New Structures of the O-Specific Polysaccharides of Proteus. 2. Polysaccharides Containing O-Acetyl Groups
A. N. Kondakova1, F. V. Toukach1, S. N. Senchenkova1, N. P. Arbatsky1, A. S. Shashkov1, Y. A. Knirel1*, K. Zych2, A. Torzewska2, K. Kolodziejska2, A. Rozalski2, and Z. Sidorczyk2
1Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninskii pr. 47, Moscow, 119991 Russia; fax: (095) 135-5328; E-mail: knirel@ioc.ac.ru2Institute of Microbiology and Immunology, University of Lodz, Banacha 12/16, 90-237 Lodz, Poland; fax: (48) 42-6784932; E-mail: zsidor@biol.uni.lodz.pl
* To whom correspondence should be addressed.
Received July 19, 2001
Structures of five new O-specific polysaccharides of Proteus bacteria were established. Four of them, Proteus penneri 4 (O72), Proteus vulgaris 63/57 (O37), Proteus mirabilis TG 277 (O69), and Proteus penneri 20 (O17), contain O-acetyl groups in non-stoichiometric quantities, and the polysaccharide of P. penneri 1 is structurally related to that of P. penneri 4. The structures were elucidated using NMR spectroscopy, including one-dimensional 1H- and 13C-NMR spectroscopy, two-dimensional 1H,1H correlation (COSY, TOCSY), H-detected 1H,13C heteronuclear multiple-quantum coherence (HMQC), heteronuclear multiple-bond correlation (HMBC), and nuclear Overhauser effect spectroscopy (NOESY or ROESY), along with chemical methods. The structural data obtained are useful as the chemical basis for the creation of the classification scheme for Proteus strains.
KEY WORDS: lipopolysaccharide, O-antigen, bacterial polysaccharide, structure, O-acetyl groups, Proteus
Abbreviations: COSY) correlation spectroscopy; Etn-P) ethanolamine phosphate; FucNAc) 2-acetamido-2,6-dideoxygalactose; Fuc3NHb) 3,6-dideoxy-3-[(R)-3-hydroxybutyramido]galactose; GlcA) glucuronic acid; Hb) 3-hydroxybutyryl group; HMBC) heteronuclear multiple-bond correlation; HMQC) heteronuclear multiple-quantum coherence; NOESY) nuclear Overhauser effect spectroscopy; ROESY) rotating-frame nuclear Overhauser effect spectroscopy; TOCSY) total correlation spectroscopy.
Although enterobacteria of the genus Proteus make up a part of
the normal flora of the human gastrointestinal tract, under favorable
conditions they cause urinary tract infections that can lead to severe
complications, such as acute and chronic pyelonephritis and formation
of kidney stones. The bacteria frequently cause diseases in patients
with urinary tract abnormalities, with urinary catheter in place or
after surgical intervention in the urogenital system [2, 3]. Recently, it has been
suggested that Proteus mirabilis plays an etiopathogenic role in
rheumatoid arthritis [4].
Currently, Proteus rods are subdivided into four species, three of which, P. mirabilis, P. vulgaris, and P. penneri, are of clinical importance. The species P. vulgaris is heterogeneous with two biogroups, 2 and 3, the latter being further subdivided into genomospecies 3 (Proteus hauseri), 4, 5 and 6 (all unnamed) [5, 6]. Based on the immunospecificity of the cell-surface lipopolysaccharides, two species, P. mirabilis and P. vulgaris,are classified into 60 O-serogroups [7, 8], and recently new O-serogroups have been proposed for P. penneri strains [9, 10].
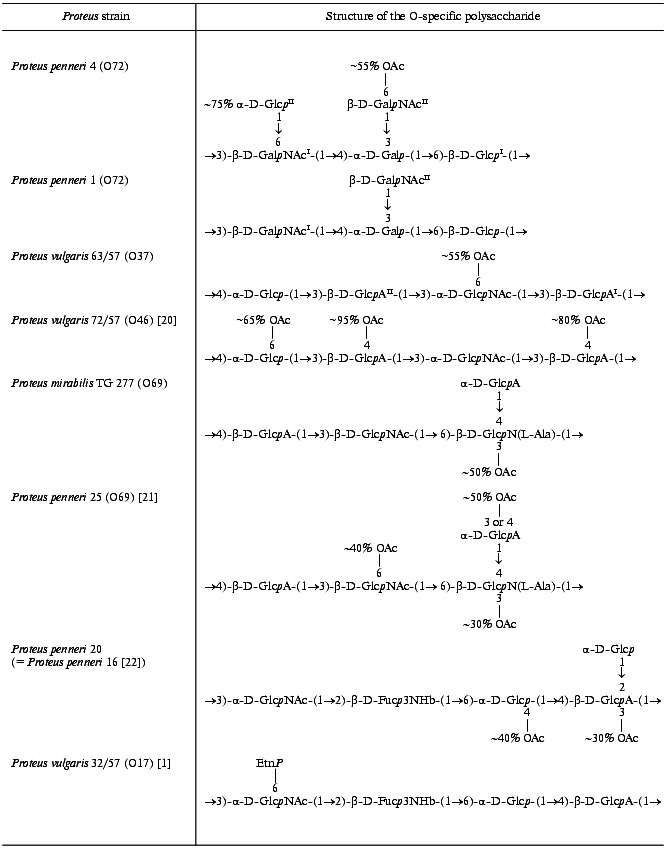
Aiming at creation of the molecular basis for classification of Proteus strain, we have been undertaking immunochemical studies of the polysaccharide chains of the lipopolysaccharides, which define the O-specificity of the bacteria [9]. Most polysaccharides studied contain acidic or both acidic and basic components, such as amino acids and phosphate and ethanolamine phosphate groups [1]. Although all of the polysaccharides have regular structures, the regularity of some polysaccharides is masked by the presence of ethanolamine phosphate or O-acetyl groups in non-stoichiometric quantities. We now report the determination of new structures of four O-specific polysaccharides containing O-acetyl groups, which were isolated from P. vulgaris 63/57 (O37), P. mirabilis TG 277 (O69), P. penneri 4 (O72)and 20 (O17), as well as that of P. penneri 1 (O72), which is structurally related to the polysaccharide of P. penneri 4.
