Some Properties of Site-Specific Nickase BspD6I and the Possibility of Its Use in Hybridization Analysis of DNA
L. A. Zheleznaya1, T. A. Perevyazova1, E. N. Zheleznyakova2, and N. I. Matvienko2*
1Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, 142290 Russia; fax: (0967) 790-5532Institute of Protein Research, Russian Academy of Sciences, Pushchino, Moscow Region, 142290 Russia; fax: (095) 924-0493; E-mail: nikmatv@vega.protres.ru
* To whom correspondence should be addressed.
Received November 5, 2001; Revision received November 30, 2001
A new method for hybridization analysis of nucleic acids is proposed on the basis of the ability of site-specific nickases to cleave only one DNA strand. The method is based on the use of a labeled oligonucleotide with the recognition site of the nickase hybridized with the target (DNA or RNA) at an optimal temperature of the enzyme (55°C). The two shorter oligonucleotides formed after the cleavage with the nickase do not complex with the target. Thus, a multiple cleavage of the labeled oligonucleotide takes place on one target molecule. The cleavage of the nucleotide is recorded either by polyacrylamide gel electrophoresis (when a radioactive labeled oligonucleotide is used) or by fluorescence measurements (if the oligonucleotide has the structure of a molecular beacon). The new method was tested on nickase BspD6I and a radioactive oligonucleotide complementary to the polylinker region of the viral DNA strand in bacteriophage M13mp19. Unfortunately, nickase BspD6I does not cleave DNA in the RNA-DNA duplexes and therefore cannot be used for detection of RNA targets.
KEY WORDS: site-specific nickases, hybridization of nucleic acids, restriction endonucleases, molecular beacons
Site-specific nickases are a new class of enzymes. Nickases recognize a specific sequence (site) on a double-stranded DNA and cleave only one of the DNA strands in a strictly determined manner. This class of enzymes includes only three members so far. The first nickase (N. BstSE) was discovered by Abdurashitov et al. [1], who suggested the name and nomenclature for the new enzyme. The second nickase (N. BstNBI) was found by Morgan et al. [2]. The third nickase (N. BspD6I) was discovered by us [3]. Though the three nickases were purified from different sources, they have the same specificity: they recognize site GAGTC on a double-stranded DNA and at a distance of four nucleotides from the site towards the 3´-end cleave only the strand containing sequence 5´-GAGTC-3´.
The fact that all the nickases found so far recognize the same site can be explained by the peculiarity of fragmentation with nickases of a double-stranded DNA: the nickases cleave DNA in fragments only if two oppositely oriented sites are positioned at a short distance from each other. In screening strains for the presence of site-specific endonucleases, phage T7 DNA is widely used as a substrate DNA. In this DNA, closely positioned inverse sequences GAGTC occur quite often (4 times), which is caused by the presence of sequence GAGTC in phage-specific promoters [4]. Thus it can be assumed that in nature there is a significant number of nickases having a different specificity, which however cannot be detected with the used substrate DNAs.
By some of their properties, the discovered site-specific nickases can be regarded as a new component of the modification-restriction system. First, two restriction endonucleases of type IIs are known, PleI and MlyI [5], that recognize the same site as the nickases do, but cleave both DNA strands, one of them being cleaved at the same distance from the site as the nickases. Second, it has been shown that, like restriction endonucleases, nickase BspD6I is sensitive to methylation [3] and, the same as in modification-restriction systems, the methylase gene is positioned side by side with the gene of nickase BstNBI [6].
It is not clear yet whether nickases appeared by mutation of the gene of restriction endonuclease capable of cleaving two DNA strands or, on the contrary, these restriction endonucleases evolved from nickases.
In this paper we describe the principle of the new method of hybridization analysis of nucleic acids that employs site-specific nickases and study the properties of nickase BspD6I important for the method.
MATERIALS AND METHODS
Phage M13mp19 DNA and plasmid pSPT18 DNA [7] as well as enzymes (phage T4 polynucleotide kinase, SP6 RNA polymerase, T7 RNA polymerase, EcoRI and HindIII endonucleases) used in the investigations were purified by us.
DNA cleavage. This was done with restriction endonucleases in conditions optimal for them. The products of cleavage were analyzed by agarose gel (1.0%) electrophoresis in 1×TBE buffer (0.089 M Tris, 0.089 M boric acid, 1 mM EDTA, pH 8.3).
