Tungsten-Containing Enzymes
N. P. L'vov, A. N. Nosikov, and A. N. Antipov*
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninskii pr. 33, Moscow, 117071 Russia; fax: (095) 954-2732; E-mail: lvov@mx.inbi.ras.ru* To whom correspondence should be addressed.
Received July 12, 2001; Revision received October 30, 2001
The biological importance of tungsten has been fully proved in the last decade due to isolation of a number of tungsten-containing enzymes (W-enzymes) from hyperthermophilic archaea. Tungsten was previously considered only as an antagonist of molybdenum, because the replacement of molybdenum by tungsten (due to their chemical similarity) leads to inactivation of molybdenum-containing enzymes (Mo-enzymes). In addition to the “true W-enzymes” in which tungsten cannot be replaced by molybdenum, recently some enzymes have been isolated which can use either molybdenum or tungsten in the catalytic process. This review briefly summarizes data on the participation of tungsten in catalysis by some enzymes and the structure of the active sites of W-enzymes.
KEY WORDS: tungsten, molybdenum, enzymatic catalysis, pterin cofactor
In most of eukaryotes and prokaryotes tungsten is an antagonist of molybdenum and during growth of organisms the latter is easily replaced by tungsten due to their chemical similarity. Inactive analogs of molybdenum-containing enzymes (Mo-enzymes) are thus formed. The “true tungsten-containing enzymes” (W-enzymes”)--formate dehydrogenase, aldehyde:ferredoxin-oxidoreductase, formaldehyde:ferredoxin-oxidoreductase, etc.--in which tungsten cannot be replaced by molybdenum were isolated from hyperthermophilic archaea cells discovered in the last decade. Pterin-containing tungsten cofactor, an analog of molybdenum cofactor of Mo-containing enzymes, is the active site of these enzymes. However, there are some enzymes that exhibit catalytic activity with both molybdenum and tungsten (formylmethanofurane dehydrogenase, trimethylamine-N-oxide-reductase, etc.). Molybdopterin able to coordinate both molybdenum and tungsten is an active site of these enzymes.
CHEMICAL NATURE OF TUNGSTEN COMPARED TO MOLYBDENUM
Together with chromium and molybdenum, tungsten is an element of group VI of the periodic table of elements and is similar to molybdenum in its chemical properties; the important role of the latter in biological processes is well known [1].
Tungsten and molybdenum have equal atomic (1.40 Å) and ionic (0.68 Å) radii and similar electronegativity (1.4 and 1.3 for W and Mo, respectively); some other coordination characteristics are also very close [2]. Both metals can be in various oxidation states (from +2 to +6) and are able to form polynucleotide complexes, but only the oxidation states +4, +5, and +6 and mononucleotide systems are biologically important [3, 4].
Tungsten and molybdenum are rare in occurrence (the Clark of both elements is about 1.2 µg/ml) and they rank 54th and 53rd in natural abundance, respectively [5]. The solubility of tungsten salts is less than that of molybdenum salts, thus its concentration in fresh water rarely exceeds 20 nM and is usually less than 0.5 nM, whereas the concentration of molybdenum is two or more orders of magnitude higher than this value. The concentration of tungsten is sea water is extremely low--5*105 times lower than that of molybdenum [2].
BIOLOGICAL ACTION OF TUNGSTEN
For many years tungsten was considered to be a biological antagonist of molybdenum and was used for study of the properties and functions of molybdenum in Mo-enzymes [6]. This was due to the fact that tungsten is able to replace molybdenum in Mo-enzymes, forming catalytically inactive (or possessing very low activity) analogs.
Together with the antagonistic action of tungsten on Mo-enzymes, a positive effect of tungsten on nitrate reductase activity of plants and bacteria has long been known. Addition of small quantities of tungsten (0.25-1.0 µg/liter) to the growth medium stimulates the nitrate reductase activity of plants [7].
