.
11.Pepper, M. S., Sappino, A.-P., Stöcklin, R.,
Montesano, R., Orci, L., and Vassalli, J.-D. (1993) J. Cell.
Biol., 122, 673-684.
12.Vassalli, J.-D., Dayer, J.-M., Wohlwend, A., and
Belin, D. (1984) J. Exp. Med., 159, 1653-1668.
13.Canipari, R., O'Connell, M. L., Meyer, G., and
Strickland, S. (1987) J. Cell. Biol., 105, 977-981.
14.Leslie, N. D., Kessler, C. A., Bell, S. M., and
Degen, J. L. (1990) J. Biol. Chem., 265, 1339-1344.
15.Sappino, A.-P., Huarte, J., Vassalli, J.-D., and
Belin, D. (1991) J. Clin. Invest., 87, 962-970.
16.Ossowski, L., Clunie, G., Masucci, M.-T., and
Blasi, F. (1991) J. Cell. Biol., 115, 1107-1112.
17.Huarte, J., Belin, D., Bosco, D., Sappino, A.,
and Vassalli, J. D. (1987) J. Cell. Biol., 104,
1281-1289.
18.Mazar, A. P., Henkin, Y., and Goldfarb, R. H.
(1999) Angiogenesis, 153, 15-32.
19.Andreasen, P. A., Kjoller, L., Christensen, L.,
and Duffy, M. (1997) Int. J. Cancer, 72, 1-22.
20.Holmes, W. E., Pennica, D., Blaber, M., Rey, M.
W., Guenzler, W. A., Steffens, G. L., and Heyneker, H. L. (1985)
Biotechnology, 3, 923-929.
21.Appella, E., Robinson, E. A., and Ullrich, S. J.
(1987) J. Biol. Chem., 262, 4437-4440.
22.Stephens, R. W., Bokman, A. M., Myohanen, H. T.,
Reisberg, T., Tapiovaara, H., Pedersen, N., Gröndahl-Hansen, J.,
Llinás, M., and Vaheri, A. (1992) Biochemistry,
31, 7572-7579.
23.Mukhina, S., Stepanova, V., Traktouev, D.,
Poliakov, A., Beabealashvilly, R., Gursky, Ya., Minashkin, M.,
Shevelev, A., and Tkachuk, V. (2000) J. Biol. Chem., 275,
16450-16458.
24.Madison, E. L. (1994) Fibrinolysis,
8, 221-236.
25.Besser, D., Verde, P., Nagamine, Y., and Blasi,
F. (1996) Fibrinolysis, 10, 215-237.
26.Ried, S., Jager, C., Jeffers, M., van de Woude,
G. F., Graeff, H., Schmitt, M., and Lengyel, E. (1999) J. Biol.
Chem., 274, 16377-16386.
27.Stoppelli, M. P., Verde, P., Grimaldi, G.,
Locatelli, E. K., and Blasi, F. (1986) J. Cell. Biol.,
102, 1235-1241.
28.Pearson, D., Altus, M. S., Horiuchi, A., and
Nagamine, Y. (1987) Biochem. Biophys. Res. Commun., 143,
329-336.
29.Kenagy, R. D., and Clowes, A. W. (1995)
Thromb. Res., 77, 55-61.
30.Van Leeuwen, R. T., Kol, A., Andreotti, F.,
Kluft, C., Maseri, A., and Sperti, G. (1994) Circulation,
90, 362-368.
31.Irigoyen, J., Munoz-Canoves, P., and Montero, L.
(1999) Cell. Mol. Life Sci., 56, 104-132.
32.Lijnen, H. R., Zamarron, C., Blaber, M., Winkler,
M. E., and Collen, D. (1990) J. Biol. Chem., 265,
5232-5236.
33.Moller, L. B., Pollanen, J., Ronne, E., Pedersen,
N., and Blasi, F. (1993) J. Biol. Chem.,
268,11152-11159.
34.Andreasen, P. A., Kjoller, L., Christensen, L.,
and Duffy, M. J. (1997) Int. J. Cancer, 72, 1-22.
35.Ellis, V., Scully, M. F., and Kakkar, V. V.
(1989) J. Biol. Chem., 264, 2185-2188.
36.Gliemann, J. (1998) J. Biol. Chem.,
379, 951-964.
