Glutamate Decarboxylase: Computer Studies of Enzyme Evolution
B. S. Sukhareva* and O. K. Mamaeva
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow, 119991 Russia; fax: (095) 135-1405; E-mail: suhareva@eimb.ru* To whom correspondence should be addressed.
Received April 19, 2002; Revision received June 5, 2002
The homology of subunit primary sequence of 40 glutamate decarboxylases (GAD) of different origin was analyzed by multiple alignment. A phylogenetic tree was designed on the basis of the resulting data. The following groups are distinguished in the consensus tree: archeans, bacteria, plant eukaryotes, and animal eukaryotes. The latter are clearly divided into two branches according to two enzyme isoforms. Borders of PLP domains in each enzyme were detected. The consensus phylogenetic tree for PLP domains is structurally rather similar to that obtained for subunits. Twenty homologous motifs of from 15 to 87 amino acid residues were revealed in all GAD studied. The results revealed the division of all of the enzymes into groups with characteristic sets of motifs in each and a fixed order of their arrangement along the sequence. Thus, we can show the divergent evolution of the enzyme. The results of multiple alignments during structural analysis of the 40 GAD confirmed and extended our previous data on conserved residues that arrange the position of the coenzyme (PLP) in the enzyme active center. The following residues should be noted: lysine forming a Schiff base with the PLP aldehyde group, an adjacent histidine, and aspartic acid that establishes a link with nitrogen of the PLP pyridine ring. The homology of the primary sequence fragments was also found in the residues in contact with the PLP phosphate group. Comparison of the GAD amino acid sequence with that of another PLP enzyme, aspartate aminotransferase, revealed a binding site for carboxylic group of the substrate--glutamic acid. The structures carrying out a particular catalytic function of all GAD studied were detected, i.e., convergent evolution of the enzyme was revealed.
KEY WORDS: glutamate decarboxylase, pyridoxal phosphate, phylogeny, conserved residues, divergent and convergent evolution
Abbreviations: GAD) glutamate decarboxylase; PLP) pyridoxal phosphate; PLP domains) PLP-binding domains.
Studies of the structure and catalytic properties of bacterial glutamate
decarboxylase began under the supervision of A. E. Braunstein in the
frame of the general problem of pyridoxal catalysis [1]. Glutamate decarboxylase
(L-glutamate-1-carboxy-lyase, EC 4.1.1.15) catalyzes the conversion of
glutamic acid to gamma-aminobutyric acid and carbon dioxide
according to the following equation:
HOOC-CH2-CH2-CH(NH2)-COOH --> CO2 + HOOC-CH2-CH2-CH2-NH2.
gamma-Aminobutyric acid formed during further metabolic transformations is converted to succinic semialdehyde and then to succinic acid, which is involved in Krebs cycle.
Glutamate decarboxylase has been found in animal and higher plant tissues as well as in yeasts and microorganisms. In animals the enzyme plays an important role in central nervous system activity because the enzyme substrate glutamic acid is a mediator of excitation process and the product, gamma-aminobutyric acid, is the most important mediator of inhibition process in the central nervous system. Besides, the physiological role of glutamate decarboxylase is connected with the problem of insulin-dependent diabetes; it is an autoantigen reacting with antibodies emerging upon autoimmune destruction of insulin-producing pancreatic beta cells. The enzyme of E. coli controls the pH level in the intestines, thus preventing acidification of the medium [2].
Different approaches have been used for structural investigations of glutamate decarboxylase. Preliminary X-ray experiments were performed on preparations of natural and recombinant enzymes of E. coli, but the spatial structure of the enzyme is still not established [2]. Secondary and quaternary structures of glutamate decarboxylase from E. coli and rat brain were studied by electron microscopy and circular dichroism; the role of coenzyme PLP in the formation and organization of the enzyme macromolecular structure was revealed [3-5]. We considered studying the molecular evolution of the enzyme as modern approach to the investigation of its structure. Computer studies of the primary structure homology of the isozyme GADalpha and other glutamate decarboxylases by the pair alignment technique were carried out both at the subunit and PLP domains levels [6].