More than 35% of nitrate reductase (NR) of plant tissue is in the form of holoenzyme containing metal-lacking molybdenum cofactor retaining, however, the ability to coordinate metals. It is precisely this molybdenum-lacking part of NR that can be “stabilized” (i.e., protected from proteolysis and other enzyme-inactivating processes) by tungsten. Although this part of NR remains inactive after inclusion of tungsten, it is able to replace tungsten by molybdenum when the trace quantities of the latter appear in the medium, as studies on spinach and cauliflower demonstrated [7]. Thus, the “tungsten-stabilized” enzyme is activated. This stabilization effect of tungsten can be significant: for example, growth of cauliflower on medium containing equimolar concentrations of tungsten and molybdenum (0.005 µg/liter) caused 20% increase in NR activity, and plant growth was significantly accelerated. Hypersynthesis of NR apoprotein of tobacco (immunochemically detected) is another possible reason for the increase in NR activity of plants growing on medium with equimolar concentrations of tungsten and molybdenum [8].
Growth of the salt-tolerant yeast Rhodotorula glutinis on medium containing molybdenum and tungsten in equimolar concentrations (1 mM) caused stimulation of synthesis of molybdenum cofactor and decrease in NR activity. It is interesting to note that tungsten was not included in NR composition and its action manifested itself only on the level of molybdenum cofactor expression [9].
In recent years, tungsten was reported to be able to cause hyperexpression of the structural gene of some other enzymes; for example, tungsten inhibited anaerobic growth of Escherichia coli on glycerol-dimethylsulfoxide medium, and this inhibition was partly compensated by hypersynthesis of dimethylsulfoxide reductase [10]. Formate dehydrogenase (FDH) from Methylobacterium sp. RXM is another example of an analogous effect of tungsten [11]. Addition of tungsten (to 0.6 µM) to Mo-containing medium (0.6-0.9 µM) caused increase in FDH activity by stimulation of enzyme synthesis.
TUNGSTEN-CONTAINING ENZYMES
In relation to molybdenum and tungsten, all organisms can be divided into at least three groups.
1. Organisms preferring molybdenum to tungsten. In most prokaryotes and eukaryotes in vivo, tungsten easily replaces molybdenum in Mo-enzymes; inactive tungsten analogs of Mo-enzymes are thus formed [6, 12-14]. For NR, this was shown for plants [15-17], fungi [18], algae [19], and bacteria [20]. For sulfite oxidase and xanthene oxidase, this was found for animal cells [21-23].
Ability of a microorganism to grow on medium with nitrate and 1 mM sodium tungstate in the absence of molybdenum indicates that NR of this microorganism does not contain molybdenum. Inactivation of Mo-enzymes by tungsten is reversible; for example, on transfer of bacterial W-cells on a molybdenum-containing medium, NR is converted into an active Mo-form, and inactive tungsten nitrate reductase isolated from E. coli W-cells can restore activity of NR defective in molybdenum cofactor of the fungus Neurospora crassa nit-1 mutant in the presence of 1 mM sodium molybdate [24].
In all the above-mentioned studies, tungsten could be incorporated into Mo-enzymes only during growth. Only in the case of animal sulfite oxidase was it possible to replace tungsten by molybdenum in vitro and thus activate the W-containing enzyme [21].
2. Organisms preferring tungsten to molybdenum. The properties of the main “true W-enzymes” so far described in the literature, in which tungsten cannot be replaced by molybdenum or vanadium, are presented in the table.
Tungsten-containing enzymes
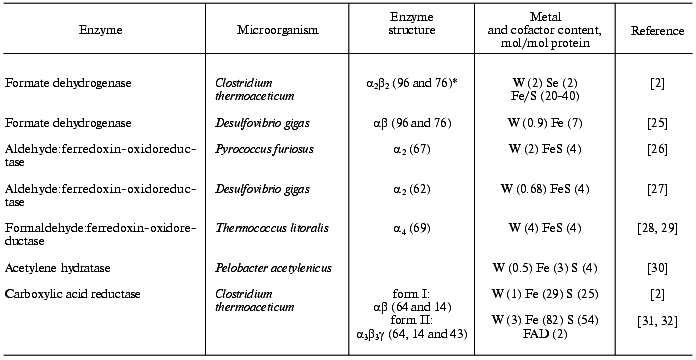
*Molecular mass of subunits (kD) is given in brackets.