Oligonucleotide SSN (5´-TCTAGAGTCGACCTGCAGGCATG-3´) was synthesized by Sintol (Russia). It was labeled at the 5´-end using [gamma-32P]ATP (Cluster Scientific Production Association, Russia) and phage T4 polynucleotide kinase as described earlier [8]. The non-incorporated label was removed from the labeled oligonucleotide by column C-18 reverse-phase chromatography [9].
Transcription in vitro was performed in 50 µl on a linearized DNA of pSPT18 (50 µg/ml) with SP6 RNA-polymerase (at 40°C) or T7 RNA-polymerase (at 37°C) for 2 h in a buffer containing 10 mM HEPES/KOH, pH 7.5, 12 mM MgCl2, 40 mM DTT, 2 mM spermidine, 5 mM of each ribonucleoside triphosphate, 200 units/ml of human placental ribonuclease inhibitor (Promega, USA) and 100 to 500 units of SP6 or T7 RNA polymerase. Then the mixture was treated with pancreatic DNAse I (Koch Light, England) at a final concentration of 0.05 mg/ml for 30 min at 37°C, phenol deproteinization was made and RNA precipitated with ethanol. The precipitate was dissolved in a buffer containing 10 mM Tris-HCl, pH 7.5, 0.1 mM EDTA. The obtained transcripts were analyzed in a denaturing (7 M urea) 12% polyacrylamide gel. The concentration of RNA transcripts was determined by the electrophoresis data compared to the RNA of the known concentration (16S RNA from E. coli).
RNA-DNA hybrids were obtained by incubation of the labeled oligonucleotide with the RNA transcript at 95°C for 5 min with the following cooling to 55°C during 40 min. The hybrid formation was monitored by electrophoresis in 8% polyacrylamide gel in 1×.BE buffer with subsequent radioautography of the gel on a Retina X-ray film (Germany).
Hydrolysis of DNA-RNA hybrids and DNA-DNA duplexes with nickase BspD6I was performed in buffer containing 10 mM Tris-HCl, pH 7.5, 100 mM KCl, 10 mM MgCl2, 10 units of nickase at 55°C for 1 h.
The analysis of the products of hydrolysis was made by electrophoresis in 20% polyacrylamide gel containing 7 M urea, 1×.BE buffer at 50°C on a Macrophor device (LKB, Sweden). On termination of the electrophoresis, the analyzed products were transferred onto a DEAE cellulose filter (BioRad, USA) using a Transblot-cell (BioRad). The transfer was performed in a 0.05×.AE buffer (1×.AE: 50 mM Tris-Ac, pH 8.0, 2 mM EDTA) for 30 min, at the current strength of 250 mA and voltage of 50 V. The position of the bands on the filter was determined using radioautography.
RESULTS
1. Method of utilization of nickases for hybridization analysis of nucleic acids. DNA-DNA and DNA-RNA hybridization techniques are the basic methods for detecting the presence of the required nucleic acid in a composite mixture of fragments. It should be emphasized that in all hybridization techniques, from the very first that employ nitrocellulose filters [10] to contemporary ones with the use of molecular beacons [11], one labeled molecule of a probe is hybridized with a target molecule, which limits the sensitivity of the methods. The method with the use of site-specific nickases suggested by us can significantly raise the sensitivity of the hybridization analysis.
The idea is to employ for hybridization a labeled oligonucleotide containing a site recognized by nickase, where the oligonucleotide should be a cleaved DNA strand and be stably hybridized with the target DNA in a reaction buffer for nickase and at an incubation temperature optimal for its work. Since the known nickases recognize the pentanucleotide sequence, the latter is often found on DNA. Therefore in general the choice of an oligonucleotide for hybridization is a quite simple task. In a rather elongated target DNA, in most cases there is a site recognized by nickase. After hybridization of the oligonucleotide with a preliminarily denatured DNA, the nickase existing in the reaction mixture would cleave the oligonucleotide, with two shorter oligonucleotides being formed. Their length should be such that they could not form stable duplexes with the target DNA at the temperature necessary for the nickase work. As a result of melting of the duplexes, the target DNA can hybridize with the following labeled oligonucleotide and the process of the oligonucleotide cleavage will be repeated. Thus, the hybridization signal will increase as much as the number of labeled oligonucleotides cleaved upon incubation. The cleavage of oligonucleotides can be monitored with electrophoresis in polyacrylamide gel.