These investigations were continued and extended in this work, where multiple alignments were used to establish the primary structure homology for 40 glutamate decarboxylases of different origin, to design a phylogenetic tree, distinguish motifs, and detect conserved amino acid residues in the PLP- and substrate-binding sites of the enzyme active center. These data allowed us to elucidate the paths of divergent and convergent evolution of the enzyme.
METHODS OF INVESTIGATION
Multiple alignment. Primary amino acid sequences for glutamate decarboxylases of different origin were obtained from the SWISS-PROT/TrEMBL data bank using software supported at the server http://www.eimb.ru/BIT/, from the NCBI (National Center for Biotechnology Information): http://www.ncbi.nlm.nih.gov/entrez/, and from KEGG data bank (Kyoto Encyclopedia of Genes and Genomes): http://www.genome.ad.jp/kegg/. Forty proteins with fully established primary structure were chosen for investigation. The only exclusion was the amino acid sequence of GAD from the fish Fugu rubripes which was obtained from an unfinished genome using the BLAST program (Basic Local Alignment Search Tool): http://www.ncbi.nlm.nih.gov/BLAST/fugu.html. Multiple alignment was carried out using the CLUSTALW program [7] with following manual adjustment. In determination of structural homology of subunits and PLP domains the sum of identical, similar, and interchangeable amino acid residues was considered.
Phylogenetic analysis. Phylogenetic trees were designed in accordance with the maximal probability, maximal parsimony, and long-distance methods using the software packages PHYLIP 3.6a2 [8] (PROTPARS, PROTDIST, NEIGHBOR) and MOLPHY 2.2 [9]. For each tree, 100 bootstrapped alignments were generated using the SEQBOOT program, and the consensus trees were constructed using the CONSENSE program. The branch lengths were obtained using FITCH algorithm. Borders of PLP domains were determined as described by Momany et al. [10] by comparison with PLP-enzymes of a known spatial structure, such as aspartate aminotransferase (PDB ID 8AAT) and ornithine decarboxylase (PDB ID 1ORD).
Search for motifs. A search of homologous motifs in a block of GAD was carried out in the Supercomputer Center, San-Diego, on the web-site Multiple Em for Motif Elicitation, http://meme.sdsc.edu/meme/website/meme.html.
RESULTS AND DISCUSSION
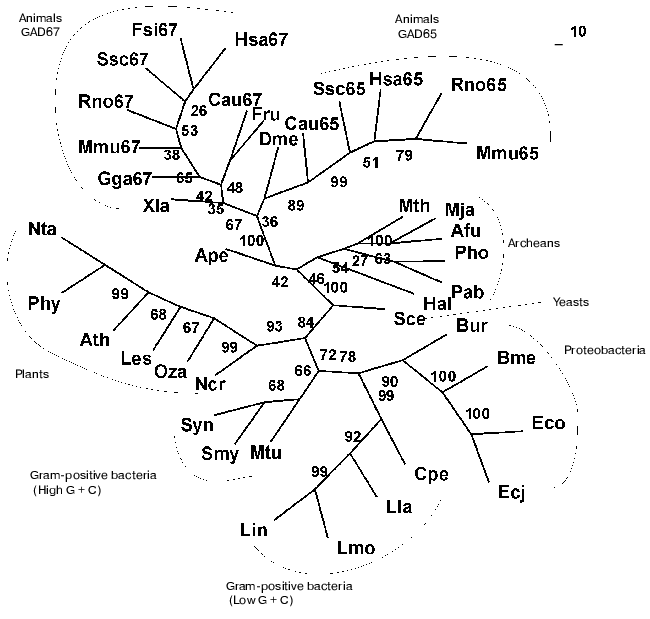