37.Stefansson, S., Kounnas, M. Z., Henkin, J.,
Mallampalli, R. K., Chappell, D. A., Strickland, D. K., and Argraves,
W. S. (1995) J. Cell. Sci., 108, 2361-2368.
38.Argraves, K. M., Battey, F. D., MacCalman, C. D.,
McCrae, K. R., Gafvels, M., Kozarsky, K. F., Chappell, D. A., Strauss,
J. F., III, and Strickland, D. K. (1995) J. Biol. Chem.,
270, 26550-26557.
39.Pepper, M. S., Ferrora, N., Orci, I., and
Montesano, R. (1991) Biochem. Biophys. Res. Commun., 181,
902-906.
40.Ellis, V., and Whawell, S. A. (1997)
Blood, 90, 2312-2322.
41.Fears, R. (1989) Biochem. J., 261,
313-324.
42.Saksela, O., and Rifkin, D. B. (1990) J. Cell.
Biol., 110, 767-775.
43.Naldini, L., Tamagnone, L., and Vigna, E. (1992)
EMBO J., 11, 4825-4833.
44.Plouet, J., Moro, F., and Bertagnolli, S. (1997)
J. Biol. Chem., 272, 13390-13396.
45.Godar, S., Horejsi, V., and Weidle, U. H. (1999)
Eur. J. Immunol., 29, 1004-1013.
46.Lijnen, H. R., van Hoef, B., Lupu, F., Moons, L.,
Carmeliet, P., and Collen, D. (1998) Arterioscler. Thromb. Vasc.
Biol., 18, 1035-1045.
47.Dumler, I., Weis, A., Mayboroda, O. A., Maasch,
C., Jerke, U., Haller, H., and Gulba, D. C. (1998) J. Biol.
Chem., 273, 315-321.
48.Dumler, I., Kopmann, A., Weis, A., Mayboroda, O.
A., Wagner, K., Gulba, D. C., and Haller, H. (1999) Arterioscler.
Thromb. Vasc. Biol., 19, 290-297.
49.Resnati, M., Guttinger, M., Valcamonica, S.,
Sidenius, N., Blasi, F., and Fazioli, F. (1996) EMBO J.,
15, 1572-1582.
50.Konakova, M., Hucho, F., and Schleuning, W. D.
(1998) Eur. J. Biochem., 253, 421-429.
51.Nguyen, D. H., Hussaini, I. M., and Gonias, S. L.
(1998) J. Biol. Chem., 273, 8502-8507.
52.Tang, H., Kerins, D. M., Hao, Q., Inagami, T.,
and Vaughan, D. E. (1998) J. Biol. Chem., 273,
18268-18272.
53.Dumler, I., Petri, T., and Schleuning, W.-D.
(1993) FEBS Lett., 322,37-40.
54.Dear, A. E., Costa, M., and Medcalf, R. L. (1997)
FEBS Lett., 402, 265-272.
55.Rabbani, S. A., Gladu, J., Mazar, A. P., Henkin,
J., and Goltzman, D. (1997) J. Cell. Physiol., 172,
137-145.
56.Goretzki, L., and Mueller, B. M. (1998)
Biochem. J., 336, 381-386.
57.Simon, D. I., Wei, Y., Zhang, L., Rao, N. K., Xu,
H., Chen, Z., Liu, Q., Rosenberg, S., and Chapman, H. A. (2000) J.
Biol. Chem., 275, 10228-10234.
58.Kanse, S. M., Kos, C., Wilhelm, O. G., Andreasen,
P. A., and Preissner, K. T. (1996) Exp. Cell. Res., 224,
344-353.
59.Deng, G., Curriden, S. A., Wang, S., Rosenberg,
S., and Loskutoff, D. J. (1996) J. Cell. Biol.,
134,1563-1571.
60.Hoyer-Hansen, G., Behrendt, N., Ploug, M., Dano,
K., and Preissner, K. T. (1997) FEBS Lett., 420,
79-85.
61.Sitrin, R. G., Pan, P. M., Harper, H. A., Todd,
R. F., III, Harsh, D. M., and Blackwood, R. A. (2000) J.
Immunol., 165, 3341-3349.
62.May, A. E., Kanse, S. M., Lund, L. R., Gisler, R.
