Functional Analysis of the Na+/H+ Antiporter Encoding Genes of the Cyanobacterium Synechocystis PCC 6803
I. V. Elanskaya1*, I. V. Karandashova1, A. V. Bogachev2, and M. Hagemann3
1Department of Genetics, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 138-5207; E-mail: irina@selansky.home.bio.msu.ru2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-0338; E-mail: sodiumc@genebee.msu.su
3Universität Rostock, FB Biowissenschaften, A.-Einstein-Str. 3, D-18057 Rostock, Germany; fax: +49 (0) 381-498-6172; E-mail: martin.hagemann@biologie.uni-rostock.de
* To whom correspondence should be addressed.
Received October 25, 2001; Revision received December 3, 2001
The role of putative Na+/H+ antiporters encoded by nhaS1 (slr1727), nhaS3 (sll0689), nhaS4 (slr1595), and nhaS5 (slr0415) in salt stress response and internal pH regulation of the cyanobacterium Synechocystis PCC 6803 was investigated. For this purpose the mutants (single, double, and triple) impaired in genes coding for Na+/H+ antiporters were constructed using the method of interposon mutagenesis. PCR analyses of DNA demonstrated that mutations in nhaS1, nhaS4, and nhaS5 genes were segregated completely and the mutants contained only inactivated copies of the corresponding genes. Na+/H+ antiporter encoded by nhaS3 was essential for viability of Synechocystis since no completely segregated mutants were obtained. The steady-state intracellular sodium concentration and Na+/H+ antiporter activities were found to be the same in the wild type and all mutants. No differences were found in the growth rates of wild type and mutants during their cultivation in liquid media supplemented with 0.68 M or 0.85 M NaCl as well as in media buffered at pH 7.0, 8.0, or 9.0. The expression of genes coding for Na+/H+ antiporters was studied. No induction of any Na+/H+ antiporter encoding gene expression was found in wild type or single mutant cells grown under high salt or at different pH values. Nevertheless, in cells of double and triple mutants adapted to high salt or alkaline pH some of the remaining Na+/H+ antiporter encoding genes showed induction. These results might indicate that some of Na+/H+ antiporters can functionally replace each other under stress conditions in Synechocystis cells lacking the activity of more than one antiporter.
KEY WORDS: cyanobacterium Synechocystis PCC 6803, Na+/H+ antiporters, salt stress, pH adaptation, interposon mutagenesis, gene expression
Cyanobacteria are photoautotrophic prokaryotes exhibiting plant-like oxygen-evolving photosynthesis. It has long been known that Na+ is essential for many cyanobacteria, especially if they are growing at alkaline pH [1, 2]. A low concentration of sodium appears to be essential for uptake of several inorganic nutrients (e.g., carbon [3]) and even for the photosynthetic electron transport at the O2 evolving complex [4]. While low concentrations of Na+ are required by cyanobacteria, higher concentrations of NaCl may be harmful. The salt tolerance levels of cyanobacteria differ widely. Low salt tolerance strains tolerate a maximum of 0.5 M NaCl, while strains from hypersaline environments depend on enhanced Na+ contents in the medium and tolerate up to 3 M NaCl. In contrast to purely osmotic stress caused by high concentrations of non-permeable organic agents, an increase in salinity not only diminishes the water potential but also means a dramatic increase in inorganic ions (especially Na+ and Cl-) entering the cell along electrochemical gradients. Successful salt adaptation includes two main processes: 1) synthesis and accumulation of compatible solutes; and 2) maintenance of low internal concentrations of sodium ions by the use of active export mechanisms.
Salt acclimation, particularly the biosynthesis of compatible solutes, has been extensively studied in cyanobacteria [5]. Most studies have been performed with the moderately halotolerant cyanobacterial strain Synechocystis PCC 6803 (henceforth referred to as Synechocystis), a cyanobacterial strain whose genome is completely known [6]. However, processes involved in cyanobacterial ion export have been less intensively analyzed. Like most living cells, at high salt concentrations cyanobacteria actively extrude Na+, accumulate K+, and maintain internal ion concentrations comparable to cells grown under low salt concentrations [7]. The extrusion of Na+ in cyanobacteria has been explained by Na+/H+ exchange, since increased Na+/H+ antiporter activities has been detected in salt-adapted cyanobacterial cells. These transporters use DeltaµH+ established by primary H+ pumps like H+-ATPases or respiratory cytochrome oxidase [8-10]. The main role of Na+/H+ antiporters for ion export to achieve high salt tolerance has been clearly shown in Escherichia coli [11]. Alternatively, from bioenergetic studies the action of a primary Na+-ATPase was predicted to serve as the main source for active Na+-extrusion in cyanobacteria [12], since at least under alkaline conditions the adverse transmembrane pH difference prevents generation of substantial DeltaµH+.