Present-day data banks contain about two hundred documents describing amino acid sequences of glutamate decarboxylases. Forty proteins from different sources whose primary structure was fully established were chosen for analysis (Table 1). It is seen that the enzyme is widespread in living nature, it is present in animals, higher plants, fungi, yeasts, bacteria, and archeans. In vertebrates the enzyme was found in the brain and pancreas in two forms of different molecular mass, 67 and 65 kD (GAD67 and GAD65). Amino acid sequence of pancreatic and brain GAD differ by only 1% [11], and these differences do not include conserved residues; that is why the pancreatic enzyme was not considered separately. The vertebrate GAD subunits consist of 555-594 amino acid residues, in the drosophila enzyme there are 510 residues, in higher plants 496-502, in yeasts 585, and in bacteria 460-475 amino acid residues. The Crenarchaeota member Ape is distinguished among archeans; here the subunit consists of 454 residues, which is close to plant and bacterial enzymes. In archeans Euryarchaeota GAD subunits contain only 355-396 amino acid residues, these subunits being the shortest. The amino acid chain probably elongated during evolution. The yeast enzyme is an exception from the rule: it consists of 585 residues, resembling the animal enzyme. The length of PLP domains in the studied proteins varies to a significantly lower extent: they contain 254 residues in animals, 244 in higher plants, 242-246 in bacteria, and 221-228 in archeans. In this case the yeast enzyme also is an exception, its PLP domains consisting of 262 amino acids (Table 1). It can be supposed that PLP domains were more conserved during evolution than other domains. It appeared to be possible to design a phylogenetic tree of glutamate decarboxylases on the basis of data obtained by multiple alignment of amino acid sequences of the enzyme subunits (Fig. 1). The following groups can be distinguished in the branching: enzymes from animal tissues, plants, bacteria, and finally, from archeans. Irrespective of the source, the separation to two groups GAD67 and GAD65 of different molecular mass is clearly seen within the group of enzymes of animal origin. A high structural homology characterizes enzymes from higher plants and bacteria, including different Gram-positive bacteria and Proteobacteria. The enzymes from Archeans form a separate group.
Table 1. Glutamate decarboxylases from
different sources
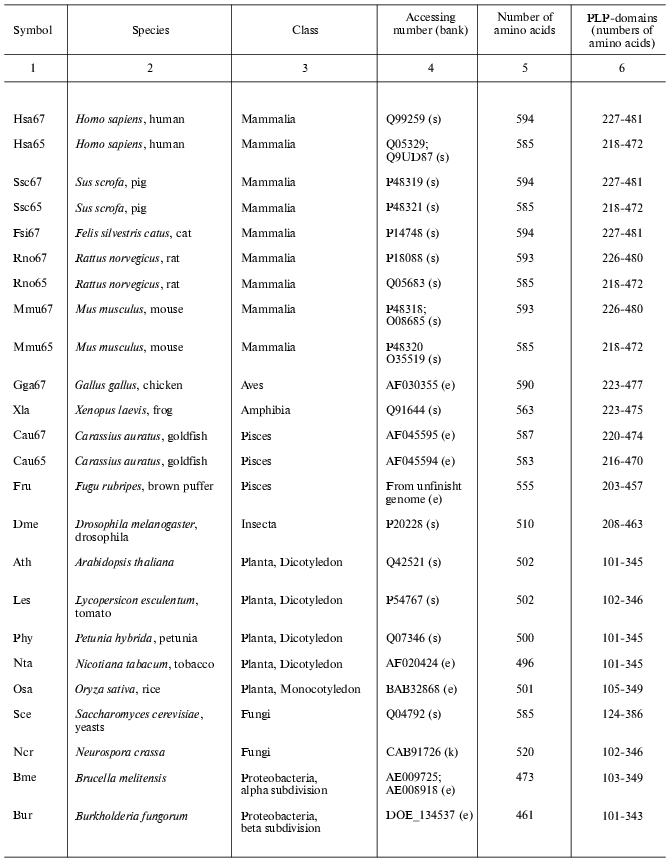
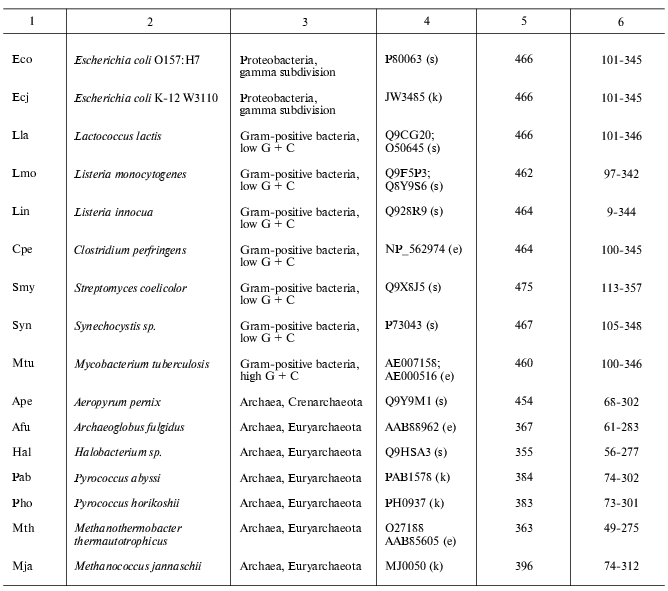