H., Imhof, B. A., and Preissner, K. T. (1998) J. Exp. Med.,
188,1029-1037.
63.Ghosh, S., Brown, R., Jones, J. C., Ellerbroek,
S. M., and Stack, M. S. (2000) J. Biol. Chem., 275,
23869-23876.
64.Carmeliet, P., and Collen, D. (1994)
Fibrinolysis, 8, 269-276.
65.Estreicher, A., Muhlhauser, J., Carpentier,
J.-L., Orci, L., and Vassalli, J.-D. (1990) J. Cell. Biol.,
111, 783-792.
66.Mawatari, O., Okamura, K., Matsuda, T., Hamanaka,
R., Mizoguchi, H., Higashio, K., Kohno, K., and Kuwano, M. (1991)
Exp. Cell. Res., 192, 574-580.
67.Odekon, L. E., Sato, Y., and Rifkin, D. B. (1992)
J. Cell. Physiol., 150, 258-263.
68.Morimoto, K., Mishima, H., Nishida, T., and
Otori, T. (1993) Thromb. Haemost., 69, 387-391.
69.Okada, S. S., Tomaszewski, J. A., and Barnathan,
E. S. (1995) Exp. Cell. Res., 217, 180-187.
70.Quax, P. H. A., Boxman, I. L. A., van Kestereen,
C. A. M., Verheuen, J. H., and Ponec, M. (1994) Fibrinolysis,
8, 221-228.
71.Pepper, M. S., Sappino, A.-P., Stocklin, R.,
Montesano, R., Orci, L., and Vassalli, J.-D. (1993) J. Cell.
Biol., 122, 673-684.
72.Reidy, M. A., Colleen, I., and Lindner, V. (1996)
Circ. Res., 78, 405-414.
73.Carmeliet, P., and Callen, D. (1996)
Haemostasis, 26, 132-153.
74.Ossowski, L. (1992) Cancer Res.,
52, 6754-6760.
75.Okada, S. S., Grobmeyer, S. R., and Barmathan, E.
S. (1996) Arterioscler. Thromb. Vasc. Biol., 16,
1269-1276.
76.Wang, N., Planus, E., Pouchelet, M., Fredberg, J.
J., and Barlovatz-Meimon, G. (1995) Am. J. Physiol., 268,
C1062-C1066.
77.Jackson, C. L., and Reidy, M. A. (1992) Ann.
N. Y. Acad. Sci., 667, 141-150.
78.Clowes, A. W., Clowes, M. M., Kirkman, T. R.,
Jackson, C. L., Au, Y. P. T., and Kenagy, R. (1992) Circ. Res.,
70, 1128-1136.
79.Lu, H., Mabilat, C., Yeh, P., Guitton, J.-D., Li,
H., Pouchelet, M., Shoevaert, D., Legrand, Y., Soria, J., and Soria, C.
(1996) FEBS Lett., 380, 21-24.
80.Stepanova, V., Bobik, A., Bibilashvily, R.,
Belogurov, A., Rybalkin, I., Domogatsky, S., Little, P. J., Goncharova,
E., and Tkachuk, V. (1997) FEBS Lett., 414, 417-474.
81.Gyetko, M. P., Todd, R. R., Wilkinson, C. C., and
Sitrin, R. G. (1994) J. Clin. Invest., 93, 1380-1387.
82.He, C. J., Rebibou, J. M., Peraldy, M. N.,
Meulders, Q., and Rondeau, E. (1991) Biochem. Biophys. Res.
Commun., 176, 1408-1416.
83.Rabbani, S. A., Desjardin, J., Bell, A. W.,
Banville, D., Mazar, A., Henkin, J., and Goltzman, D. (1990)
Biochem. Biophys. Res. Commun., 173, 1058-1064.
84.Anichini, E., Fibbi, G., Pucci, M., Chevanne, M.,
and del Rosso, M. (1994) Exp. Cell. Res., 213,
438-448.
85.De Petro, G., Copeta, A., and Barlati, S. (1994)
Exp. Cell. Res., 213, 286-294.
86.Shetty, S., Kumar, A., Johnson, A., Pueblitz, S.,
and Idell, S. (1995) Am. J. Physiol., 268, L972-L982.