MATERIALS AND METHODS
P. vulgaris 63/57 (O37) was from the Czech National Collection of Type Cultures (CNCTC, Institute of Epidemiology and Microbiology, Prague, Czech Republic). P. mirabilis TG 277 (O69) was kindly provided by Prof. J. L. Penner (Department of Medical Genetics, Toronto University, Ontario, Canada). Strains of P. penneri were from the collection of the Institute of Microbiology and Immunology (University of Lodz, Poland). The bacteria were grown on nutrient broth (Warsaw Laboratory of Sera and Vaccines) supplemented with 1% glucose. Bacterial cells were separated by centrifugation, washed with distilled water, and lyophilized.
Lipopolysaccharides were isolated by extraction of dried cells with aqueous phenol [11] and purified by treatment with ribonuclease and deoxyribonuclease and by ultracentrifugation as described [12]. The lipopolysaccharides were degraded with 2% acetic acid at 100°C until the precipitation of lipid. High-molecular-mass O-specific polysaccharide was isolated from the water-soluble portion by gel-permeation chromatography on a Sephadex G-50 column in pyridine-acetate buffer pH 4.5 as described [12] using a Knauer differential refractometer (Germany) for monitoring.
NMR spectra were recorded on Bruker WM-250 and Bruker DRX-500 spectrometers (Germany) in solutions in D2O (internal standard acetone, deltaH 2.225 ppm, deltaC 31.45 ppm, or 85% aqueous H3PO4, deltaP 0 ppm). NMR spectra were assigned as described previously [1].
O-Deacetylation of the polysaccharides was performed with 12% aqueous ammonia at 60°C for 2 h, and the modified polysaccharides were isolated by gel-permeation chromatography on a Sephadex G-50 column.
The polysaccharides were hydrolyzed with 2 M trifluoroacetic acid (120°C, 2 h). Methanolysis of the polysaccharides was performed with 1 M hydrogen chloride in methanol and followed by acetylation with acetic anhydride in pyridine (100°C, 1 h). A portion of the acetylated methanolysis products was subjected to (+)-2-octanolysis in the presence of anhydrous trifluoroacetic acid (120°C, 16 h) followed by acetylation [13]. The acetylated derivatives were analyzed by GLC and GLC/mass spectrometry on a Hewlett Packard 5890 chromatograph (USA) with a DB-5 capillary column equipped with a Nermag R10-10L mass spectrometer (France), using a temperature gradient of 160°C (1 min) to 290°C at 3°C/min.
RESULTS
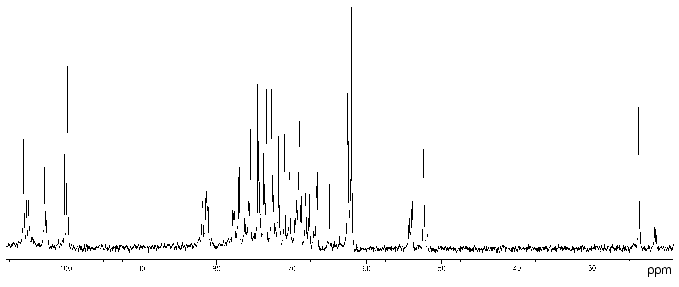
Proteus penneri strains 4 and 1 (serogroup O72). The 13C- (Fig. 1) and 1H-NMR spectra of the polysaccharide of P. penneri 4 demonstrated structural heterogeneity, most likely owing to non-stoichiometric O-acetylation (there were signals for CH3 of O-acetyl groups at deltaH 2.14 and 2.15 ppm, deltaC 21.7 (major), 21.4 and 21.5 ppm (both minor)). After O-deacetylation with aqueous ammonia, the spectra showed a higher degree of regularity but a number of minor signals were still present, which were due to non-stoichiometric glucosylation (see below).
The 13C-NMR spectrum of the modified polysaccharide showed a pentasaccharide repeating unit containing two amino sugars. There were signals for five anomeric carbons at delta 99.8-105.6 ppm, two nitrogen-bearing carbons at delta 52.4 and 54.1 ppm, three unsubstituted HOCH2 groups at delta 62.0, 62.5, 62.6 ppm, and two substituted HOCH2 groups at delta 66.6 and 68.1 ppm (data of an attached-proton test experiment [14]), other sugar ring carbons in the region delta 68.9-81.4 ppm, and two N-acetyl groups at delta 23.7 (CH3) and 176.0 ppm (CO). Accordingly, the 1H-NMR spectrum contained, inter alia, signals for five anomeric protons at delta 4.61-4.99 ppm and two N-acetyl groups at delta 2.05 and 2.06 ppm.Fig. 1. 13C-NMR spectrum of the polysaccharide of P. penneri 4 (O72) (the resonance region of CO groups is not shown).