Note: s, SWISS-PROT/TrEMBL; e, NCBI: National Center for Biotechnology
Information, Entrez, BLAST; k, KEGG: Kyoto Encyclopedia of Genes and
Genomes.
Then multiple alignment was performed for amino acid sequences of PLP domains of all studied proteins, and a corresponding phylogenetic tree was designed. As soon as PLP domains include over a half of the subunit, the phylogenetic tree obtained was rather similar to that designed for the subunit. The preservation of branching order and only a slight shift of two-three positions within the GAD67 branch should be noted.Fig. 1. Phylogenetic tree of glutamate decarboxylase (complete subunit). The consensus tree presented was calculated by methods of maximal parsimony (MP), maximal likelihood (ML), and by methods based on distant matrices (DM). Confidence interval values (%) obtained by the DM method, are shown in nodes. Abbreviations as in Table 1.
Further analysis revealed 20 motifs of a repeated amino acid sequence in the studied proteins; these are peptides of different composition and length (Table 2).
Table 2. Homologous motifs found for 40
glutamate decarboxylases from different sources

Note: (K), lysine forming Schiff 's base with PLP.
Motifs characteristic of animal enzymes are shown in the diagram in Fig. 2. These enzymes contain a set of seven motifs: 9, 5, 6, 3, 1, 14, and 4. Only these motifs are present in the GAD sequence of drosophila, frog, and fish Fugu rubripes. In organisms of a higher organization level motif 15 is added to the N-terminal part of seven mentioned motifs, this being characteristic of all GAD65 as well as of GAD67 from goldfish. Motif 11 is added at the N-terminus and supplements the same seven motifs in another GAD67. Motif 1, present in all sequences, contains a lysine forming a Schiff's base with the PLP aldehyde group.
Quite a different set of motifs is characteristic of GAD from archeans, bacteria, and plants (Fig. 3). Motifs 12, 13, and 16 are common for all these proteins. Enzymes from archeans Euryarchaeota contain only six motifs: 19, 12, 20, 13, 17, and 16. In this group of proteins the PLP-binding lysine is located in motif 17. The situation is more complicated in GAD from Aeropyrum pernix (Ape, Crenarchaeota) where a more extended PLP-binding motif 2 occupies the place of motif 17, and motif 10 is added at the C-end.Fig. 2. Diagram of motifs found for sequences of GAD from vertebrates and invertebrates (Dme). The amino acid composition of each motif is shown in Table 2.