87.Stepanova, V., Mukhina, S., and Kohler, E. (1999)
Mol. Cell. Biochem., 195, 199-206.
88.Rabbani, S. A., Mazar, A., Bernier, S. M., Haq,
M., Bolivar, I., Henkin, J., and Goltzman, D. (1992) J. Biol.
Chem., 267, 14151-14156.
89.Ossowski, L., Aguirre Ghiso, J., Liu, D., Yu, W.,
and Kovalski, K. (1999) Medicina, 59, 547-552.
90.Dumler, I., Stepanova, V., Jerke, U., Mayboroda,
O. A., Vogel, F., Bouvet, P., Tkachuk, V., Haller, H., and Gulba, D. C.
(1999) Curr. Biol., 9, 1468-1476.
91.Langille, B. L. (1993) J. Cardiovasc.
Pharmacol., 21, S11-S17.
92.Cowan, D. B., and Langille, B. L. (1996) Curr.
Opin. Lipidol., 7, 94-100.
93.Schwartz, R. S. (1998) Am. J. Cardiol.,
81, 14E-17E.
94.Lupu, F., Bergonzelli, G. E., Heim, D. A.,
Cousin, E., Genton, C. Y., Bachmann, F., and Kruithof, E. K. (1993)
Arterioscler. Thromb., 13, 1090-1100.
95.Lupu, F., Heim, D. A., Bachmann, F., Hurni, M.,
Kakkar, V. V., and Kruithof, E. K. (1995) Arterioscler. Thromb.
Vasc. Biol., 15, 1444-1455.
96.Raghunath, P. N., Tomaszewski, J. E., Brady, S.
T., Caron, R. J., Okada, S. S., and Barnathan, E. S. (1995)
Arterioscler. Thromb. Vasc. Biol., 15, 1432-1443.
97.Agrotis, A., and Bobik, A. (1996) Clin. Exp.
Pharmacol. Physiol., 23, 363-380.
98.Galis, Z. S., Sukhova, G. K., Lark, M. W., and
Libby, P. (1994) Clin. Invest., 94, 2493-2503.
99.Van der Bom, J. G., de Knijff, P., Haverkate, F.,
Bots, M. L., Meijer, P., de Jong, P. T., Hofman, A., Kluft, C., and
Grobbee, D. E. (1997) Circulation, 95, 2623-2627.
100.Hamsten, A., Eriksson, P., Karpe, F., and
Silveira, A. (1994) Curr. Opin. Lipidol., 5, 382-389.
101.Zateishchikov, D. A., Averkov, O. V.,
Gratsiansky, N. A., Dobrovolsky, A. B., and Storozhilova, A. N. (1992)
Kardiologiya, 32, 9-12.
102.Juhan-Vague, I., and Alessi, M. C. (1997)
Thromb. Haemost., 78, 656-660.
103.Krasnikova, T. L., Parfyonova, Ye. V.,
Alekseeva, I. A., Arefieva, T. I., Mukhina, S. A., Dobrovolsky, A. B.,
Titaeva, Y., Lyakishev, A. A., Resink, T. J., Erne, P., and Tkachuk, V.
A. (1999) Clin. Exp. Pharmacol. Physiol., 26,
354-357.
104.Schneiderman, J., Sawdey, M. S., Keeton, M. R.,
Bordin, G. M., Bernstein, E. F., Dilley, R. B., and Loskutoff, D. J.
(1992) Proc. Natl. Acad. Sci. USA, 89, 6998-7002.
105.Chomiki, N., Henry, M., Alessi, M. C., Anfosso,
F., and Juhan-Vague, I. (1994) Thromb. Haemost., 72,
44-53.
106.Plekhanova, O. S. (2000) Author's abstract of
Candidate's dissertation [in Russian], Russian Cardiology Research
Center, Moscow.
107.Kugivama, K., Sakamoto, T., Misumi, I.,
Sugiyama, S., Ohgushi, M., Ogawa, H., Horiguchi, M., and Yasue, H.
(1993) Circ. Res., 73, 335-343.
108.Tremoli, E., Camera, M., Maderna, P., Sironi,
L., Prati, L., Colli, S., Piovella, F., Bernini, F., Corsini, A., and
Mussoni, L. (1993) Atheroscler. Thromb., 13, 338-346.