The major series in the two-dimensional homonuclear and heteronuclear shift-correlated spectra (COSY, TOCSY, ROESY, 1H,13C HMQC) of the O-deacetylated polysaccharide of P. penneri 4 was similar to those of the polysaccharide of P. penneri 1. Assignment of the spectra (Tables 1 and 2) revealed the presence in both polysaccharides of the same sugar residues, which were identified as one residue each of Galp and Glcp and two residues of GalpNAc. The spin system for Glcp was distinguished from those for Galp and GalpNAc on the basis of the 3JH,H coupling constants values [15], and the GalpNAc spin systems were identified by lower field positions of the signals for H2 (delta 4.02-4.07 versus delta 3.76 ppm, respectively) as well as by their correlation to the corresponding nitrogen-bearing carbons at delta 52.5 and 54.1 ppm revealed by a 1H,13C HMQC experiment.
Table 1. 1H-NMR data
(delta, ppm)
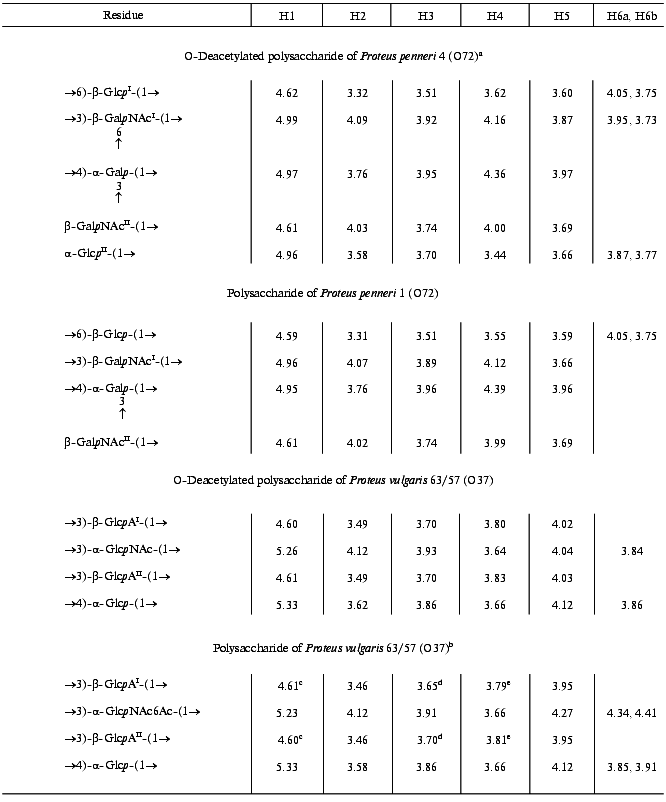
Note: Additional chemical shifts for the N-acetyl groups are delta 2.01-2.07 ppm.
aData for the major repeating unit containing the terminal glucose residue.
bData for the O-acetylated repeating unit.
c-eAssignment could be interchanged.
Table 2. 13C-NMR data
(delta, ppm)
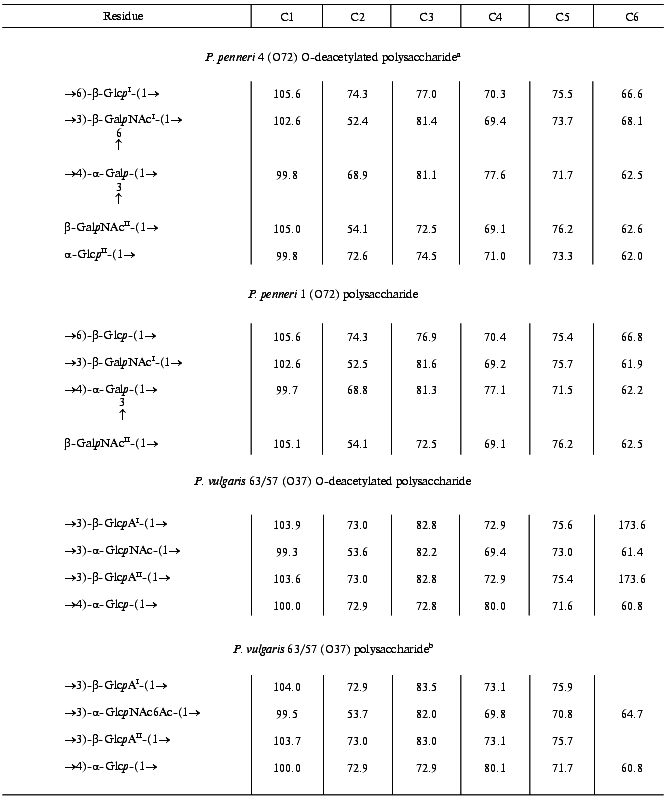
Note: Additional chemical shifts for NAc groups are delta 23.3-23.7 (CH3) and 175.1-176.3 ppm (CO).
aData for the major repeating unit, bearing terminal Glcp residue.
bData for the O-acetylated repeating unit.
In addition to these four monosaccharides, the repeating unit of the P. penneri 4 polysaccharide contained another Glcp residue. Sugar analysis confirmed the composition of the P. penneri 1 and 4 polysaccharides and demonstrated the D-configuration of all monosaccharides. Therefore, the repeating unit of the P. penneri 1 polysaccharide is a tetrasaccharide containing two residues of D-GalNAc and one residue each of D-Glc and D-Gal, and that of the P. penneri 4 is a pentasaccharide containing two residues each of D-Glc (I and II) and D-GalNAc (I and II), one residue of D-Gal, and O-acetyl groups.
The structure of the simpler polysaccharide from P. penneri 1 was elucidated first. The JH1,H2 coupling constant values of 7-8 Hz for Glcp, GalpNAcI and GalpNAcII were characteristic of beta-linked sugars, whereas the JH1,H2 value of 4 Hz showed that Galp is alpha-linked. The 1JC1,H1 coupling constant values of 162.5-165.4 Hz, which were determined from the non-decoupled HMQC spectrum, confirmed the beta-configuration of Glcp and both GalpNAc residues, and the value of 171.1 Hz was consistent with the alpha-configuration of Galp [16].