The proposed method can be used also for detecting target RNAs if nickases are capable of cleaving the DNA within the DNA-RNA hybrid.
For the discussed scheme to work, the nickase should satisfy two requirements. First, it should cleave only one strand and only in a duplex. Second, it should be capable of turnovers of the cleavage acts. Earlier we demonstrated that nickase BspD6I isolated by us meets the first requirement: it cleaves only one strand and only in a double-stranded DNA. The aim of this work is to clarify whether our nickase satisfies the second requirement and whether it is capable of cleaving the DNA within the RNA-DNA hybrid.
2. Analysis of the ability of nickase BspD6I for turnover of the cleavage acts. Similarly to type IIs restriction endonucleases, nickases recognize the asymmetric sequence and cleave DNA apart from the recognition site. However, there are literature data that type IIs endonucleases remain bound to the site after cleavage of DNA, i.e., they are incapable of multiple recurrence of DNA cleavage (cited in [12]). Therefore it was necessary to clarify whether nickase BspD6I is capable of multiple recurrences of the cleavage acts.
The work was carried out on a model system using a single-stranded DNA of phage M13mp19 and a labeled oligonucleotide SSN complementary to the polylinker region (target) of this DNA. The oligonucleotide contained sequence 5´-GAGTC-3´ and, upon hybridization with the DNA, formed a duplex with the site recognized by nickase at such an orientation that the nickase cleaved the oligonucleotide. The oligonucleotide also satisfied the following requirements: it formed a stable duplex with the nickase at an incubation temperature (55°C) and its fragments melted after cleavage with the nickase. The cleavage was monitored by the appearance of a cleaved, labeled fragment on a radioautograph.
A series of tests was performed in which the amount of oligonucleotide remained unchanged (1 pmol) and the amount of DNA in different tests varied from 1 to 10-6 pmol. The samples were incubated with a standard amount of nickase (10 units). The results given in Fig. 1 show that cleavage of the total oligonucleotide in the reaction mixture occurs at a molar ratio of DNA to oligonucleotide 1 : 10. This demonstrates that during the reaction the following processes are multiply repeated (no less than 10 times): hybridization of the oligonucleotide with the DNA, cleavage of the oligonucleotide within the duplex, removal of cleaved parts of the oligonucleotide from the target DNA and attachment of the next oligonucleotide.
The other task was to clarify whether the suggested method is valid for work with a composite mixture of DNA fragments. The main problem that could arise during the work was renaturation of DNA during incubation with the nickase and, as a result, the appearance of double-stranded DNA sites recognized by the nickase; in this case the nickase would be “diverted” to cleavage of the renatured DNA. To answer this, a preliminarily denatured (95°C, 5 min) DNA from calf thymus (2 µg) was added to the reaction mixture containing the oligonucleotide, the target DNA (0.15 µg) and the nickase. The same mixture, but without the target DNA, was used as a control. An additional control was the test in which the oligonucleotide was incubated with the nickase in the presence of a preliminarily denatured double-stranded DNA from phage M13mp19 (a replicative form). The results showed that the oligonucleotide is cleaved only in the presence of the target DNA (Fig. 1b, lanes 3, 4, and 6) and is not cleaved when it is absent (Fig. 1b, lane 5), i.e., the proposed method works in a composite mixture of DNA fragments.Fig. 1. a) Cleavage of labeled oligonucleotide with nickase at different ratios of oligonucleotide and target RNA (single-stranded DNA of phage M13mp19). I, initial oligonucleotide (1 pmol); II, the same preparation with added target DNA. Numbers show the amount of target DNA (pmol) in the reaction mixture. b) Cleavage of oligonucleotide with nickase in the presence of calf thymus DNA: 1) initial oligonucleotide; 2) oligonucleotide with added nickase; 3-6) preparations containing oligonucleotide, nickase, and the following components: 3) target DNA; 4) target DNA and thymus DNA; 5) thymus DNA; 6) double-stranded DNA from phage M13mp19. Electrophoresis in 20% polyacrylamide gel (7 M urea).