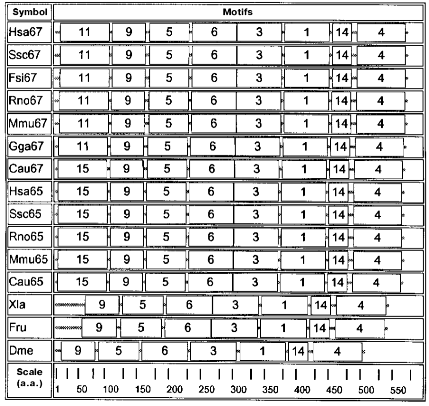
As compared with Ape, motifs 7 and 8 appear in the enzyme from Mtu and Smy (Gram-positive bacteria), the structure seeming to be transitive from GAD of Archeans to GAD of bacterial origin.Fig. 3. Diagram of motifs found for sequences of GAD from plants, bacteria, and Archeans. The amino acid composition of each motif is shown in Table 2. Homologous motifs found for 40 glutamate decarboxylases from different sources.
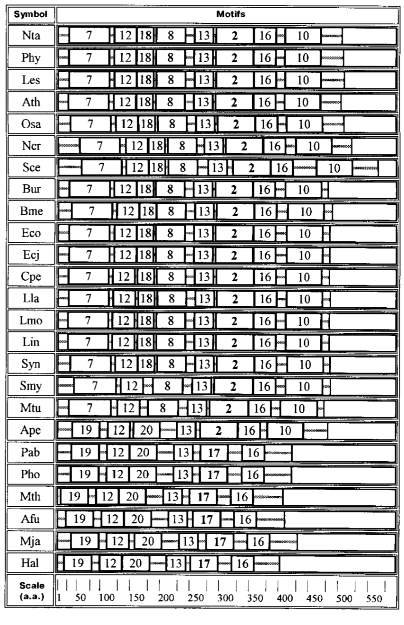
Bacterial and plant enzymes contain eight motifs: 7, 12, 18, 8, 13, 2, 16, 10; the PLP-binding lysine is located in motif 2.
The data described support the division of glutamate decarboxylases into three groups by their amino acid sequences: GAD from Archeans, GAD from bacteria together with plants, and GAD from animals. Each group is characterized by its own set of motifs, their lengths, and motifs containing the PLP-binding lysine (motifs 17, 2, 1). Especially strong are distinctions between animal GAD, on one hand, and enzymes of bacterial and plant origin, on the other: no identical motifs were found in them. Thus, the motif diagrams clearly reveal divergent evolution of the enzyme.
Carrying out catalytic function is connected with the structure of the enzyme active center, thus it was of interest to reveal the localization of functionally important residues forming the GAD active center. Previously, a reference point for detection of localization of these amino acid residues served for structural comparison of GAD and aspartate aminotransferase, because both enzymes have one and the same substrate and coenzyme [6, 10]. These investigations were continued for 40 GAD analyzed in this work (Table 3). Lysine that binds aldehyde group of PLP and aspartic acid, forming a salt bridge with nitrogen of the PLP pyridinium ring should be distinguished first among all amino acid residues. A histidine residue adjacent to PLP-lysine is a specific guide for decarboxylases for localization of the latter. Table 3 also shows amino acid residues that are in contact with the PLP phosphate group. These are G, A, and S in GAD of vertebrates; S, S, and A in most plant and bacterial GAD; and G, T, and A in GAD of Archeans. All these residues are responsible for the coenzyme position in the enzyme active center. In addition, there is also an arginine residue located in the ERL or DRL sequence (ERL in aspartate aminotransferase) and probably involved in binding of carboxyl of glutamic acid (substrate). The participation of arginine in substrate binding was shown previously by chemical modification of glutamate decarboxylase [12].