In the 13C-NMR spectrum of the polysaccharide, significant low-field displacements of the signals for C6 of Glcp, C3 and C4 of Galp, and C3 of GalpNAcI to delta 66.8, 81.3, 77.1, and 81.6 ppm, as compared with their positions in the spectra of the corresponding unsubstituted monosaccharides at delta 61.9, 70.4, 70.6, and 72.4 ppm [17], respectively, were due to the known alpha-effects of glycosylation. These data showed that the polysaccharide is branched, Glcp is 6-substituted, Galp 3,4-disubstituted, and GalpNAcI 3-substituted. No significant displacements were observed for the C2-C6 signals of GalpNAcII and, hence, it occupies the non-reducing end of the side chain.
A ROESY experiment with the P. penneri 1 polysaccharide revealed the following correlations between the transglycosidic protons: Galp H1/Glcp H6b at delta 4.95/3.75, Glcp H1/GalpNAcI H3 at delta 4.59/3.89, GalpNAcI H1/Galp H4 at delta 4.96/4.39, and GalpNAcII H1/Galp H3 at delta 4.61/3.96 ppm. These data were in accordance with the substitution pattern as determined by the 13C-NMR chemical shift data and revealed the full sequence of the monosaccharide residues in the repeating unit.
Therefore, the O-specific polysaccharide of P. penneri 1 has the structure shown in Table 3.
Table 3. Structures of the O-specific
polysaccharides of Proteus determined in this work and
structurally related polysaccharides
The major series of the signals in the 13C-NMR spectrum of the O-deacetylated polysaccharide of P. penneri 4 was close to that of the P. penneri 1 polysaccharide (Table 2), except for the signals for C5 and C6 of GalpNAcI, which were shifted from delta 75.7 and 61.9 to delta 73.7 and 68.1 ppm, respectively. Such displacements are typical of the effects of glycosylation by alpha-linked monosaccharides at position 6 [17] and were in accordance with the presence of an additional alpha-GlcpII residue. The terminal position and the alpha-configuration of GlcpII were additionally confirmed by the chemical shifts for C2-C5, which were close to those for alpha-glucopyranose [17]. In the 1H-NMR spectrum, marked displacements were also observed for the signals of GalpNAcI (in particular, the H5 signal shifted from delta 3.67 to 3.87 ppm), whereas the chemical shifts of the signals for the other sugar residues common for both polysaccharides were essentially the same. Therefore, the major repeating unit of the O-deacetylated polysaccharide of P. penneri 4 differs from the repeating unit of the polysaccharide of P. penneri 1 only in the presence of one additional alpha-glucopyranose residue attached at position 6 of GalNAcI.
A minor series of the signals in the NMR spectra of the P. penneri 4 polysaccharide belonged to a tetrasaccharide repeating unit lacking the terminal GlcpII residue and, thus, having the same structure as the polysaccharide of P. penneri 1. Hence, GlcpII is present in the P. penneri 4 polysaccharide in a non-stoichiometric amount. As judged by the ratio of the integral intensities of the signals in the major and minor series, the degree of alpha-glucosylation is ~75-80%.
Comparison of the 13C-NMR spectra of the initial and O-deacetylated polysaccharides of P. penneri 4 showed that in the former the signals for C5 and C6 of GalNAcII are partially shifted from delta 76.2 and 62.6 to delta 73.6 and 65.0 ppm, respectively. These displacements indicated O-acetylation of this monosaccharide at position 6 [18]. This conclusion was confirmed by a down-field displacement in the 1H-NMR spectrum of the signals for H6a and H6b of GalNAcII from delta 3.70-3.85 to delta 4.70 and 4.62 ppm due to a deshielding effect of the O-acetyl group. The degree of O-acetylation at position 6 of GalNAcII was estimated as ~60%, and, as judged by the ratio of the integral intensities of the signals for the O-acetyl and N-acetyl groups in the 1H-NMR spectrum, the total degree of O-acetylation in the repeating unit is ~75%. The position of minor O-acetyl groups (~15%) was not determined.
The data obtained showed that the major repeating unit of the O-specific polysaccharide of P. penneri 4 has the structure shown in Table 3.
Proteus vulgaris 63/57 (serogroup O37). Sugar analysis after full acid hydrolysis of the polysaccharide revealed the presence of Glc, GlcN, and GlcA, all having the D-configuration.
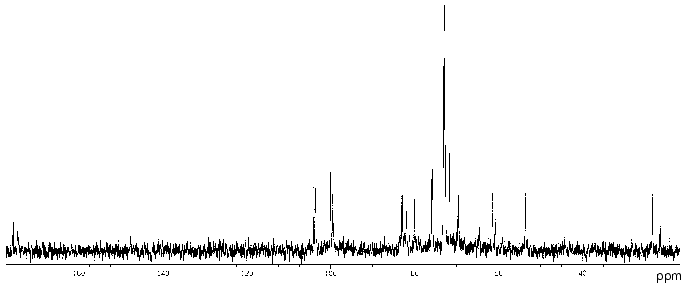
The 13C-NMR spectrum of the polysaccharide (Fig. 2) contained signals of different intensities, most likely, owing to non-stoichiometric O-acetylation (there was a signal for CH3 of an O-acetyl group at 21.5 ppm). The 13C-NMR spectrum of the O-deacetylated polysaccharide contained a major series of signals consisting of signals for four anomeric carbons at delta 99.3-103.9 ppm, one nitrogen-bearing carbon (C2 of GlcpN) at delta 53.6 ppm, two HOCH2 groups (C6 of Glcp and GlcpN) at delta 61.4 and 60.8 ppm, two COOH groups (C6 of GlcpA) at delta 173.6 ppm, 15 other sugar ring carbons at delta 69.4-82.8 ppm, and one N-acetyl group (CH3 at delta 23.2 ppm, CO at delta 175.5 ppm). The absence from the 13C-NMR spectrum of signals in the region of delta 83-88 ppm characteristic for furanosides [19] showed that all monosaccharides are in the pyranose form.
