Synthetic Peptide VKGFY and Its Cyclic Analog Stimulate Macrophage Bactericidal Activity through Non-Opioid beta-Endorphin Receptors
E. V. Navolotskaya1*, A. A. Kolobov2, E. A. Kampe-Nemm2, T. A. Zargarova1, N. V. Malkova1, S. B. Krasnova1, Yu. A. Kovalitskaya1, V. P. Zav'yalov3, and V. M. Lipkin1
1Branch of Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Pushchino, Moscow Region, 142290 Russia; fax: (0967) 79-0527; E-mail: navolots@fibkh.serpukhov.su2State Research Institute for Highly Pure Biopreparations, Ministry of Health of the Russian Federation, ul. Pudozhskaya 7, St. Petersburg, 197110 Russia; fax: (812) 235-5504
3Center for Immunological Engineering “Biokad”, Lyubuchany, Moscow Region, 142380 Russia; fax: (095) 797-5689
* To whom correspondence should be addressed.
Received July 18, 2002
We synthesized linear and cyclic pentapeptides corresponding to the sequence 369-373 of human immunoglobulin G heavy chain--VKGFY (referred to as pentarphin and cyclopentarphin, respectively). The effect of pentarphin and cyclopentarphin on phagocytosis of Salmonella typhimurium virulent 415 strainbacteria by mouse peritoneal macrophages in vitro was studied. Control experiments showed that macrophages actively captured these bacteria, but did not digest them: the captured microbes were viable and continued to proliferate inside the phagocytes; within 12 h all macrophage monolayer was destroyed (incomplete phagocytosis). If 1 nM pentarphin or cyclopentarphin was added to the cultivation medium, macrophage bactericidal activity was significantly increased and they digested all captured microorganisms within 6 h (complete phagocytosis). To study the receptor binding properties of pentarphin and cyclopentarphin we prepared 125I-labeled pentarphin (179 Ci/mmol specific activity). The binding of 125I-labeled pentarphin to mouse peritoneal macrophages was high-affinity (Kd = 3.6 ± 0.3 nM) and saturable. Studies on binding specificity revealed that this binding was insensitive to naloxone and [Met5]enkephalin, but completely inhibited by unlabeled cyclopentarphin (Ki = 2.6 ± 0.3 nM), immunorphin (Ki = 3.2 ± 0.3 nM), and beta-endorphin (Ki = 2.8 ± 0.2 nM). Thus, the effects of pentarphin and cyclopentarphin on macrophages are mediated by naloxone-insensitive receptors common for pentarphin, cyclopentarphin, immunorphin, and beta-endorphin.
KEY WORDS: phagocytosis, macrophage, beta-endorphin, tuftsin, naloxone, immunoglobulin G, peptides, receptors, immune system
In 1970 Najiar's discovery of the endogenous tetrapeptide tuftsin (Thr-Lys-Pro-Arg, fragment 289-292 of the CH2 domain of human immunoglobulin G (IgG) H-chain) which has both immunostimulatory and neurotropic activities [1-4], provided the motivation for the search for other bioactive peptides of the same origin. In 1980 Julliard et al. found adrenocorticotropic hormone-like and beta-endorphin-like sequences in the IgG H-chain [5]. The tetradecapeptide SLTCLVKGFYPSDI corresponding to the beta-endorphin-like sequence of IgG (fragment 364-377 of the H-chain CH3 domain) was synthesized by Houck et al. and shown to compete with 125I-labeled beta-endorphin for binding to rat brain membranes [6]. We have synthesized the decapeptide SLTCLVKGFY corresponding to the sequence 364-373 of human IgG H-chain (referred to as immunorphin) and found it to inhibit 125I-labeled beta-endorphin binding to mouse peritoneal macrophages (Ki 5.9 nM) [7] and T-lymphocytes from human blood (Ki 0.6 nM) [8, 9]. Studies on the specificity of the binding sites, which bind both beta-endorphin and immunorphin, revealed that they are insensitive to natural opioid peptides [Met5]enkephalin and [Leu5]enkephalin as well as to the antagonist of opioid receptors naloxone, that is, they are non-opioid ones. We also showed that beta-endorphin and immunorphin stimulate concanavalin A (Con A)-induced proliferation of human T-lymphocytes in vitro [10]. [Met5]enkephalin and naloxone tested in parallel were inactive. Naloxone did not inhibit the stimulating effect of beta-endorphin and immunorphin on T-lymphocyte proliferation. We then prepared 125I-labeled immunorphin (232 Ci/mmol) and shown that it binds to human T-lymphocytes with high affinity (Kd 7.4 nM) [7, 8]. The binding of the radiolabeled immunorphin was naloxone-insensitive and completely inhibited by unlabeled beta-endorphin (Ki1.1 nM) [11]. Studies on the inhibition of 125I-labeled beta-endorphin binding to T-lymphocytes by synthetic immunorphin fragments demonstrated that the pentapeptide VKGFY (fragment 6-10, referred to as pentarphin) was the shortest active fragment of immunorphin [8].