Increased Na+/H+ antiporter activities have been shown in cyanobacterial strains grown at enhanced sodium concentrations [8, 10]. Even after cultivation at alkaline pH cyanobacteria maintain a relatively neutral cytoplasm. A sodium cycle involving Na+/H+ antiporters seems to be also involved in maintenance of pH homeostasis in cyanobacteria growing at high pH [12-14] in a similar fashion to heterotrophic bacteria [11]. Interestingly, a variety of Na+/H+ antiporters seems to cooperate in cyanobacteria, since after the analysis of the complete genome sequence of Synechocystis, five genes were identified encoding putative Na+/H+ antiporters [6]. According to sequence similarities, two of these antiporters (Slr1727 = NhaS1; Sll0273 = NhaS2) seem to be of eukaryotic type, while the remaining three proteins (Sll0689 = NhaS3; Slr1595 = NhaS4; Slr0415 = NhaS5) are obviously of prokaryotic type (Fig. 1). For two of these putative transport proteins (NhaS1 and NhaS3) Na+/H+ antiporter activities were successfully shown after the functional expression of their coding genes in E. coli [15]. One of the eukaryotic-type proteins (NhaS2) was found to be essential for the uptake of Na+ at low external concentrations, while it is probably not involved in ion export at high salt concentrations [16].
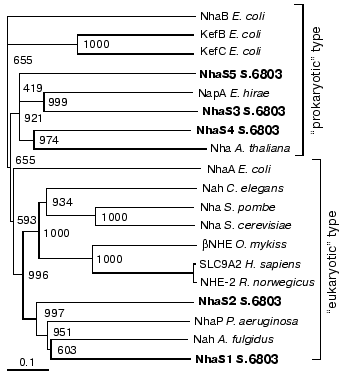
To analyze the importance of these different Na+/H+ antiporters for salt acclimation and also pH regulation of Synechocystis, mutants affected in single Na+/H+ antiporters as well as double and triple mutants affected in different combinations of these genes were generated. These mutants were grown at different salt concentrations and pH values. With the exception of the NhaS3 mutant that could not be segregated, none of the mutants showed a significant growth depression under these conditions. However, the characterization of the expression of antiporter encoding genes showed that some of these transporters were expressed under certain conditions, particularly on the background of different mutations.Fig. 1. Phylogenetic relationships among cyanobacterial Na+/H+ antiporters and functionally related proteins from Archaea, Bacteria, and Eukarya. Cluster analysis was performed using the CLUSTAL X software package. The Na+/H+ antiporter NhaB from Escherichia coli was used as outgroup. Sequences were extracted from the EBI sequence database and CyanoBase. Bootstrap values after 1000 replications are indicated.
MATERIALS AND METHODS
Strains and culture conditions. A derivative of the Synechocystis sp. strain PCC 6803 with enhanced transforming capacity used in all experiments was from the collection of the Department of Genetics of Moscow State University, Russia. Axenic cells were cultured on plates at 30°C under constant illumination using the mineral medium BG-11 [17]. For the physiological characterization, axenic cultures of the cyanobacteria were grown photoautotrophically in batch cultures [18]. The Escherichia coli strain TG1 was used for routine DNA manipulations [19].
DNA manipulations. Total DNA from Synechocystis was isolated according to the method described earlier [20]. All other techniques such as plasmid isolation, transformation of E. coli, and ligation and restriction analysis were standard methods [19] or were performed according to the instructions of the manufacturer of the chemicals used (New England Biolabs). The plasmid vector pGEM-T (Promega, USA) was used to clone the genes of interest after amplification of DNA fragments by PCR. Besides, pUC18 vector plasmid [21] was used for cloning of DNA fragments. The primers for amplification of selected genes (Table 1) were designed using the complete genome sequence of Synechocystis [6].
Table 1. Primers used to amplify
Synechocystis DNA

Generation of insertion mutants. For the generation of mutations in specific ORFs of Synechocystis, the gene conferring kanamycin-resistance (Kmr, 1.2 kb) from pUC4K [21], chloramphenicol-resistance (Cmr, 1.4 kb) from pACYC184 [22], or streptomycin-resistance (Smr, 2.1 kb) from pBSL130 [23] was integrated at selected unique restriction sites into the ORFs cloned into E. coli vectors. Plasmid DNA of these constructs was isolated from E. coli and about 1 µg of DNA was used for transformation of Synechocystis to antibiotic-resistance (Kmr, Cmr, or Smr). Transformants were initially selected on media containing 10 µg*ml-1 Km, 10 µg*ml-1 Cm, or 2 µg*ml-1 Sm while the segregation of clones and cultivation of mutants was performed at 100 µg*ml-1 Km, 20 µg*ml-1 Cm, or 4 µg*ml-1 Sm.