The 1H-NMR spectrum of the initial polysaccharide contained a major series of signals for four anomeric protons at delta 4.60-5.33 ppm, one N-acetyl group at delta 2.03 ppm, and other protons at delta 3.46-4.41 ppm. Both 13C- and 1H-NMR spectra of the O-deacetylated polysaccharide contained also a minor series of signals, which was absent from the spectra of the initial polysaccharide and, therefore, reflected a structural heterogeneity, which resulted from degradation in the course of alkaline O-deacetylation.Fig. 2. 13C-NMR spectrum of the polysaccharide of P. vulgaris 63/57 (O37).
The 1H-NMR spectrum of the O-deacetylated polysaccharide was assigned using two-dimensional COSY, TOCSY, NOESY, and 1H,13C HMQC experiments (Table 1). The spin system of GlcpNAc was identified by a correlation of the H2 proton at delta 4.12 ppm with a nitrogen-bearing carbon at delta 53.6 ppm observed in the HMQC spectrum. The J1,2 coupling constant value of ~3 Hz indicated that Glcp and GlcpNAc are alpha-linked, whereas the J1,2 value of ~8 Hz showed that both GlcpA residues (I and II) are beta-linked.
The 1H,13C HMQC experiment enabled full assignment of the 13C-NMR spectrum of the O-deacetylated polysaccharide (Table 2). Relatively low-field positions at delta 80-84 ppm of the signals for C3 of GlcpNAc, GlcpAI and GlcpAII and C4 of Glcp, as compared with their positions in the spectra of the corresponding nonsubstituted monosaccharides [6], revealed the glycosylation pattern in the repeating unit.
In the NOESY spectrum of the O-deacetylated polysaccharide, the direct assignment of cross-peaks between the transglycosidic protons was complicated by a coincidence of the H1 and H3 signals of GlcAI and GlcAII at delta ~4.60 and 3.70 ppm, respectively. This problem could be overcome by taking into account the modes of substitution of the monosaccharide residues, which were determined independently by the 13C-NMR chemical shift data (see above). Therefore, the cross-peaks at delta 4.60/3.66, 5.33/3.70, 4.61/3.93, and 5.26/3.70 ppm could be assigned to GlcAI H1/Glc H4, Glc H1/GlcAII H3, GlcAII H1/GlcNAc H3, and GlcNAc H1/GlcAI H3 correlations, respectively. These data confirmed the structure of the repeating unit of the polysaccharide.
The position of the O-acetyl group was determined by comparison of the 13C-NMR spectra of the initial and O-deacetylated polysaccharides. A lower field position of the H6a,6b signals of GlcNAc at delta 4.34, 4.41 ppm in the former as compared to that at delta 3.84, 3.84 ppm in the latter could only be caused by O-acetylation of GlcNAc at O6. Differences in the chemical shifts of GlcpNAc C5 and C6 (delta 70.8 and 64.7 versus 73.0 and 61.9 ppm; beta- and alpha-effects of O-acetylation [18], respectively) confirmed the location of the O-acetyl group at position 6. Relative integral intensities of the signals for the O-acetylated and non-O-acetylated GlcNAc residues showed that the degree of O-acetylation is ~55%.
The structure of the repeating units of the O-specific polysaccharide of P. vulgaris 63/57 (O37) and a related polysaccharide of P. vulgaris Î46 [20] are shown in Table 3.
Proteus mirabilis TG 277 (serogroup O69). Sugar analysis of the polysaccharide showed the presence of GlcA and GlcN.
The 13C-NMR spectrum (Fig. 3) suggested that the polysaccharide has a tetrasaccharide repeating unit that contains an O-acetyl group in a non-stoichiometric quantity (there was a signal at delta 21.7 ppm). The spectrum of the O-deacetylated polysaccharide was typical of a regular polymer. It contained signals for four anomeric carbons (delta 101.9-104.0 ppm), three nitrogen-bearing carbons (delta 50.8-56.0 ppm), one substituted (delta 68.8 ppm) and one unsubstituted HOCH2 group (delta 61.9 ppm, data of an INEPT experiment), two methyl groups at delta 17.6 and 23.7 ppm, and four carbonyl groups at delta 172.5-176.0 ppm.
The 13C-NMR spectrum of the initial polysaccharide resembled that of Proteus penneri 25 from serogroup O69, which was studied by us earlier [21]. For instance, regions for the anomeric carbons (delta 102-104 ppm) and nitrogen-bearing carbons (delta 55-56 ppm) were similar. Moreover, the spectra of the O-deacetylated polysaccharides from the two strains were practically identical. Therefore, the two polysaccharides differed in the position of the O-acetyl groups only. The structure of the repeating unit and the O-acetylation pattern of the polysaccharide of P. penneri 25 is shown in Table 3. A peculiar feature of this polysaccharide is the presence of a glucosamine residue N-substituted by L-alanine (GlcNAla) and three sites of non-stoichiometric O-acetylation.Fig. 3. 13C-NMR spectrum of the polysaccharide of P. mirabilis TG 277 (O69).