109.Loskutoff, D. J., Curriden, S. A., Hu, G., and
Deng, G. (1999) APMIS, 107, 54-61.
110.Stary, H. C. (1989) Arteriosclerosis,
9, 119-132.
111.Blasi, F. (1999) Thromb. Haemost.,
82, 298-304.
112.Falcone, D. J., and Ferenc, M. J. (1988) J.
Cell. Physiol., 135, 387-396.
113.Falkenberg, M., Giglio, D., Bjornheden, T.,
Nygren, H., and Risberg, B. (1998) J. Vasc. Res., 35,
318-324.
114.Steins, M. B., Padro, T., Li, C. X., Mesters,
R. M., Ostermann, H., Hammel, D., Scheld, H. H., Berdel, W. E., and
Kienast, J. (1999) Atherosclerosis, 145, 173-180.
115.Copeta, A., Tavian, D., Marchina, E., de Petro,
G., and Barlati, S. (2000) Growth Factors, 17,
249-268.
116.Davies, M. J., and Thomas, A. C. (1985) Br.
Heart J., 53, 363-373.
117.Haley, K. J., Lilly, C. M., Yang, J. H., Feng,
Y., Kennedy, S. P., Turi, T. G., Thompson, J. F., Sukhova, G. H.,
Libby, P., and Lee, R. T. (2000) Circulation, 102,
2185-2189.
118.Galis, Z. S., Sukhova, G. K., Lark, M. W., and
Libby, P. (1994) Clin. Invest., 94, 2493-2503.
119.Kumamoto, M., Nakashima, Y., and Sueishi, K.
(1995) Hum. Pathol., 26, 450-456.
120.Inoue, M., Itoh, H., Ueda, M., Naruko, T.,
Kojima, A., Komatsu, R., Doi, K., Ogawa, Y., Tamura, N., Takaya, K.,
Igaki, T., Yamashita, J., Chun, T. H., Masatsugu, K., Becker, A. E.,
and Nakao, K. (1998) Circulation, 98, 2108-2116.
121.Serruys, P. W., and Breeman, A. (1992) Eur.
Heart J., 13, 76-88.
122.Levine, G. N., Chodos, A. P., and Loscalzo, J.
(1995) Clin. Cardiol., 18, 693-703.
123.Nikol, S., Huehns, T., and Höfling, B.
(1996) Atherosclerosis, 123, 17-31.
124.Tkachuk, V., Stepanova, V., Little, P. J., and
Bobik, A. (1996) Clin. Exp. Pharmacol. Physiol., 23,
759-765.
125.Plekhanova, O., Parfyonova, Ye., Bibilashvily,
R., Stepanova, V., Erne, P., Bobik, A., and Tkachuk, V. (2000) J.
Hypertension, 18, 1065-1069.
126.Plekhanova, O., Parfyonova, Ye., Bibilashvily,
R., Stepanova, V. V., Domogatskii, S., Gulba, D., Agrotis, A., Bobik,
A., and Tkachuk, V. A. (2001) Atherosclerosis, in press.
127.Parfyonova, E. V., Plekhanova, O. S., Kalinina,
N. I., Bibilashvily, R. Sh., Bobik, A., and Tkachuk, V. A. (2000)
Kardiologiya, 9, 69-77.
128.Mukhina, S. A., Stepanova, V. V., Matveev, M.
Yu., Domogatsky, S. P., and Tkachuk, V. A. (1998) Vopr. Med.
Khim., 44, 84-90.
129.Carmeliet, P., Moons, L., Dewerchin, M.,
Rosenberg, S., Herbert, J. M., Lupu, F., and Collen, D. (1998) J.
Cell. Biol., 140, 233-245.
130.Carmeliet, P. F. (1995) Baillieres Clin.
Haematol., 8, 391-401.
131.Carmeliet, P., Moons, L., Lijnen, R., Baes, M.,
Lemaitre, V., Tipping, P., Drew, A., Eeckhout, Y., Shapiro, S., Lupu,
F., and Collen, D. (1997) Nat. Genet., 17, 439-444.
132.Carmeliet, P., Moons, L., Lijnen, R., Janssens,
S., Lupu, F., Collen, D., and Gerard, R. D. (1997) Circulation,
96, 3180-3191.