NhaS1 (Slr1727) mutant. APCR fragment containing part of slr1727 (1090 bp) was cloned in pGEM-T. To remove additional HincII sites, the internal DraI/PstI fragment (978 bp) was cut from recombinant plasmid and cloned into SmaI/PstI cut pUC18. For inactivation of this gene, Kmr, or Smr cassette was inserted into the HincII site.
NhaS3 (Sll0689) mutant. A 1172-bp PCR fragment was cloned into pGEM-T. A Kmr, Cmr, or Smr cassette was inserted into the unique Bpu1102 restriction site.
NhaS4 (Slr1595) mutant. A 1137-bp PCR fragment was cloned in pGEM-T. A Kmr, Cmr, or Smr cassette was inserted into the unique BstEII restriction site.
NhaS5 (Slr0415) mutant. A PCR fragment (1154 bp) was cloned in pGEM-T. A Kmr, Cmr, or Smr cassette was inserted into the unique SmaI restriction site.
For the construction of double mutants, completely segregated single mutants conferring inactivated genes with the Smr cassette were transformed by recombinant plasmids carrying the additional nhaS gene inactivated by insertion of the Kmr cassette. Transformants were initially selected on media containing 10 µg*ml-1 Km, then segregated at 100 µg*ml-1 Km and supported at media supplemented with 100 µg*ml-1 Km and 4 µg*ml-1 Sm.
For the construction of a triple mutant, the double nhaS1::Smr/nhaS4::Cmr mutant was transformed by a recombinant plasmid carrying the nhaS5 gene inactivated by insertion of the Kmr cassette.
Measurements of the expression of Na+/H+ antiporter genes. The expressionof Na+/H+ antiporter genes was measured using RNA isolated from cells cultivated at different NaCl concentrations or pH values. Salt-adapted cells were grown for four days in the presence of 0.68 M NaCl. To investigate the influence of pH, the medium was buffered to pH 7.0, 8.0, or 9.0 using MOPS, HEPES, and CHES buffers (Sigma, USA) at final concentrations of 5 mM. Total RNA was isolated from 100-ml portions of the cell cultures. The cells were harvested by centrifugation (4000g, 10 min, 2°C), immediately frozen, and stored at -80°C. RNA was extracted with a High Pure RNA isolation kit (Boehringer, Germany). The methods used for separating RNA, blotting, and hybridization were described in details [20]. A gene-specific DNA hybridization probe was obtained after PCR amplification of its coding sequence using the same primers as for the construction of antiporter mutants (Table 1). The DNA was labeled with [alpha-32P]dATP (Amersham Buchler, Germany) using the HexaLabel DNA labeling kit (MBI Fermentas, Lithuania). Hybridization signals were recorded and quantified with a Phosphoimager apparatus (model BAS1000, Fuji, Japan). To correct the quantitative data for variations in RNA loading, all calculations were related to intensities of hybridization signals obtained after applying a radiolabeled 16S-rRNA-specific probe to the same filters. Primers used for generating the 16S-rRNA specific probes [24] are shown in Table 1. The transcripts of weakly expressed genes were quantified by the RT-PCR-technique. Reverse transcription was performed using SuperScript II RNaseH Reverse Transcriptase (Gibco BRL, USA) according to instructions of the manufacturer. The reaction mixture (20 µl) contained 1 µg of RNA and 10 pmol of each RT-primer (RT-0689-Fw, RT-1727-Rev, RT-1595-Rev, RT-0415-Rev). After reverse transcription the reaction mixtures were diluted (1 : 50) and 1 µl was used for conventional PCR (35 cycles) with both RT-primers (Fw and Rev). PCR products were separated in 2% agarose gel; the optical densities of the bands were recorded and quantified using Bio1D computer software (Vilber Lourmat, France). To correct the quantitative data for variations in RNA loading, all calculations were made using the relative intensities of hybridization signals obtained after a Northern-blot hybridization of the same amount of RNA that was used for reverse transcription with radiolabeled 16S-rRNA-specific probe. The average data and standard deviations were calculated on the base of at least four or five independent PCR reactions for each primer and for each mutant.
Physiological characterization of mutants. Growth and cell density were monitored by reading the absorption of diluted cyanobacterial suspensions at 750 nm (A750) using a U2000 spectrophotometer (Hitachi, Japan).