Interest in a probable biological function was first raised in the early 70s when it was found that FDH of clostridia can use tungsten along with molybdenum, the former even being preferred [14]. Then tungsten incorporation into FDH was found in the anaerobic methanogen Methanococcus vannielii [33]. During growth of this microorganism on tungsten-free medium two isoforms of FDH were synthesized: one having molecular mass 105 kD and containing molybdenum and iron-sulfur clusters and another, high-molecular-weight and containing selenium, molybdenum, and iron-sulfur clusters. The latter isoform prevailed in the case of tungsten-containing medium.
However, spectacular progress in studies of biological function of tungsten was achieved after the discovery of hyperthermophilic archaea in the early 90s. Tungsten enzymes isolated from hyperthermophilic archaea cells--aldehyde:ferredoxin-oxidoreductase, formaldehyde:ferredoxin-oxidoreductase, acetylene hydratase, etc. (see table)--are exceptions from the group of W-enzymes because the dependence of their catalytic activity on tungsten is obligatory, this metal not being replaceable by molybdenum or vanadium [34]. Some hyperthermophilic archaea, for example, Pyrococcus furiosus, synthesize three W-enzymes--aldehyde:ferredoxin-oxidoreductase, formaldehyde:ferredoxin-oxidoreductase, and glyceraldehyde-3-phosphate dehydrogenase [34]. An essential difference between W-enzymes and their Mo-analogs is that the former are expressed constitutively in most studied microorganisms, whereas Mo-analogs are induced only in the presence of molybdenum [35, 36]. Besides this, W-enzymes catalyze oxidation-reduction reactions at much lower redox potentials than their Mo-analogs.
Thus, one can conclude that mesophilic microorganisms mainly synthesize Mo-enzymes, whereas hyperthermophilic bacteria and archaea with anaerobic metabolism synthesize W-enzymes. Since hyperthermophilic archaea are considered to be the earliest microorganisms [1, 37], cofactor (the active site) of these enzymes consisting of molybdopterin and tungsten but nucleotide-lacking is supposed to be a precursor of the modern molybdenum cofactors of oligonucleotide nature. Moreover, the presence of the same labile complex--molybdenum cofactor able to function both with molybdenum and tungsten--in various organisms from the earliest hyperthermophilic archaea to humans indicates the importance of Mo- and W-containing enzymes in the evolutionary process. It is supposed that before the appearance of molecular oxygen formed during photosynthesis, molybdenum and tungsten were present on the Earth as sulfides (MoS2 and WS2) rather than oxyanions (MoO4 and WO4). Since tungsten sulfide is better soluble in water than molybdenum sulfide, it is supposed that in the pre-oxygen epoch tungsten could be more available to organisms than molybdenum. This suggestion agrees with the fact that “true W-enzymes” are found in the strict anaerobic microorganisms, although there is an exception--FDH of methylotrophic bacteria [38].
FDH of sulfate-reducing bacteria Desulfovibrio gigas (see table) could be also related to the “true W-enzymes” [25]. This enzyme includes tungsten even during growth on Mo-containing medium. It is interesting to note that under the same conditions D. gigas cells synthesize Mo-aldehyde oxidase along with W-FDH. The authors [25] suppose that in early evolution sulfate reducers of Desulfovibrio genus were able to use both molybdenum and tungsten, but now they use mainly molybdenum.