133.Carmeliet, P., Moons, L., Ploplis, V., Plow,
E., and Collen, D. (1997) J. Clin. Invest., 99,
200-208.
134.Mintz, G. S., Popma, J. J., and Pichard, A. D.
(1996) Circulation, 94, 35-43.
135.Shi, Y., O'Brien, J. E., Jr., Fard, A., and
Zalewski, A. (1996) Arterioscler. Thromb. Vasc. Biol.,
16, 1298-1305.
136.Zalewski, A., and Shi, Y. (1997)
Arterioscler. Thromb. Vasc. Biol., 13, 417-422.
137.Plekhanova, O. S., Kalinina, N. I., Volynskaya,
E. A., and Parfyonova, E. V. (2000) Byul. Eksp. Biol. Med.,
129, 511-514.
138.Plekhanova, O. S., Solomatina, M. A.,
Domogatsky, S. P., Naumov, V. G., Tkachuk, V. A., Parfyonova, E. V.,
and Chazov, E. I. (2001) Ross. Fiziol. Zh. im. I. M. Sechenova,
87, 11-17.
139.Alekseeva, I. A. (2000) Author's abstract of
Candidate's dissertation [in Russian], Russian Cardiology Research
Center, Moscow.
140.Strauss, B. H., Lau, H. K., Bowman, K. A.,
Sparkes, J., Chisholm, R. J., Garvey, M. B., Fenkell, L. L., Natarajan,
M. K., Singh, I., and Teitel, J. (1999) Circulation, 100,
1616-1622.
141.Lamfers, V. L., Lardenove, J. H., de Vries, M.
R., Aalders, M. C., Engelse, M. A., Grimbergen, J. M., van Hinsbergh,
V. W., and Quax, P. H. (2001) Gene Ther., 8, 534-541.
142.Folkman, J. (1987) in Thrombosis and
Hemostasis (Verstate, M., Vermylen, J., Lijnen, H., and Arnout, A.,
eds.) International Society of Thrombosis and Hemostasis and Leuven
University Press, Leuven, Belgium, pp. 583-596.
143.Goldie, I. (1970) Acta Orthop. Scand.,
40, 751-764.
144.Newell, F. W. (1986) Ophthalmology
Principles and Concepts, CV Mosby Co., St Louis, MO.
145.Carmeliet, P. (2000) Nat. Med.,
6, 389-395.
146.Battegay, E. J. (1995) J. Mol. Med.,
73, 333-346.
147.Saksela, O. (1985) Biochim. Biophys.
Acta, 823, 35-65.
148.Murphy, G., Atkinson, S., Ward, R., Gavrilovic,
J., and Reynolds, J. J. (1992) Ann. N. Y. Acad. Sci.,
667, 1-12.
149.Mazar, A. P., Henkin, J., and Goklfarb, R. H.
(1999) Angiogenesis, 3, 15-32.
150.Adams, D. O., and Hamilton, T. A. (1992) in
Inflammation: Basic Principles and Clinical Correlates (Gallin,
J. I., Goldstein, M., and Snyderman, R., eds.) Raven Press, N. Y.,pp.
662-667.
151.Arras, M., Ito, W. D., Scholz, D., Winkler, B.,
Schaper, J., and Schaper, W. (1998) J. Clin. Invest.,
101, 40-50.
152.Berman, M., Winthrop, D., Ausprunk, D., Rose,
J., Langer, R., and Gage, J. (1982) Invest. Ophthalmol. Vis.
Sci., 22, 191-199.
153.Gualandris, A., Conejo, T. L., Giunciuglio, D.,
Albini, A., Sabini, E., Rusnati, M., Dell'Era, P., and Presta, M.
(1997) Vicrovae Res., 53, 254-260.
154.Graham, C. H., Fitzpatrick, T. E., and McCrae,
K. R. (1998) Blood, 91, 3300-3307.
155.Mandriota, S. J., Seghezzi, G., Vassalli, J.
D., Ferrara, N., Wasi, S., Mazzieri, R., Mignatti, P., and Pepper, M.
S. (1995) J. Biol. Chem., 270, 9709-9716.
156.Mignati, P., Mazzieri, R., and Rifkin, D. B.
(1991) J. Cell. Biol., 113, 1193-201.