Measurements of Na+/H+ antiporter activities. Na+/H+ antiporter activity was measured by the acridine orange fluorescence quenching technique [8]using an MPF-4 fluorimeter (excitation 492 nm; emission 530 nm). The assay mixture (total volume 2 ml) was composed of 0.8 M mannitol, 10 mM HEPES/Tris buffer (pH 7.5), and 1 µM acridine orange. Synechocystis cells were grown in the presence of 0.68 M NaCl for three days. The Na+-loaded cells (approximately 25 µg protein) were added to the assay mixture in a volume of 6 µl. Where indicated, 100 µl of 1 M Na2SO4 solution was added to the mixture.
Sodium and potassium steady-state levels and gradients. Cells were salt adapted for three days in BG-11 medium containing 0.68 M NaCl. To measure steady-state intracellular sodium and potassium contents 100-150 µl (13 µg protein) of growing cells were put onto Millipore nitrocellulose filters (0.45 µm) and washed three times with 1 ml of 0.5 M choline chloride. Sodium and potassium concentrations were measured by flame photometry. To estimate the sodium and potassium gradients, intracellular sodium and potassium contents of Synechocystis cells incubated in the growth medium in the presence of 100 µM CCCP ([Na]in = [Na]out and [K]in = [K]out) were also measured.
RESULTS
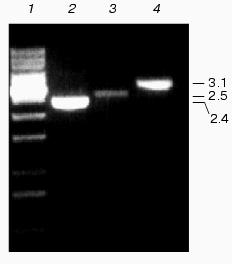
Genetic characterization of Na+/H+ antiporter mutants. Specific mutations of Na+/H+ antiporter coding genes were generated by interposon mutagenesis. Antibiotic-resistant clones were cultivated for several weeks on solid medium in the presence of increasing antibiotic concentrations to enrich the mutated gene in the multi-copy genome of Synechocystis. From selected clones, chromosomal DNA was isolated and analyzed by PCR using corresponding primers. The DNA from single mutants NhaS1, NhaS4, and NhaS5; double mutants NhaS1/NhaS4, NhaS1/NhaS5, and NhaS4/NhaS5; as well as the triple mutant NhaS1/NhaS4/NhaS5, contained only mutated copies (DNA fragments enlarged by the inserted antibiotic resistance gene cassette) of corresponding genes (Fig. 2). The complete segregation was found previously for the NhaS2 mutant [16]. Only for the nhaS3 gene no completely segregated mutant could be obtained. All antibiotic resistant clones were found to be merodiploids, which means they contained a few copies of the mutated genes in parallel to dominating wild type copies of the nhaS3 gene. All attempts to increase the proportion of mutated copies by increasing the antibiotic concentration were unsuccessful. Thus, the NhaS3 Na+/H+ antiporter seems to be essential for cell viability even under normal growth conditions, while the remaining four genes could be inactivated even combined in the double and triple mutants.
Physiological characterization of Na+/H+ antiporter mutants. To analyze the role of inactivated genes in salt or pH tolerance of Synechocystis, cells of single, double, or triple mutants were grown in the presence of 0.68 M or 0.85 M NaCl (Fig. 3) as well as in media buffered to pH 7.0, 8.0, or 9.0 (data not shown). In all cases no significant differences between wild type and mutants were found regarding their growth behavior.Fig. 2. Evidence for complete segregation of mutations in the triple mutant defective in NhaS1, NhaS4, and NhaS5. Total DNA was isolated from cells of NhaS1/NhaS4/NhaS5 mutant and analyzed by PCR using primers for nhaS5 (lane 2), nhaS4 (lane 3), and nhaS1 (lane 4). 1-kb DNA ladder (MBI Fermentas) was used as a molecular weight marker (lane 1). Sizes are given in kb.
Like the majority of organisms, Synechocystis wild type cells grown in sodium-enriched media keep high concentration of potassium and low concentration of sodium in their cytoplasm. The addition of uncoupler CCCP dissipates these cation gradients (Table 2). Essentially the same Na+ and K+ contents and gradients were detectable in wild type and mutant cells adapted to 0.68 M NaCl. This clearly indicates that despite defects of several putative Na+/H+ antiporter genes, all mutants possessed undamaged systems for potassium uptake as well as sodium export.Fig. 3. Growth of wild type (a) and NhaS1/NhaS4/NhaS5 triple mutant (b) in BG-11 medium (1) and in BG-11 supplemented with 0.68 M (2) or 0.85 M (3) NaCl.