The aim of the present study was to investigate the effect of pentarphin and cyclopentarphin on phagocytosis of S. typhimurium virulent strain415 bacteria by mouse peritoneal macrophages in vitro as well as the binding of 125I-labeled pentarphin to these cells.
MATERIALS AND METHODS
The chemicals used in this study were: [Met5]enkephalin, beta-endorphin, naloxone, tuftsin, 1,3,4,6-tetrachloro-3alpha,6alpha-diphenylglycoluril (Iodogen), cell cultivation media, and fetal calf serum (Sigma, USA); L-glutamine and Hepes (ICN, USA); penicillin and streptomycin (Gibco, USA); agarose, sucrose, BSA, EDTA, EGTA, Tris, phenylmethylsulfonyl fluoride (PMSF), and sodium azide (NaN3) (Serva, Germany); scintillator Unisolv 100 (Amersham, England). tert-Butyloxycarbonyl derivatives of amino acids were prepared as described elsewhere [12]. N-Methylpyrrolidone, diisopropylcarbodiimide, 1-hydroxybenzotriazole, and thioanisole were obtained from Merck (Germany). All other chemicals were purchased from domestic companies and additionally purified before use.
Peptides were synthesized on Applied Biosystems 430A and Vega Coupler C250 automatic peptide synthesizers (USA), using Boc/Bzl peptide chain elongation technique. Pentarphin was synthesized on phenacylamidomethyl (PAM) polymer by the in situ method [13]. 2-Chlorobenzyloxycarbonyl and dichlorobenzyl groups were used for the protection of lysine and tyrosine, respectively. At the completion of protected polypeptide chain synthesis, the end product was relieved with simultaneous detachment from polymer, using anhydrous hydrogen fluoride in the presence of scavengers.
Protected cyclopentarphin was synthesized on polymer-bound oxime [14]. For cyclization, the protected linear peptide was treated with acetic acid in dimethylformamide in the presence of diisopropylethylamine. The protected cyclopeptide was precipitated by ether, filtered, dried to constant weight, and treated with anhydrous liquid hydrogen fluoride.
Both peptides were purified to homogeneity by preparative reverse-phase chromatography (Gilson chromatograph, France) on a Waters SymmetryPrep C18 column (19 × 300 mm; Malva, Greece), 7 µm, 10 ml/min flow rate, elution with 0.1% trifluoroacetic acid, 10-40% acetonitrile gradient within 30 min). The peptides were characterized by analytic reverse-phase HPLC (Gilson chromatograph) on an XTerra RP18 column (125 Å, 3.9 × 150 mm; Malva), 5 µm, 1 ml/min flow rate, elution with 0.1% trifluoroacetic acid, 10-40 and 20-40% acetonitrile gradient within 16 min for pentarphin and cyclopentarphin, respectively), amino acid analysis (6 N HCl hydrolysis, 24 h, 110°C; amino acid analyzer LKB 4151 Alpha Plus, Sweden), and fast atom bombardment mass spectrometric analysis (Finnigan mass spectrometer, USA).