157.Olofsson, B., Korpelainen, E., Pepper, M. S.,
Mandriota, S. J., Aase, K., Kumar, V., Gunji, Y., Jeltsch, M. M.,
Shibuya, M., Alitalo, K., and Eriksson, U. (1998) Proc. Natl. Acad.
Sci. USA, 95, 11709-11714.
158.Pepper, M. S., Ferrara, N., Orci, L., and
Montesano, R. (1992) Biochem. Biophys. Res. Commun., 189,
824-831.
159.Koolwijk, P., van Erck, M. G., de Vree, W. J.,
Vermeer, M. A., Weich, H. A., Hanemaaijer, R., and van Hinsbergh, V. W.
(1996) J. Cell. Biol., 132, 1177-1188.
160.Odecon, L. E., Sato, Y., and Rifkin, D. B.
(1992) J. Cell. Physiol., 150, 258-263.
161.Lu, H., Mabilat, C., Yeh, P., Guitton, J. D.,
Li, H., Pouchelet, M., Shoevaert, D., Legrand, Y., Soria, J., and
Soria, C. (1996) FEBS Lett., 380, 21-24.
162.Min, H. Y., Doyle, L. V., Vitt, C. R.,
Zandonella, C. L., Stratton-Thomas, J. R., Shuman, M. A., and
Rosenberg, S. (1996) Cancer Res., 56, 2428-2433.
163.Lansink, M., Koolwijk, P., van Hinsbergh, V.
W., and Kooistra, T. (1998) Blood, 92, 927-938.
164.Levenson, A. S., Kwaan, H. C., Svoboda, K. M.,
Weiss, I. M., Sakurai, S., and Jordan, V. C. (1998) Br. J.
Cancer, 78, 88-95.
165.Jain, R. K., Safabakhsh, N., Sckell, A., Chen,
Y., Jiang, P., Benjamin, L., Yuan, F., and Keshet, E. (1998) Proc.
Natl. Acad. Sci. USA, 95, 10820-10825.
166.Koolwijk, P., van Erck, M., de Vree, W.,
Vermeer, M. A., Weich, H. A., Hanemaaijer, R., and van Hinsbergh, V. W.
(1996) J. Cell. Biol., 132, 1177-1188.
167.Kroon, M. E., Koolwijk, P., van Goor, H.,
Weidle, U. H., Collen, A., van der Pluijm, G., and van Hinsbergh, V. W.
(1999) Am. J. Pathol., 154, 1731-1742.
168.Evans, C. P., Elfman, F., Parangi, S., Conn,
M., Cunha, G., and Shuman, M. A. (1997) Cancer Res., 57,
3594-3599.
169.Asahara, T., Murohara, T., Sulivan, A., Silver,
M., van der Zee, R., Li, T., Witzenbichler, B., Schatteman, G., and
Isner, J. M. (1997) Science,275, 964-967.
170.Tang, H., Kerins, D. M., Hao, Q., Inagami, T.,
and Vaughan, D. E. (1998) J. Biol. Chem., 273,
18268-18272.
171.Fibbi, G., Caldini, R., Chevanne, M., Pucci,
M., Schiavone, N., Morbidelli, L., Parenti, A., Granger, H. J., del
Rosso, M., and Ziche, M. (1998) Lab. Invest., 78,
1109-1119.
172.Dubois-Stringfellow, N., Jonczyk, A., and
Bautch, V. L. (1994) Blood, 33, 3206-3217.
173.Bajou, K., Noel, A., Gerard, R. D., Masson, V.,
Brunner, N., Holst-Hansen, C., Skobe, M., Fusenig, N. E., Carmeliet,
P., Collen, D., and Foidart, J. M. (1998) Nat. Med., 4,
923-928.
174.Maehara, Y., Hasuda, S., Abe, T., Oki, E.,
Kakeji, Y., Ohno, S., and Sugimachi, K. (1998) Clin. Cancer,
4, 2129-2134.
175.Foekens, J. A., Look, M. P., Peters, H. A., van
Putten, W. L., Portengen, H., and Klijn, J. G. (1995) JNCI,
87, 751-756.
176.Roberts, A. B., Sporn, M. B., Assoian, R. K.,
Smith, J. M., Roche, N. S., Wakefield, L. M., Heine, U. I., Liotta, L.