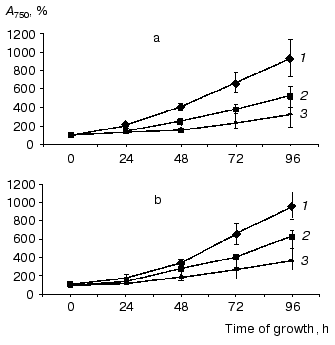
Table 2. Sodium and potassium steady-state
levels (without uncoupler CCCP) and gradients (collapsed by the
uncoupler CCCP) in growing cells of wild type and the triple
Na+/H+ antiporter mutant of
Synechocystis
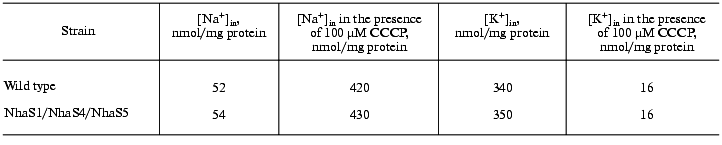
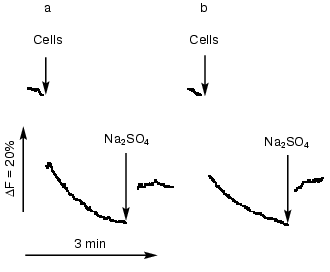
The cyanobacterial cells adapted to NaCl developed efficient systems for sodium extrusion. Changes in fluorescence quenching of acridine orange as a function of transmembrane Na+ gradients were approximately the same in wild type and all Na+/H+ antiporter mutants, providing evidence that Na+/H+ exchange activity was not changed in salt-adapted cells (Fig. 4).
Analysis of the expression of Na+/H+ antiporter genes. Changes of the expression of genes under different environmental conditions might give additional hints for the involvement of their gene products in a special physiological process. Therefore, the steady-state mRNA levels of Na+/H+ antiporter encoding genes was followed during salt- and pH-adaptation by Northern-blot hybridization experiments. The levels of expression of most antiporter encoding genes were very low. Clearly detectable hybridization signals were obtained only with probes specific for nhaS3. The expression of other antiporter genes was below the recognition ability of the Phosphoimager (data not shown).Fig. 4. Na+/H+ antiporter activity in the cells of wild type (a) and NhaS1/NhaS4/NhaS5 triple mutant (b). Na+/H+ antiporter activity was measured by the acridine orange fluorescence quenching technique using an MPF-4 fluorimeter (excitation 492 nm; emission 530 nm). The medium contained 0.8 M mannitol, 10 mM HEPES/Tris, pH 7.5, 1 µM acridine orange in 2-ml volume. As indicated by arrows, the Na+-loaded cells of the corresponding Synechocystis strain (approximately 25 µg protein) were added to the medium in the volume of 6 µl. Where indicated by arrows, 100 µl of 1 M Na2SO4 solution was added to the medium.
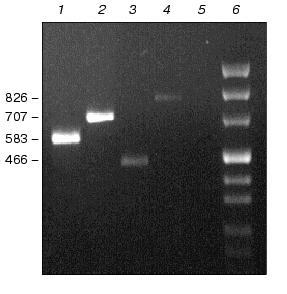
Thus, the low levels of transcription of most Na+/H+ antiporter genes prompted us to apply the more sensitive RT-PCR technique for the analysis of the expression of these genes under stress conditions. Only 1 µg of RNA isolated from salt-adapted or pH-adapted cells was sufficient to obtain enough cDNA, which could be amplified in the following PCR using primers specific for each Na+/H+ antiporter encoding genes. Using RT-PCR the expression of all Na+/H+ antiporter encoding genes could be shown. Moreover, clear quantitative differences in the amplification products of these genes were detected. The highest level of expression was characteristic for nhaS3 gene; the expression of other genes decreased in the following direction: nhaS3 > nhaS1 > nhaS4 > nhaS5 (Fig. 5). To avoid possible differences based on uneven amounts of RNA used in reverse transcription reactions, the amount of RNA used for reverse transcription was compared in Northern-blot hybridization experiments applying a radio-labeled 16S-rRNA-specific probe. These data were used to normalize all quantitative results of RT-PCR obtained by densitometry to the same RNA concentrations in the reaction. Figure 6 shows the results of expression analysis of Na+/H+ antiporter genes in wild type cells grown in the presence of 0.68 M NaCl or in the medium buffered at pH 7.0, 8.0, or 9.0. No statistically significant induction of any of the antiporter encoding genes nhaS1, nhaS3, nhaS4, or nhaS5 was observed in comparison to control cells grown in ordinary BG-11 medium. The same holds true for the expression of nhaS2 gene [16]. The same results were obtained when the expression of Na+/H+ antiporter genes was analyzed in the single mutants NhaS1, NhaS4, and NhaS5 grown under stress conditions (data not shown).