The virulent Salmonella typhimurium 415 strain with typical morphological and functional properties was used in phagocytosis studies. The LD50 was approximately 100 bacterial cells per mouse injected intraperitoneally. S. typhimurium was grown in Hottinger's broth for 4-6 h at 37°C, then transferred to beef-extract agar and incubated at 37°C for 18 h.
Mouse peritoneal macrophages were isolated and cultured as described in [15]. Phagocytic activity of macrophages was determined as we previously described [16]. Macrophage monolayers on cover glasses were grown at 37°C in sterile glass test tubes in medium 199 supplemented with streptomycin and penicillin (100 µg/ml each) and 5% inactivated fetal calf serum. In 24 h macrophages were infected with S. typhimurium 415(108 bacterial cells/ml) in medium 199 supplemented with serum. After 2 h, the contact between the bacteria and macrophages was interrupted by replacing the infectious medium with fresh one supplemented with antibiotics. To prevent the recapture of bacteria released from broken cells by other phagocytes, the culture medium was replaced with fresh medium every hour. The macrophages on cover glasses (in triplicate for every time point, in 1, 2, 4, 7, and 12 h) were fixed in methanol for 3 min. Then the preparations were stained with 0.1% aqueous azure II-eosin for 5 min. Cells (300 per cover glass) were examined under a light microscope (×1350) and analyzed for the following parameters: phagocytic activity (PA), the percentage of macrophages participating in phagocytosis; bacterial cytocidal activity (BCA), the percentage of phagocytes destroyed by intracellular bacteria; and phagocytic number (PN), the average number of bacteria per macrophage.
Pentarphin (10 µg) was labeled with 125I using 1 mCi Na125I and Iodogen [17]. The labeled peptide was purified by gel filtration on Sephadex G-10 (0.9 × 10 cm column, 50 mM phosphate buffer, pH 7.4, 5 ml/h). The volume corresponding to the labeled peptide was determined in control experiments using unlabeled peptide. Radioactivity was counted using a Mini-Gamma Counter (LKB, Sweden). Fractions with the maximum radioactivity at the positions corresponding to those of the unlabeled peptide peak in control experiments were pooled, and the total as well as the specific radioactivity of the preparation were determined. The purity of the labeled peptide was tested by TLC on aluminum oxide glass in n-butanol-CH3COOH-H2O (4 : 1 : 1 v/v) solvent system, followed by autoradiography. The specific activity of labeled pentarphin was 179 Ci/mmol.
The binding of 125I-labeled pentarphin to mouse peritoneal macrophages was assayed in medium 199 containing 25 mM Hepes, 20 mM NaN3, and 0.6 mg/ml PMSF, pH 7.4, as follows: 100 µl labeled peptide (concentration range 10-10-10-7 M, each concentration point in triplicate) plus 100 µl medium (for total binding) or 10-4 M unlabeled peptide (for nonspecific binding) were added to 800 µl cell suspension (107 cells) and incubated at 4°C for 60 min. Then the samples were filtered through GF/A glass fiber filters (Whatman, England) to separate cell-bound labeled peptide from non-bound (free) labeled peptide. Filters were washed three times with 5 ml ice-cold saline. Radioactivity was counted using the Mini-Gamma counter (LKB). The specific binding of 125I-labeled pentarphin to macrophages was determined as the difference between total and nonspecific binding. The latter was measured in the presence of 10-4 M unlabeled peptide (1000-fold excess). To determine the characteristics of labeled pentarphin specific binding to macrophages (the equilibrium dissociation constant Kd and the binding capacity n--number of binding sites per cell), the profiles of the ratio between bound (B) and free (F) labeled pentarphin molar concentrations as a function of bound labeled peptide molar concentration (B) were plotted. Binding capacity (n) was estimated from the equation: n = (R0·A)/N, where R0 is the molar concentration of the receptors, A is Avogadro's number, and N is cell number per liter [18].