A., Falanga, V., and Kehrl, J. H. (1986) Proc. Natl. Acad. Sci.
USA, 83, 4167-4171.
177.Berse, B., Hunt, J. A., Diegel, R. J.,
Morganelli, P., Yeo, K., Brown, F., and Fava, R. A. (1999) Clin.
Exp. Immunol., 115, 176-182.
178.Dvorak, H. F., Harvey, V. S., Estrella, P.,
Brown, L. F., McDonagh, J., and Dvorak, A. M. (1990) J. Cell.
Biol., 111, 743-755.
179.Bacharach, E., Itin, A., and Keshet, E. (1992)
Proc. Natl. Acad. Sci. USA, 89, 10686-10690.
180.Dvorak, H. F., Brown, L. F., Detmar, M., and
Dvorak, A. M. (1995) Am. J. Pathol., 146, 1029-1039.
181.Heymans, S., Luttun, A., Nuyens, D.,
Theilmeier, G., Creemers, E., Moons, L., Dyspersin, G. D., Cleutjens,
J. P. M., Shipley, M., Angellilo, A., Levi, M., Nube, O., Baker, A.,
Keshet, E., Lupu, F., Herbert, J.-M., Smits, J. F. M., Shapiro, S. D.,
Baes, M., Borgers, M., Collen, D., Daemen, M. J. A. P., and Carmeliet,
P. (1999) Nat. Med., 5, 1135-1142.
182.Li, H., Lu, H., Griscelli, F., Opolon, P., Sun,
L. Q., Ragot, T., Legrand, Y., Belin, D., Soria, J., Soria, C.,
Perricaudet, M., and Yeh, P. (1998) Gene Ther., 1105-1113.
183.Nakata, S., Ito, K.-I., Fujimori, M., Shingu,
K., Kajikawa, S., Adachi, W., Matsuyama, I., Tsuchiya, S., Kuwano, M.,
and Amano, J. (1998) Int. J. Cancer, 79, 179-186.
184.Duffy, M. J., Maguire, T. M., McDermott, E. W.,
and O'Higgins, N. (1999) J. Surg. Oncol., 71,
130-135.
185.Ugwu, F., van Hoef, B., Bini, A., Collen, D.,
and Lijnen, H. R. (1998) Biochemistry, 37, 7231-7236.
186.Guo, Y., Higazi, A. Al-R., Arakelian, A.,
Sachais, B. S., Cines, D., Goldfarb, R. H., Jones, T. R., Kwaan, H.,
Mazar, A. P., and Rabbani, S. A. (2000) FASEB J., 14,
1400-1410.
187.Mishima, K., Mazar, A. P., Gown, A., Skelly,
M., Ji, X.-D., Wang, X.-D., Jones, T. R., Cavenee, W. K., and Huang,
H.-J. S. (2000) Proc. Natl. Acad. Sci. USA, 97,
8484-8489.
188.O'Reilly, M. S., Holmgren, L., Shing, Y., Chen,
C., Rosenthal, R. A., Moses, M., Lane, W. S., Cao, Y., Sage, E. H., and
Folkman, J. (1994) Cell, 79, 315-328.
189.Cao, Y., Ji, R. W., Davidson, D., Schaller, J.,
Marti, D., Sohndel, S., McCance, S. G., O'Reilly, M. S., Llinas, M.,
and Folkman, J. (1996) J. Biol. Chem., 271,
29461-29467.
190.Ji, W.-R., Castellino, F. J., Chang, Y.,
DeFord, M. E., Gray, H., Villarreal, X., Kondri, M. E., Marti, D. H.,
Llinas, M., Schaller, J., Kramer, R. A., and Trail, P. A. (1998)
FASEB J., 12, 1731-1738.
191.Cao, Y., Chen, A., An, S. S. A., Ji, R. W.,
Davidson, D., and Llinas, M. (1997) J. Biol. Chem., 272,
22924-22928.
192.Ji, W.-R., Barrientos, L. G., Llinas, M., Gray,
H., Villarreal, X., DeFord, M. E., Castellino, F. J., Kramer, R. A.,
and Trail, P. (1998) Biochem. Biophys. Res. Commun., 247,
414-419.