THE ACTIVE SITE OF TUNGSTEN-CONTAINING ENZYMES
Study of the “true W-enzymes” (see table) mainly isolated from the cells of hyperthermophilic archaea proved that tungsten is bound similarly to molybdenum in Mo-enzymes with 6-substituted pterin (molybdopterin) [1, 37, 39, 40]. In contrast to Moco in Mo-enzymes, pterin in W-cofactor is not bound with oligonucleotides (GMP, CMP, AMP, or IMP). The main feature of tungsten coordination in the active site of W-enzymes probably providing their thermal stability is that every subunit contains two molecules of molybdopterin, which are coordinated by four sulfur ligands [41]. The similarity of pterin components of W-cofactor and Moco is also proved by the fact that W-FDH from Clostridium thermoaceticum restored activity of NR defective in Moco of the fungus Neurospora crassa nit-1 mutant [42]. Such reconstruction was possible only in the presence of 1 mM sodium molybdate. The possibility to use the E. coli W-substituted NR as a donor of Moco for defective NR of N. crassa nit-1 mutant in the presence of molybdenum was demonstrated earlier [24]. These studies allow the conclusion that Moco activity of NR of nit-1 mutant is exhibited only with molybdenum but not with tungsten.
3. Organisms to some extent able to use both metals. In these organisms molybdenum can be replaced by tungsten in vivo, and in this case the enzyme is not completely inactivated but only decreases its catalytic activity. Thus, in these organisms Mo-enzymes can probably use molybdenum and less efficiently tungsten for catalysis. For these organisms, tungsten seems to be a secondary element in bacterial metabolism because most W-enzymes are analogs of Mo-enzymes with the same functions in the same organisms.
Formylmethanofurane dehydrogenase (FMFDH), the first enzyme in the chain of enzymes participating in the synthesis of methane, is the most studied enzyme exhibiting activity both with molybdenum and tungsten [43, 44]. It was noted that whereas the dependence of growth of hyperthermophilic methanogenic archaea on the presence of tungsten in the medium is obligatory, thermophilic methanogenic archaea can use both tungsten and molybdenum for growth and FMFDH synthesis.
For example, the thermophilic methanogen Methanobacterium wolfei contains W- and Mo-isoenzymes of FMFDH, and the W-isoenzyme is synthesized during growth on W-containing medium, whereas the Mo-analog is synthesized during growth on Mo-containing medium [45-47]. In spite of a great similarity in the structure and amino acid sequence of the N-end of two of three subunits, these isoforms differ in some properties (chromatographic behavior, substrate affinity and specificity, sensitivity to O2 and cyanide).
For another thermophilic methanogen, Methanobacterium thermoautotrophicum, tungsten is used preferentially to molybdenum for FMFDH synthesis [43]. During growth on molybdenum-containing medium one Mo-isoenzyme is synthesized, and during growth on tungsten-containing medium two isoenzymes are synthesized: one with molybdenum and another with tungsten; in the latter case one of the FMFDH isoenzymes was identical to the molybdenum isoenzyme and thus could use both metals.
The Mo-enzyme trimethylamine-N-oxide reductase (TMAOR) (EC 1.6.6.9) isolated from E. coli cells is another example of an enzyme that can use both molybdenum and tungsten for catalysis [48]. Growing E. coli on media containing tungsten instead of molybdenum, the authors obtained an active W-TMAOR which had two times higher catalytic activity than Mo-TMAOR, higher sensitivity to high pH, and increased thermal stability.
A usual Mo-cofactor or, more precisely, its molybdenum-free precursor, molybdopterin (MPT), which although it can coordinate both metals seems to be also synthesized by enzymes able to use both molybdenum and tungsten.
There is little or no data on the possible participation of tungsten in NR catalysis in the literature. It is only known that replacement of molybdenum by tungsten in E. coli NR resulted in decrease of Km value against nitrate [49]. It is interesting that a strong inhibiting effect of 15 mM tungstate on E. coli nitrate reduction could be overcome in the presence of only 10 µM molybdate [50]. It was also shown that for hyperthermophilic archaea, a higher tungsten concentration in the medium is needed for NR synthesis, and molybdenum does not replace tungsten [51].
Thus, data obtained so far indicate a very important biological role of tungsten in various metabolic processes and the participation of this microelement in enzymes catalyzing these processes.
This work was financially supported by the Russian Foundation for Basic Research (grant Nos. 01-04-48553 and 01-04-48217).