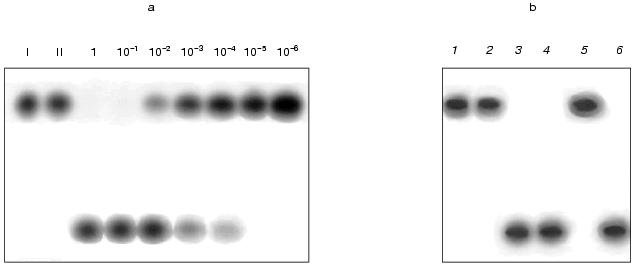
Thus, nickase BspD6I can be used in the suggested method of hybridization analysis of DNA.
3. Analysis of sensitivity of DNA-RNA hybrids to hydrolysis with nickase BspD6I. It is known that some restriction endonucleases can cleave DNA within a DNA-RNA hybrid [13]. That is why it could be expected that nickase BspD6I would also cleave DNA within the DNA-RNA hybrid, which would allow its usage in the proposed method for hybridization analysis of RNA.
The study was performed on a model system. Two transcripts, prepared on linearized plasmid pSPT18 in which the polylinker sequence of plasmid pUC18 is flanked with promoters for SP6 and T7 RNA polymerases, were used as RNA. One transcript, SP6-RNA, was prepared using SP6 RNA polymerase on a plasmid cleaved with restriction endonuclease EcoRI. The other transcript, T7-RNA, was prepared using T7 RNA polymerase on the same plasmid cleaved with restriction endonuclease HindIII. The length of the transcripts was about 60 nucleotides. A labeled SSN oligonucleotide complementary to SP6-RNA and containing the sequence 5´-GAGTC-3´ was used as DNA. The oligonucleotide was hybridized with the SP6- and T7-RNA transcripts. The formation of a hybrid was monitored by polyacrylamide gel electrophoresis (Fig. 2). Figure 2 shows that only in a reaction mixture containing the oligonucleotide and the SP6-RNA transcript was a labeled product formed (lane 2) whose position is above the initial oligonucleotide (lane 1). No such product is found in the mixture (lane 3) containing the oligonucleotide and the T7-RNA transcript. These data indicate that hybridization of the oligonucleotide with the SP6-RNA transcript does take place, while, as should be expected, hybridization with the T7-RNA transcript, which is not complementary to the oligonucleotide, does not occur. The prepared hybrid was incubated with the nickase. As a control of the hydrolysis, a duplex of the oligonucleotide with a single-stranded DNA M13mp19 was incubated with the nickase. The same duplex was incubated with the nickase in the presence of the T7-RNA transcript to verify a possible inhibiting effect of RNA on the nickase activity. Figure 3 shows the results of the experiment. It is seen that the nickase does not hydrolyze DNA within the DNA-RNA hybrid (lane 2), whereas under the same conditions of hybridization and hydrolysis, the nickase cleaves the DNA-DNA duplex (lane 3) even in the presence of RNA (lane 4).
Fig. 2. Scheme of hybridization of oligonucleotide and RNA: 1) initial oligonucleotide; 2) oligonucleotide with transcript SP6-RNA; 3) oligonucleotide with transcript T7-RNA. Electrophoresis in 8% polyacrylamide gel.

Thus, it has been established that nickase BspD6I does not hydrolyze DNA within the DNA-RNA hybrid.Fig. 3. Incapability of nickase to cleave the RNA-DNA hybrid: 1) initial oligonucleotide; 2) hybrid of oligonucleotide with transcript SP6-RNA incubated with nickase; 3) duplex of oligonucleotide and DNA from phage M13mp19 incubated with nickase; 4) the same duplex incubated with nickase in the presence of transcript T7-RNA. Electrophoresis in 20% polyacrylamide gel (7 M urea).