Fig. 5. Separation of PCR products amplified from cDNAs obtained by reverse transcription using RT-primers for nhaS3 (lane 1), nhaS1 (lane 2), nhaS4 (lane 3), and nhaS5 (lane 4). The reaction mixture for reverse transcription (20 µl) contained 1 µg of RNA and 10 pmol of each RT-primers. After reverse transcription the reaction mixtures were diluted (1 : 50) and 1 µl was used for conventional PCR (35 cycles) with both RT-primers (Fw and Rev). DNA contamination of RNA extracts was checked by PCR without adding reverse transcriptase (lane 5). 1-kb DNA ladder was used as a marker (lane 6). Sizes are given in bp.
A reliable induction of some Na+/H+ antiporter encoding genes was only observed in the background of double and triple mutants adapted to high sodium concentration or to high pH (pH 8.0 or 9.0, Fig. 7). In the cells of double mutants NhaS1/NhaS5 and NhaS4/NhaS5 and triple mutant NhaS1/NhaS4/NhaS5, respectively, a clear increase in mRNA level of the nhaS3 gene was observed under these conditions (Fig. 7). Nevertheless, the expression of nhaS3 was not induced in NhaS1/NhaS4 double mutant grown under the same stress conditions. Since no induction of nhaS3 gene expression was found in NhaS5 single mutant, these results may indicate that Na+/H+ antiporter encoded by the nhaS3 gene might be necessary to replace the function of NhaS5 (but not of NhaS4) only if some other antiporters (NhaS1, NhaS4, or both) are inactivated. No induction of Na+/H+ antiporter encoding gene expression was observed in any mutant adapted to pH 7.0 (Figs. 6 and 7). Beside the essential nhaS3 gene, also the expression of other antiporter encoding genes, nhaS1, nhaS4, or nhaS5, was increased after adaptation to high NaCl concentrations or to alkaline pH values (8.0, 9.0) in cells of double and triple mutants. The expression of the nhaS5 gene was enhanced under salt stress conditions in double mutant NhaS1/NhaS4 and, vice versa, nhaS4 expression was induced under the same conditions in the double mutant NhaS1/NhaS5. These data indicate that Na+/H+ antiporters NhaS4 and NhaS5 can probably replace each other in stressed mutant cells. The expression of nhaS1 was only observed in salt- and alkaline pH value-adapted cells lacking both NhaS4 and NhaS5. The results concerning the induction of single Na+/H+ antiporter genes in defined mutants are summarized in Table 3.Fig. 6. Relative levels of expression of nhaS1, nhaS3, nhaS4, and nhaS5 genes, respectively, in wild type cells adapted to 0.68 M NaCl (2), or grown in the medium buffered at pH 7.0 (3), pH 8.0 (4), or pH 9.0 (5). The level of expression in the corresponding strain grown in BG-11 was considered as 100% (1).
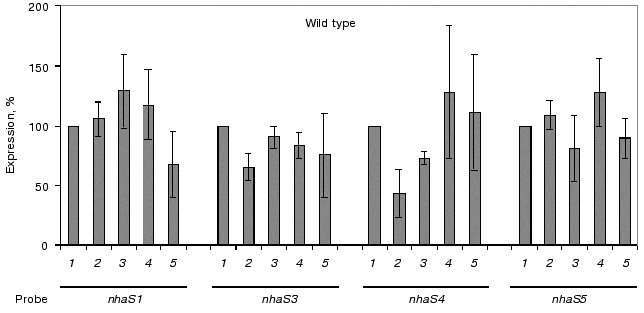
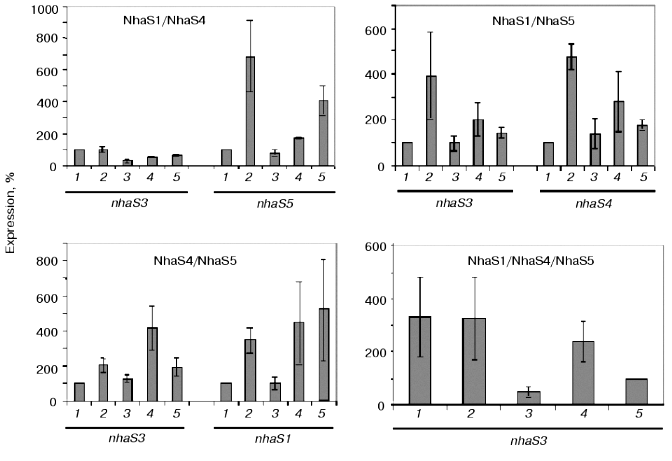
Table 3. Induction of Na+/H+ antiporter genes in wild type and antiporter mutants under different growth conditionsFig. 7. Relative levels of expression of nhaS1, nhaS3, nhaS4, and nhaS5 genes in double and triple Na+/H+ antiporter mutants adapted to 0.68 M NaCl (2), or grown in the medium buffered at pH 7.0 (3), pH 8.0 (4), or pH 9.0 (5). The level of expression in the corresponding strain grown in BG-11 was considered as 100% (1).