REFERENCES
1.Johnson, M. K., Rees, D. C., and Adams, M. W. W.
(1996) Chem. Rev., 96, 2817-2839.
2.Kletzin, A., and Adams, M. W. W. (1996) FEMS
Microbiol. Rev.,18, 5-63.
3.Pope, M. T. (1987) in Comprehensive Coordination
Chemistry (Wilkinson, Sir G., ed.) Pergamon Press, Oxford-New York,
pp. 1023-1060.
4.McCleverty, J. A. (1994) in Encyclopedia of
Inorganic Chemistry (King, R. B., ed.) John Wiley and Sons, New
York, pp. 2304-2330.
5.Greenwood, N. N., and Earnshaw, A. (1984) in
Chemistry of the Elements, Pergamon Press, Oxford, pp.
1167-1168.
6.L'vov, N. P. (1989) Molybdenum in Nitrogen
Assimilation in Plants and Microorganisms, 43rd A. N. Bach
Memorial Lecture [in Russian], Nauka, Moscow.
7.Notton, B. A. (1983) in Metals and
Micronutrients. Uptake and Utilization by Plants (Robb, D. A., and
Pierpoint, W. S., eds.) Academic Press, pp. 219-239.
8.Deng, M., Moureaux, T., and Caboche, M. (1989)
Plant Physiol., 91, 304-309.
9.Nosikov, A. N., Chichikalo, E. V., Golubeva, L. I.,
Zvyagil'skaya, R. A., and L'vov, N. P. (2000) Biochemistry
(Moscow), 65, 204-207.
10.Rothery, R. A., Simala Grant, J. L., Johnson, J.
L., Rajagopalan, K. V., and Weiner, J. H. (1995) J.
Bacteriol.,177, 2057-2063.
11.Girio, F. M., Roseiro, J. C., and Silva, A. I.
(1998) Curr. Microbiol., 36, 337-340.
12.L'vov, N. P., Alikulov, Z. A., Kil'dibekov, N.
A., and Kretovich, V. L. (1981) Izv. Akad. Nauk SSSR, Ser.
Biol., 2, 219-236.
13.Kil'dibekov, N. A., Omarov, R. T., and L'vov, N.
P. (1996) Usp. Biol. Khim., 36, 136-186.
14.Ljungdahe, L. G. (1976) Trends Biochem.
Sci., 1, 63-65.
15.Heimer, Y. N., Wray, J. L., and Filner, P. (1969)
Plant Physiol., 44, 1197-1199.
16.Wray, J. L., and Filner, P. (1970) Biochem.
J., 119, 715-725.
17.Notton, B. A., and Hewitt, E. J. (1971)
Biochem. Biophys. Res. Commun., 44, 702-710.
18.Lee, K. Y., Erickson, R., Pan, S. S., Jones, G.,
May, F., and Nason, A. (1974) J. Biol. Chem., 249,
3953-3959.
19.Guerrero, M. G., and Vega, J. M. (1975) Arch.
Microbiol., 102, 91-94.
20.Enoch, H. G., and Lester, R. L. (1972) J.
Bacteriol., 110, 1032-1040.
21.Johnes, H. P., Johnson, J. L., and Rajagopalan,
K. V. (1977) J. Biol. Chem., 252, 4988-4993.
22.Johnson, J. L., Cohen, H. J., and Rajagopalan, K.
V. (1974) J. Biol. Chem., 249, 5040-5055.
23.Johnson, J. L., Wand, W. R., Cohen, H. J., and
Rajagopalan, K. V. (1977) J. Biol. Chem., 249,
5056-5061.
24.Amy, N. K., and Rajagopalan, K. V. (1979) J.
Bacteriol., 140, 114-124.
25.Almendra, M. J., Brondino, C. D., Gavel, O.,
Pereira, A. S., Tavares, P., Bursakov, S., Duarte, R., Caldeira, J.,
Moura, J. J. G., and Moura, I. (1999) Biochemistry,38,
16366-16372.