Table 3. Conserved amino acid residues in
GAD
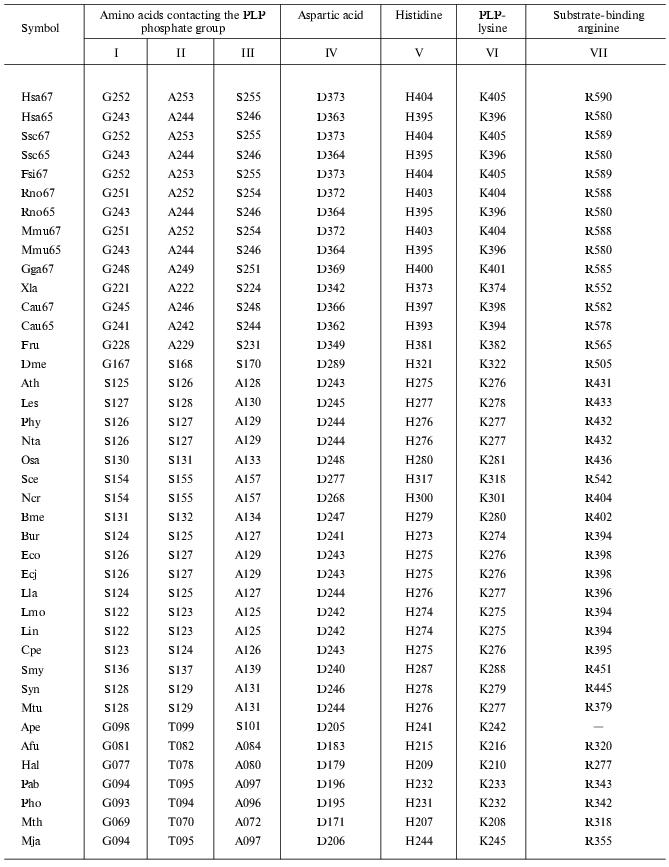
The carrying out one and the same enzymatic reaction of glutamic acid decarboxylation is due to identical (or similar) residues in all GAD irrespective of the source of their preparation. This reveals enzyme convergent evolution in which function predetermines necessary structural peculiarities. Thus, both divergent and convergent types of enzyme evolution were revealed by the example of glutamate decarboxylase.
This work was supported by the Russian Foundation for Basic Research (project No. 02-04-49106).
REFERENCES
1.Braunstein, A. E. (1981) in Of Oxygen, Fuels and
Living Matter (Semenza, G. N. Y., ed.) Pt. 2, J. Wiley & Sons,
pp. 251-313.
2.Sukhareva, B. S., Darii, E. L., and Khristoforov,
R. R. (2001) Uspekhi Biol. Khim., 41, 131-162.
3.Tikhonenko, A. S., Sukhareva, B. S., and
Braunstein, A. E. (1968) Biochim. Biophys. Acta, 167,
476-479.
4.Chen, C.-W., Wu, S. J., and Martin, D. I. (1998)
Arch. Biochem. Biophys., 349, 175-182.
5.Bazhulina, N. P., Darii, E. L., Lobachev, V. M.,
Stel'mashchuk, V. Ya., and Sukhareva, B. S. (2002) J. Biomol.
Struct. Dyn., 19, 999-1006.
6.Areshev, A. G., Mamaeva, O. K., Andreeva, N. S.,
and Sukhareva, B. S. (2000) J. Biomol. Struct. Dyn., 18,
127-136.
7.Thompson, J. D., Higgins, D. G., and Gibson, T. J.
(1994) Nucleic Acids Res., 22, 4673-4680.
8.Felsenstein, J. (1996) Meth. Enzymol.,
26, 418-427.
9.Adachi, J., and Hasegawa, M. (1992) Computer
Science Monographs, No. 27, Institute of Statistical Mathematics,
Tokyo.
10.Momany, C., Ghosh, R., and Hackert, M. (1995)
Prot. Sci., 4, 849-854.
11.Kawasaki, E., Moriuchi, R., Watanabe, M., Saitoh,
K., Brunicardi, F. C., Watt, P. C., Yamaguchi, T., Mullen, Y., Akazawa,
S., and Miyamoto, T. (1993) Biochem. Biophys. Res. Commun.,
192, 1353-1359.
12.Vospelnikova, N. D., Darii, E. L., and Sukhareva,
B. S. (1983) Bioorg. Khim., 9, 1026-1031.