Comparison of the 13C-NMR spectra of the initial and O-deacetylated polysaccharides of P. mirabilis TG 277 showed that the O-acetyl group is located at position 3 of about half GlcNAla residues. This followed from a splitting of the C2 signal of GlcN (delta 56.1 ppm in the non-acetylated residue and delta 55.1 ppm in the O-acetylated residue; beta-effect of O-acetylation [18]). The non-stoichiometric O-acetylation of GlcNAla was confirmed by a splitting of the signals for C2 and C3 of alanine (delta 50.5/50.8 and 17.1/17.6 ppm, respectively) owing to its attachment to N2 of GlcN located near the O-acetylation site. The signals of the other sugar residues, including those of GlcNAc and GlcA, which are partially O-acetylated in the polysaccharide of P. penneri 25 [21], were not split and their chemical shifts were the same in the initial and O-deacetylated polysaccharides of P. mirabilis TG 277. Therefore, whereas there are three O-acetylation sites in the polysaccharide of P. penneri 25, the polysaccharide of P. mirabilis TG 277 has only one of them. The full structure of the P. mirabilis TG 277 polysaccharide is shown in Table 3.
Proteus penneri 20 (serogroup O17). The O-antigen of P. penneri 20 was found to be serologically identical to that of P. penneri 16, whose structure has been established earlier [22]. The P. penneri 16 polysaccharide includes a rarely occurring component, 3,6-dideoxy-3-[(R)-3-hydroxybutyramido]-D-galactose [D-Fuc3NHb] (Table 3). It also contains O-acetyl groups, but their positions have not been determined. The 13C-NMR spectra of the polysaccharides of P. penneri 16 and 20 were identical and, hence, the polysaccharides have the same structure. This was additionally confirmed by the identity of the 1H- and 13C-NMR spectra of the O-deacetylated polysaccharides from both strains.
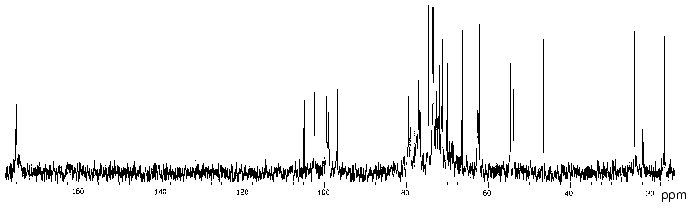
In order to determine the location of the O-acetyl groups, the polysaccharide of P. penneri 20 was studied in detail using two-dimensional NMR spectroscopy. Comparison of the 13C-NMR spectra of the initial (Fig. 4) and O-deacetylated polysaccharides showed no significant difference in chemical shifts of GlcNAc, Fuc3NHb and the lateral Glc residue, whereas those of GlcA and 6-substituted Glc were markedly shifted. As compared with the spectrum of the O-deacetylated polysaccharide, in the spectrum of the initial polysaccharide the intensities of the signals of GlcA and 6-substituted Glc decreased and new signals for the corresponding O-acetylated residues appeared.
Position 3 is the only possible site of O-acetylation in GlcA since positions 2 and 4 are glycosylated. Acetylation of this residue at O3 was confirmed by characteristic upfield displacements, due to beta-effects of O-acetylation [18], of the signals for C2 and C4 from delta 78.6 and 77.3 to delta 77.5 and 76.6 ppm, respectively. Determination of the effects of O-acetylation in 6-substituted Glc was complicated by a coincidence of many signals for the O-acetylated residue with signals of other monosaccharides. However, the location of acetyl groups at O2 and O3 could be excluded since neither beta-effect on C1 and C4 nor alpha-effect on C3 was observed. Hence, the 6-substituted Glc residue is O-acetylated at position 4. As judged by the relative integral intensities of the signals in the O-acetylated and non-acetylated residues, the degree of O-acetylation of GlcA and Glc is ~30 and ~40%, respectively. The full structure of the repeating unit of the polysaccharide of P. penneri 20 is shown in Table 3.Fig. 4. 13C-NMR spectrum of the polysaccharide of P. penneri 20 (O17).
DISCUSSION
O-Acetylation in bacterial glycopolymers plays an important biological role. For instance, 3-O-acetylation of 2-acetamido-2-deoxy-D-mannuronic acid in the capsular polysaccharide of Staphylococcus aureus type 5 increased significantly the resistance of the bacterium to opsonophagocytic killing and the ability to colonize the kidneys in a mouse model [23]. 6-O-Acetylation of N-acetylmuramic acid increased the resistance of peptidoglycan of various human pathogens (Neisseria gonorrhoeae, Proteus mirabilis, Staphylococcus aureus) to hydrolytic degradation by lysozyme and muramidases [24]. A biosynthetic modification of extracellular bacterial polysaccharides that contributes to virulence (e.g., alginate of Pseudomonas species [25, 26] and Vi antigen of Salmonella typhi and Citrobacter freundii [27]) by O-acetylation of D-mannuronic acid and 2-acetamido-2-deoxy-D-galacturonic acid, respectively, has important implications, e.g., affects the binding properties of carboxyl groups. 8-O-Acetylation of legionaminic acid in the O-specific polysaccharide chain of the lipopolysaccharide increases the hydrophobicity of Legionella pneumophila cell surface and thus enhances the ability of the microorganism to form stable aerosols, which may promote the spread of the bacterium during outbreaks [28].
O-Acetylation in specific bacterial polysaccharides creates the antigenic diversity, which enhances the survival of the bacteria by promotion of immune avoidance. For instance, O-acetyl groups significantly contribute to the epitope specificity of L. pneumophila lipopolysaccharide, particularly to its recognition by monoclonal antibodies [29, 30]. O-Acetylation is in part responsible for serotype-conversion by modification of the O-specific polysaccharide of Shigella flexneri [31]. O-Acetyl transferase of this and some other bacteria has a bacteriophage origin. Lysogenic conversion in Salmonella enterica sv. typhimurium by specific bacteriophages A3 and A4 caused O-acetylation of the O-polysaccharide, which altered the binding behavior of the bacteriophages and prevented subsequent infection [32].