DISCUSSION
At least five genes encoding putative Na+/H+ antiporters have been assigned in the genome of Synechocystis [6]. Detailed sequence comparisons and phylogenetic analyses (see Fig. 1) showed that the proteins encoded by these genes contain all features known from functionally characterized Na+/H+ antiporters, which make their physiological function very likely [15]. Furthermore, for two of these proteins, NhaS1 and NhaS3, one from the “eukaryotic” as well as one from the “prokaryotic” group, Na+(Li+)/H+ antiporter activity became detectable after their expression in a corresponding E. coli mutant [15]. With this study, as well as with the characterization of NhaS2 mutant [16], we provide clear evidence that all five Na+/H+ antiporter encoding genes are active, which means they are all transcribed even under control conditions in wild type cells. Using mutants affected in single, two or even three of the Na+/H+ antiporter encoding genes, we tried to investigate the role of these transporters in the salt adaptation and/or internal pH regulation of Synechocystis cells.
The present work shows that most of the antiporters are not essential for viability of Synechocystis, since completely segregated single mutants defective in NhaS1, NhaS4, as well as NhaS5 were isolated in the present work. The same was found previously for the NhaS2 mutant [16]. Moreover, in double mutants NhaS1/NhaS4, NhaS1/NhaS5, and NhaS4/NhaS5, as well as the triple mutant NhaS1/NhaS4/NhaS5, all mutations were completely segregated. Only for the nhaS3 gene no completely segregated mutant could be obtained, indicating that the NhaS3 Na+/H+ antiporter seems to be essential for cell viability even under normal growth conditions. Also, Inaba et al. [15] were not able to obtain a mutant affected in this gene. This gives a clear indication that NhaS3 might be the most important Na+/H+ antiporter in Synechocystis, which can at least functionally replace all other antiporters. Our finding that the nhaS3 gene showed the highest expression level supports the high importance of NhaS3. The nhaS3 gene was the only detectable one in Northern blot experiments and led also to the highest signal intensity in RT-PCR. Furthermore, it was demonstrated [15] that the NhaS3 showed the highest affinity for Na+ and Li+.
In the present work it was demonstrated that single, double, and triple mutants affected in the nhaS1, nhaS4, and nhaS5 genes did not differ from wild type cells in the ability to grow at high salt concentrations and alkaline pH values. Thus, the activity of the corresponding proteins is not necessary for the adaptation of Synechocystis cells for growth under these conditions. This conclusion is supported as well by the fact that all mutant strains keep the same Na+ and K+ gradients at high NaCl concentrations as wild type cells. These results can be interpreted in two ways. First, according to the initial hypothesis that Na+/H+ antiporters are responsible for such purposes it can be concluded that the activity of NhaS3 is fully sufficient to fulfill these tasks. Second, nevertheless, we cannot rule out that alternative mechanisms like primary Na+ exporting mechanisms [12, 25] are responsible for these functions in the mutants.
However, the present data provide some indications that the Na+/H+ antiporters are necessary for special purposes, since on the background of specific mutants the upregulation of certain Na+/H+ antiporter encoding genes was observed. In wild type cells and single mutants no induction of nhaS1, nhaS3, nhaS4, or nhaS5 genes was observed under high salt or alkaline pH. This might indicate that in spite of the low expression of these genes the corresponding Na+/H+ antiporters can manage the removal of surplus Na+ or the adjusting of the pH. Furthermore, there are several data that the transport activity of the existing Na+/H+ antiporters is directly stimulated by environmental factors. Immediately after the addition of Na+ ions in salt-shocked cells a dramatically increase in Na+ extrusion and Na+/H+ antiporter activity has been measured [7-10], which is obviously sufficient to deal with the new environmental conditions. A direct influence on transport activity rather then gene expression has been also shown in salt-stressed bacteria for proteins involved in transport of compatible solutes. Nevertheless, in double and triple mutants we could observe the increased expression of some Na+/H+ antiporter encoding genes. Particularly, the induction of the nhaS3 gene expression was observed in salt- or high-pH-adapted cells of double and triple mutants lacking functional NhaS5. This seems to indicate that NhaS5 and NhaS3 are functional closely related as one can also predict from their close vicinity in the phylogenetic tree (Fig. 1). Moreover, the study of expression of nhaS1, nhaS4, and nhaS5 genes in different double mutants gave additional indications that corresponding Na+/H+ antiporters might probably replace each other under salt-stress conditions in Synechocystis cells lacking the activity of more than one antiporter.