To test the inhibitory effects of cyclopentarphin, immunorphin, beta-endorphin, naloxone, and [Met5]enkephalin on the specific binding of 125I-labeled pentarphin to macrophages, 107 cells/ml were incubated with 1 nM 125I-labeled pentarphin and one of the potential competitors at various concentrations (10-10-10-6 M, each concentration point in triplicate) as described above. The inhibition constant (Ki) was calculated using the equation: Ki = [I]50/(1 + [L]/Kd) [19], where [L] is the molar concentration of 125I-labeled pentarphin, Kd is the equilibrium dissociation constant of the 125I-labeled pentarphin/receptor complex, and [I]50 is the concentration of the competing ligand giving half-maximal inhibition of 125I-labeled pentarphin binding. [I]50 was estimated graphically from the inhibition curve (plot of the inhibition (%) as a function of inhibitor molar concentration). The Kd value was determined in a preliminary experiment as described earlier.
Statistical significance was analyzed using the Microsoft Excel MS 2000 electronic table system. The data are presented as the means ± SEM of at least three independent experiments.
RESULTS
Synthesis of pentarphin and cyclopentarphin. The peptides were synthesized with automatic synthesizers and purified to homogenous state by preparative reversed-phase chromatography. The synthesized compounds were characterized by preparative reversed-phase high performance liquid chromatography (HPLC), amino acid analysis, and mass-spectrometry. The characteristics of the peptides are presented in Table 1. Pentarphin and cyclopentarphin yields were 33 and 10%, respectively.
Table 1. Main characteristics of the
peptides

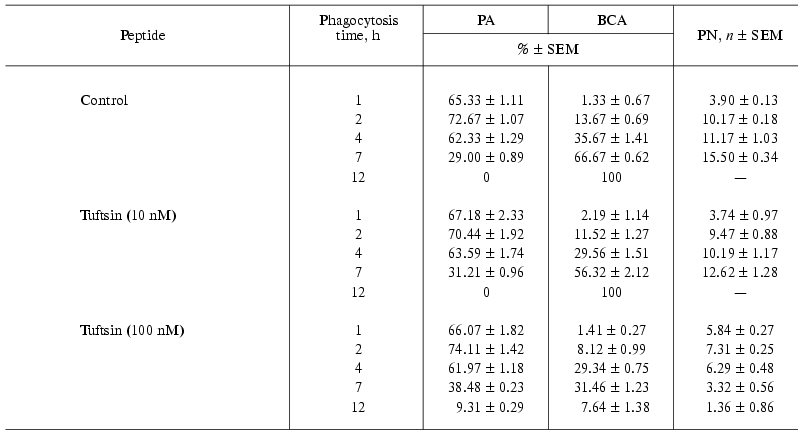
Effects of pentarphin and cyclopentarphin on the activity of mouse peritoneal macrophages in vitro. Pentarphin and cyclopentarphin stimulated peritoneal macrophage bactericidal activity against S. typhimurium 415 bacteria in vitro. Tuftsin served as a positive control. The values of the main phagocytosis characteristics (PA, PN, BCA) in the absence (control) and in the presence of pentarphin (0.1, 1, and 10 nM) or cyclopentarphin (0.1 and 1 nM) are given in Table 2. The results of control experiments showed that macrophages captured actively bacteria of this strain: in 2 h, more than 2/3 of the total number of macrophages participated in phagocytosis (PA = 72.67 ± 1.07%), and each phagocyte contained an average of 10 microorganisms (PN = 10.17 ± 0.18). But captured microbes were not digested. Moreover, they continued to proliferate inside of phagocytes, as PN increased from 10.17 ± 0.18 to 15.50 ± 0.34 between the 2nd and 7th hours of the process. The macrophage monolayer was infected for 2 h, then the infection medium was replaced by fresh cultivation medium, and thus, beginning from this moment, the growth of PN can occur only by multiplication of previously captured microbes. By 7 h of phagocytosis, macrophage destruction on a mass scale was observed (BCA = 66.67 ± 0.62%), and within 12 h all of the macrophage monolayer was destroyed (BCA = 100%). Thus, in control, the interaction between bacteria and macrophages resulted in phagocyte destruction (incomplete phagocytosis).