26.Mukund, S., and Adams, M. W. W. (1991) J.
Biol. Chem., 266, 14208-14216.
27.Hensgens, C. M. H., Hagen, W. R., and Hansen, T.
A. (1995) J. Bacteriol.,177, 6195-6200.
28.Roy, R., Mukund, S., Schut, G. J., Dunn, D. M.,
Weiss, R., and Adams, M. W. W. (1999) J. Bacteriol., 181,
1171-1180.
29.Mukund, S., and Adams, M. W. W. (1993) J.
Biol. Chem., 268, 13592-13600.
30.Meckenstock, R. V., Krieger, R., Ensigh, S.,
Kroneck, P. M. H., and Schink, B. (1999) Eur. J. Biochem.,
264, 176-182.
31.White, H., Feicht, R., Huber, C., Lottspeich, F.,
and Simon, H. (1991) Biol. Chem. Hoppe-Seyler, 372,
999-1005.
32.White, H., Strobl, G., Feicht, R., and Simon, H.
(1989) Eur. J. Biochem., 184, 89-96.
33.Jones, J. B., and Stadtman, T. (1981) J. Biol.
Chem.,256, 656-663.
34.Mukund, S., and Adams, M. W. W. (1996) J.
Bacteriol., 178, 163-167.
35.Hochheimer, A., Hedderich, R., and Thauer, R. K.
(1998) Arch. Microbiol., 170, 389-393.
36.Hochheimer, A., Linder, D., Thauer, R. K., and
Hedderich, R. (1996) Eur. J. Biochem.,242, 156-162.
37.Johnson, J. L., Rajagopalan, K. V., Mukund, S.,
and Adams, M. W. W. (1993) J. Biol. Chem., 268,
4848-4852.
38.Girio, F. M., Marcos, J. C., and Amarall-Collaco,
M. T. (1992) FEMS Microbiol. Lett., 97, 161-166.
39.Chan, M. K., Mukund, S., Kletzin, A., Adams, M.
W. W., and Rees, D. C. (1995) Science, 267,
1463-1469.
40.McMaster, J., and Enemark, J. H. (1998) Curr.
Opin. Chem. Biol., 2, 201-207.
41.Fischer, B., Enemark, J. H., and Basu, P. (1998)
J. Inorg. Biochem., 72, 13-21.
42.Deaton, J. C., Solomon, E. J., Durfor, C. N.,
Wetherbee, P. J., Burgess, B. K., and Jacobs, D. B. (1984) Biochem.
Biophys. Res. Commun., 121, 1042-1047.
43.Bertram, P. A., Schmitz, R. A., Linder, D., and
Thauer, R. K. (1994) Arch. Microbiol., 161, 220-228.
44.Vorholt, J. A., Vaupel, M., and Thauer, R. K.
(1997) Mol. Microbiol., 23, 1033-1042.
45.Schmitz, R. A., Albracht, S. P. J., and Thauer,
R. K. (1992) Eur. J. Biochem., 209, 1013-1018.
46.Schmitz, R. A., Richter, M., Linder, D., and
Thauer, R. K. (1992) FEBS Lett., 309, 78-81.
47.Schmitz, R. A., Richter, M., Linder, D., and
Thauer, R. K. (1992) Eur. J. Biochem., 207, 559-565.
48.Buc, J., Santini, C. L., Giordani, R., Czjzek,
M., Wu, L. F., and Giordano, G. (1999) Mol. Microbiol.,
32, 159-168.
49.Buc, J., Santini, C. L., Blasco, F., Giordani,
R., Cardenas, M. L., and Chippaux, M. (1995) Eur. J.
Biochem.,234, 766-772.
50.Afshar, S., Kim, C., Monbouquette, H. G., and
Schroder, J. (1998) Appl. Environ. Microbiol., 64,
3004-3008.
51.Potter, L. C., Millington, P. D., Thomas, G. H.,
Rothery, R. A., Giordano, G., and Cole, J. A. (2000) FEMS Microbiol.
Lett.,185, 51-57.