This work was supported by a grant of the Deutsche Forschungsgemeinschaft (DFG), a grant of the Russian Ministry of Science and Technology (project “Phytobiotechnology”), and a travel grant to I. V. Elanskaya from the Deutscher Akademischer Austauschdienst (DAAD).
REFERENCES
1.Allen, M. B. (1952) Arch. Microbiol.,
17, 34-53.
2.Kratz, W. A., and Myers, J. (1955) Am. J.
Botany, 4, 282-287.
3.Kaplan, A., Volokita, M., Zenvirth, D., and
Reinhold, L. (1984) FEBS Lett., 176, 166-168.
4.Zhao, J., and Brand, J. J. (1988) Arch. Biochem.
Biophys., 264, 657-664.
5.Hagemann, M., Schoor, A., Mikkat, S., Effmert, U.,
Zuther, E., Marin, K., Fulda, S., Vinnemeier, J., Kunert, A.,
Milkowski, C., Probst, C., and Erdmann, N. (1999) in Microbiology
and Biogeochemistry of Hypersaline Environments (Oren, A., ed.) CRC
Press LLC, Boca Raton, pp. 177-186.
6.Kaneko, T., Sato, S., Kotani, H., Tanaka, A.,
Asamizu, E., Nakamura, Y., Miyajima, N., Hirosawa, M., Sugiura, M.,
Sasamoto, S., Kimura, T., Hosouchi, T., Matsuno, A., Muraki, A.,
Nakazaki, N., Nruo, K., Okumura, S., Shimpo, S., Takeuchi, C., Wada,
T., Watanabe, A., Yamada, M., Yasuda, M., and Tabata, S. (1996) DNA
Res., 3, 109-136.
7.Reed, R. H., and Stewart, W. D. P. (1985) Mar.
Biol., 88, 1-9.
8.Blumwald, E., Wolosin, J. M., and Packer, L. (1984)
Biochem. Biophys. Res. Commun., 122, 452-459.
9.Molitor, V., Erber, W., and Peschek, G. A. (1986)
FEBS Lett., 204, 251-256.
10.Nitschmann, W. H., and Packer, L. (1992) Arch.
Biochem. Biophys., 294, 347-352.
11.Padan, E., and Schuldiner, S. (1993) J.
Bioenerg. Biomembr., 25, 647-669.
12.Ritchie, R. J. (1992) J. Plant Physiol.,
139, 320-330.
13.Miller, A. G., Turpin, D. H., and Canvin, D. T.
(1984) J. Bacteriol., 159, 100-106.
14.Buck, D. P., and Smith, G. D. (1995) FEMS
Microbiol. Lett., 128, 315-320.
15.Inaba, M., Sakamoto, A., and Murata, N. (2001)
J. Bacteriol., 183, 1376-1384.
16.Mikkat, S., Milkowski, C., and Hagemann, M.
(2000) Plant Cell Environ., 23, 549-559.
17.Rippka, R., Deruelles, J., Waterbury, J. B.,
Herdman, M., and Stanier, R. Y. (1979) J. Gen. Microbiol.,
111, 1-61.
18.Erdmann, N., Fulda, S., and Hagemann, M. (1992)
J. Gen. Microbiol., 138, 363-368.
19.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: a Laboratory Manual, 2nd ed., Cold
Spring Harbour Laboratory Press, Cold Spring Harbour, New York.
20.Hagemann, M., Richter, S., and Mikkat, S. (1997)
J. Bacteriol., 179, 714-720.
21.Vieira, J., and Messing, J. (1982) Gene,
19, 259.
22.Chang, A. C. Y., and Cohen, S. N. (1978) J.
Bacteriol., 134, 1141-1156.
23.Alexeyev, M. F., Shokolenko, I. N., and Croughan,
T. P. (1995) Gene, 160, 63-67.
24.Lane, D. J. (1991) in Nucleic Acid Techniques
in Bacterial Systematics (Stackebrandt, E., and Goodfellow, M.,
eds.) John Wiley & Sons, Chichester, United Kingdom, pp.
115-175.
25.Ritchie, R. J. (1998) Can. J. Botany,
76, 1127-1145.