DISCUSSION
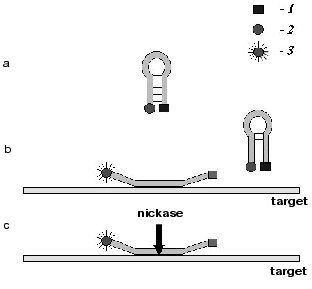
The results have shown that nickase BspD6I can be used in the proposed method for hybridization analysis of DNA. Of special promise for the hybridization analysis in solution is a combined use of nickases and molecular beacons [11]. A molecular beacon is an oligonucleotide with mutually complementary ends (Fig. 4a). In the free state it has the structure of a stem with a loop. The sequence of the loop is complementary to the target DNA. A fluorophore is attached to one end of the oligonucleotide and a fluorescence quencher to the other. Thus, in the free state the beacon does not fluoresce. Upon hybridization of the beacon to the target DNA, its ends diverge and the quencher no longer scatters the energy absorbed by the fluorophore (Fig. 4b), and by the fluorescence intensity one can judge the presence and amount of the target DNA in the sample.
As early as in the first experiments an exciting circumstance was revealed. It appeared that the temperature of the stem melting in the beacon is much higher (by more than 10°C) than that at hybridization of the same sequences within two different molecules. It is this circumstance that gives hope for a development of the hybridization analysis with the use of beacons and nickases. The stability of the stem in the beacon should be chosen so that the hairpin would be stable in the entire beacon at the incubation temperature. However upon the oligonucleotide cleavage in the loop and removal of the target DNA, the two formed oligonucleotides should not hybridize in solution (Fig. 4c). Therefore the fluorophore and the quencher will dissociate. Sensitization of many fluorophores will occur on one target molecule as a result of multiple cleavage acts of the beacon. Thus, in contrast to the traditional modes of beacon activation due to a single hybridization, with the use of nickases the activation is realized mainly at the expense of cleavage of many molecules of the beacon.Fig. 4. Scheme demonstrating the use of a molecular beacon in hybridization analysis of nucleic acids with the use of site-specific nickases: a) beacon structure; b) binding of beacon to the target; c) cleavage with nickase of several beacon molecules; 1) fluorescence quencher; 2) fluorophore whose emission is absorbed by the quencher; 3) emitting fluorophore.
The nickase isolated by us cannot cleave DNA within the DNA-RNA hybrid and consequently cannot be used for hybridization analysis of the RNA. We do not know whether nickases BstSE and BstNBI are capable of cleaving DNA-RNA hybrids. However, if they have no such capability either, probably, as we have noted, nickases recognizing sites different from the recognition sites of the currently known nickases may be found in future. Among the novel nickases there might be those that would cleave DNA-RNA hybrids.
The study was supported by the Russian Foundation for Basic Research, grant No. 99-04-48272.
REFERENCES
1.Abdurashitov, M. A., Belichenko, O. A., Shevchenko,
A. V., and Degtyarev, S. Kh. (1996) Mol. Biol. (Moscow),
30, 1261-1267.
2.Morgan, R. D., Calvet, C., Demeter, M., Agra, R.,
and Kong, H. (2000) Biol. Chem.,381, 1123-1125.
3.Zheleznaya, L. A., Perevyazova, T. A., Alzhanova,
D. V., and Matvienko, N. I. (2001) Biochemistry (Moscow),
66, 989-994.
4.Dunn, J. J., and Studier, F. W. (1983) J. Mol.
Biol., 166, 477-535.
5.Roberts, R. J., and Macelis, D. (2000) Nucleic
Acids Res., 28, 306-307.
6.Higgens, L. S., Besnier, C., and Kong, H. (2001)
Nucleic Acids Res., 29, 2492-2501.
7.Boehringer Mannheim Catalog (1987) Mannheim,
Germany.
8.Maniatis, T., Fritsch, E., and Sambrook, J. (1984)
Molecular Cloning [Russian translation], Mir, Moscow.
9.Matvienko, N. N., Kramarov, V. M., Zheleznaya, L.
A., and Matvienko, N. I. (1993) Biochemistry (Moscow),
58, 803-814.
10.Prives, S. L., Aviv, N., Paterson, V. M.,
Roberts, B. E., Rozenblatt, S., Revel, M., and Winocour, E. (1974)
Proc. Natl. Acad. Sci. USA, 71, 3020-3025.
11.Tyagi, S., and Kramer, F. R. (1996) Nature
Biotech., 14, 303-306.
12.Kong, H., Morgan, R. D., Maunus, R. E., and
Schidkraut, I. (1993) Nucleic Acids Res., 21,
987-991.
13.Molloy, P. L., and Symons, R. H. (1980)
Nucleic Acids Res., 8, 2939-2946.