In the O-specific polysaccharides of the bacteria of the genus Proteus, the O-acetyl groups are rather common and also contribute to the immunospecificty of strains. For instance, the O-acetylation pattern of the O-polysaccharide served as the basis for the classification of P. vulgaris strains 72/57 and 63/57 into two different serogroups, O37 and O46, respectively [7]. Their O-specific polysaccharides are built up of tetrasaccharide repeating units having the same structure except that the former has three O-acetylation sites (at Glc and two GlcA residues) and the latter only one O-acetylation site at another sugar residue (GlcNAc) (Table 3). Studies with O-antisera against P. vulgaris 63/57 (O37) and 72/57 (O46) revealed close serological relatedness of the lipopolysaccharides of these strains [33]. The O-acetyl groups themselves were not found to form particular epitopes but rather to interfere with binding of anti-P. vulgaris 63/57 (O37) serum to P. vulgaris 72/57 (O46) antigen. Based on these data, we propose to exclude P. vulgaris 72/57 from serogroup O46 and to classify it into Proteus serogroup O37 as the second subgroup.
In several other Proteus serogroups there are pairs of structurally similar O-specific polysaccharides that differ in the presence or the absence of O-acetyl groups either alone or along with other structural features. For instance, the O-specific polysaccharide of P. mirabilis TG 277 studied in this work is a lower O-acetylated variant of the polysaccharide of P. penneri 25 [21] from serogroup O69 [10], which lacks two of three sites of O-acetylation present in the P. penneri 25 polysaccharide (Table 3). These data show that P. mirabilis TG 277 should be classified into the same Proteus serogroup O69 as P. penneri 25.
The trisaccharide repeating units of the polysaccharides of P. penneri strains 19 and 62 have the same topology and sugar composition and differ only in the O-acetylation of a GlcNAc residue in strain 19 but not in strain 62, as well as in the mode of substitution of N-acetylisomuramic acid (at position 4 or 6, respectively) [9, 10]. Hence, these strains, along with the P. penneri 71 strain, which contains N-acetylglucosamine instead of N-acetylisomuramic acid and lacks O-acetyl groups, were classified intro serogroup O64 as three subgroups [34].
The O-specific polysaccharides of P. penneri strains 1 and 4 studied in this work differ in glucosylation of one of the GalNAc residues and O-acetylation of the other residue in P. penneri 4 (Table 3). Based on these data, we propose to combine P. penneri strains 1 and 4 in one Proteus serogroup, O72, as two subgroups.
Similarly, the polysaccharide of P. penneri 16 and 20 on one hand and P. vulgaris 32/57 from serogroup O17 [1] on the other hand have the same structure of the main carbohydrate chain but differ in the presence of a terminal glucose residue and an O-acetyl group in P. penneri and the presence of ethanolamine phosphate in P. vulgaris. These data and the data of serological studies, which will be published elsewhere, showed that it is reasonable to include strains P. penneri 16 and 20, as well as serologically identical strains P. penneri 10, 18, 32, 45, 108 and 111, into Proteus serogroup O17 as the second subgroup.
Therefore, differences in the O-acetylation pattern contribute to the differentiation between Proteus strains having structurally and serologically related O-antigens. On the other hand, the structural data of this work, along with serological data [10], showed that it is reasonable to combine such strains in one serogroup, which improves the existing classification of Proteus strains.
It should be noted that in the most Proteus O-specific polysaccharides O-acetylation is non-stoichiometric and thus masks the regularity of the polymers. For instance, the degree of O-acetylation at each site in the O-specific polysaccharides studied in this work varies from 30 to 95% (Table 3). This peculiar feature enabled the suggestion that O-acetylation is a post-polymerization modification of the O-specific polysaccharides, but the exact mechanisms of the Proteus O-antigen biosynthesis have not been clarified yet.
In some polysaccharides the degree of O-acetylation is so low that the exact location of the O-acetyl groups could be determined only tentatively, if at all. For instance, in previous studies of the polysaccharide of P. penneri 16 (O17) the positions of O-acetyl groups, which are present in a minority of the repeating units, were not determined [22]. In this work, the polysaccharide of P. penneri 20 was found to have the same structure as that of P. penneri 16, including the O-acetylation pattern, and further studies showed that O-acetyl groups in this polysaccharide substitute ~30% GlcA residues and ~40% Glc residues (Table 3). In the O-specific polysaccharides of P. mirabilis O23 [35] and P. vulgaris O32 [36], the location of the O-acetyl groups was determined only tentatively. The position of only one, major O-acetyl group was determined in the polysaccharide of P. penneri 4 (this work), whereas the attachment sites of the other, minor O-acetyl groups remain unknown.
This work was supported by the Russian Foundation for Basic Research (grants 99-04-48279 and 00-04-06599) and the Sciences Research Committee (KBN, Poland, grant 6 PO4A 074 20).
REFERENCES
1.Toukach, F. V., Kondakova, A. N., Arbatsky, N. P.,
Senchenkova, S. N., Shashkov, A. S., Knirel, Y. A., Zych, K., and
Sidorczyk, Z. (2002) Biochemistry (Moscow), 67,
265-276.
2.Warren, J. W. (1996) in Urinary Tract
Infections, Molecular Pathogenesis and Clinical Management
(Mobley, H. L. T., and Warren, J. W., eds.) ASM Press, Washington
DC, pp. 2-28.
3.Stamm, W. E. (1999) in Clinical Infection
Diseases: a Practical Approach (Root, R. K., ed.)Oxford University
Press, New York, pp. 549-656.
4.Wilson, C., Thakore, A., Isenberg, D., and
Ebringer, A. (1997) Rheumatol. Int., 16, 187-189.
5.O'Hara, C. M., Brenner, F. W., Streigerwalt, A. G.,
Hill, B. C., Holmes, B., Grimont, P. A. D., Hawkey, P. M., Penner, J.
L., Miller, J. M., and Brenner, D. J. (2000) Int. J. Syst. Evol.
Microbiol., 50, 1869-1875.
6.O'Hara, C. M., Brenner, F. W., and Miller, J. M.
(2000) Clin. Microbiol. Rev., 13, 534-546.
7.Larsson, P. (1984) Meth. Microbiol.,
14, 187-214.
8.Penner, J. L., and Hennessy, J. N. (1980) J.
Clin. Microbiol., 12, 304-309.
9.Knirel, Y. A., Kaca, W., Rozalski, A., and
Sidorczyk, Z. (1999) Pol. J. Chem., 73, 895-907.
10.Zych, K., Kowalczyk, M., Knirel, Y. A., and
Sidorczyk, Z. (2000) in Genes and Proteins Underlying Microbial
Urinary Tract Virulence. Basic Aspects and Applications (Hacker,
J., Blum-Ochtev, G., Pal, T., and Emody, L., eds.) Kluwer
Academic/Plenum Publishers, New York, pp. 339-344.
11.Westphal, O., and Jann, K. (1965) Meth.
Carbohydr. Chem.,5, 83-91.
12.Shahskov, A. S., Toukach, F. V., Paramonov, N.
A., Ziolkowski, A., Senchenkova, S. N., Kaca, W., and Knirel, Y. A.
(1996) FEBS Lett., 386, 247-251.
13.Leontein, K., Lindberg, B., and Lönngren, J.
(1978) Carbohydr. Res., 62, 359-362.
14.Patt, S. L., and Shoolery, J. N. (1982) J.
Magn. Reson.,46, 535-539.
15.Altona, C., and Haasnoot, C. A. G. (1980) Org.
Magn. Reson.,13, 417-429.
16.Bock, K., and Pedersen, C. (1974) J. Chem.
Soc. Perkin Trans. 2,293-297.
17.Lipkind, G. M., Shashkov, A. S., Knirel, Y. A.,
Vinogradov, E. V., and Kochetkov, N. K. (1988) Carbohydr.
Res.,175, 59-75.
18.Jansson, P.-E., Kenne, L., and Schweda, E. (1987)
J. Chem. Soc. Perkin Trans. 1,377-383.
19.Bock, K., and Pedersen, C. (1983) Adv.
Carbohydr. Chem. Biochem.,41, 27-66.
20.Perepelov, A. V., Babicka, D., Shashkov, A. S.,
Senchenkova, S. N., Rozalski, A., and Knirel, Y. A. (2000)
Carbohydr. Res.,328, 229-234.
21.Arbatsky, N. P., Shashkov, A. S., Widmalm, G.,
Knirel, Y. A., Zych, K., and Sidorczyk, Z. (1997) Carbohydr.
Res.,298, 229-235.
22.Vinogradov, E. V., Sidorczyk, Z., Swierzko, A.,
Rozalski, A., Daeva, E. D., Shashkov, A. S., Knirel, Y. A., and
Kochetkov, N. K. (1991) Eur. J. Biochem.,197, 93-103.
23.Bhasin, N., Albus, A., Michon, F., Livolsi, P.
J., Park, J. S., and Lee, J. C. (1998) Mol.
Microbiol.,27, 9-21.
24.Clarke, A. J., and Dupont, C. (1992) Can. J.
Microbiol.,38, 85-91.
25.Skjak-Braek, G., Larsen, B., and Grasdalen, H.
(1985) Carbohydr. Res.,145, 169-174.
26.Skjak-Braek, G., Grasdalen, H., and Larsen, B.
(1986) Carbohydr. Res.,154, 230-250.
27.Bystricky, S., and Szu, S. C. (1994) Biophys.
Chem.,51, 1-7.
28.Lück, P. C., Freier, T., Steudel, C.,
Knirel, Y. A., Lüneberg, E., Zähringer, U., and Helbig, J. H.
(2001) Int. J. Med. Microbiol.,291, 345-352.
29.Helbig, J. H., Lück, P. C., Knirel, Y. A.,
and Zähringer, U. (1995) Epidemiol. Infect.,115,
71-78.
30.Kooistra, O., Lüneberg, E., Lindner, B.,
Knirel, Y. A., Frosch, M., and Zähringer, U. (2001)
Biochemistry,40, 4630-4640.
31.Allison, G. E., and Verma, N. K. (2000) Trends
Microbiol.,8, 17-23.
32.Wollin, R., Stocker, B. A., and Lindberg, A. A.
(1987) J. Bacteriol.,169, 1003-1009.
33.Torzewska, A., Kondakova, A. N., Perepelov, A.
V., Senchenkova, S. N., Shashkov, A. S., Rozalski, A., and Knirel, Y.
A. (2001) FEMS Immunol. Med. Microbiol.,31, 227-234.
34.Zych, K., Kocharova, N. A., Kowalczyk, M.,
Toukach, F. V., Kaminska, D., Shashkov, A. S., Knirel, Y. A., and
Sidorczyk, Z. (2000) Eur. J. Biochem., 267, 808-814.
35.Cedzynski, M., Swierzko, A. S., Ziolkowski, A.,
Rozalski, A., Kaca, W., Paramonov, N. A., Vinogradov, E. V., and
Knirel, Y. A. (1998) Microbiol. Immunol.,42, 7-14.
36.Bartodziejska, B., Shashkov, A. S., Babicka, D.,
Grachev, A. A., Torzewska, A., Paramonov, N. A., Chernyak, A. Y.,
Rozalski, A., and Knirel, Y. A. (1998) Eur. J.
Biochem.,256, 488-493.