Table 2. Effect of pentarphin and
cyclopentarphin on phagocytosis of S. typhimurium virulent 415
strain bacteria by mouse peritoneal macrophages in vitro

In the presence of 1 nM pentarphin or cyclopentarphin the character of bacteria-macrophage interaction changed completely: the phagocytic capacity of macrophages was augmented to the extent that by 12 h of phagocytosis they digested all captured microbes (BCA = 2.35 ± 1.21% and 0% in the presence of pentarphin and cyclopentarphin, respectively) (Table 2). A comparison between phagocytosis characteristics of pentarphin and cyclopentarphin shows that the latter is much more active. For instance, in the presence of pentarphin, by 7 h of phagocytosis BCA was 10.24 ± 1.17 and by 12 h this index decreased to 2.35 ± 1.21, whereas in the presence of cyclopentarphin, at a phagocytosis period from 1 to 7 h, BCA did not exceed 2.5%; by 12 h there were no destroyed cells in the monolayer and the macrophages were free of digested microbes.
Tuftsin tested in parallel also enhanced phagocytic capacity of macrophages, but its active concentration was 100 times larger than that of pentarphin or cyclopentarphin. As evident from Table 3, in the presence of 100 nM tuftsin the number of phagocytes destroyed by intracellular bacteria by 7 h of phagocytosis was half less than that of control (BCA = 31.46 ± 1.23 and 66.67 ± 0.62%, respectively). By 12 h of the process, BCA was 7.64 ± 1.38% and approximately 10% of the phagocytes contained an average of 1.36 ± 0.86% non-digested microorganisms. Thus, the activity of tuftsin was at least 100 times lower than that of pentarphin and cyclopentarphin.
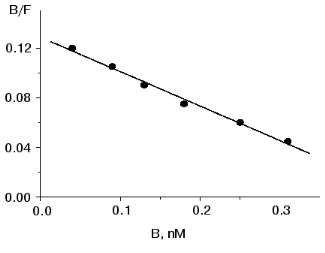
Binding of 125I-labeled pentarphin to mouse peritoneal macrophages. 125I-labeled pentarphin is able to bind specifically to mouse peritoneal macrophages. Figure 1a shows the values of total (curve 1), specific (curve 2), and nonspecific (curve 3) 125I-labeled pentarphin binding to macrophages at 4°C as a function of incubation time. It is evident that the labeled peptide-receptor system reached dynamic equilibrium in approximately 1 h and this state was kept at least for 2 h; therefore, to assess the equilibrium dissociation constant (Kd), the reaction of 125I-labeled pentarphin binding to macrophages was carried out for 1 h. Saturation curves (binding isotherms) showing the values of total (curve 1), specific (curve 2), and nonspecific (curve 3) 125I-labeled pentarphin binding as a function of its concentration are presented in Fig. 1b. The fact that curve 2 flattens out indicates that the labeled pentarphin specific binding is saturable (the number of sites for peptide specific binding on the cell surface is limited). Scatchard analysis of 125I-labeled pentarphin specific binding to macrophages (linear plot, Fig. 2) indicates that these cells possess only one type of binding site (receptor) for this peptide. The Kd value (3.6 ± 0.3 nM) suggests that pentarphin binds to the receptor with high affinity. The number of binding sites for the labeled peptide specific binding per macrophage (receptor density) was 28,000 ± 3,000.
Fig. 1. Dependence of total (1), specific (2) and nonspecific (3) 125I-labeled pentarphin binding to mouse peritoneal macrophages on incubation time (a) and peptide concentration in the medium (b). Specific binding was calculated by subtracting nonspecific binding from total binding.
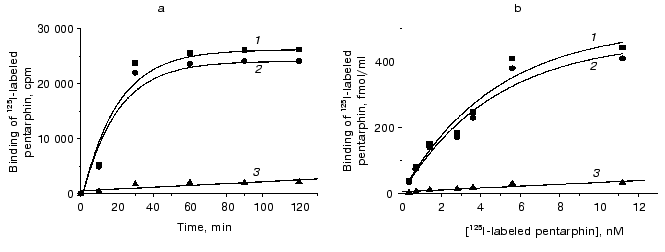
Table 3. Effect of tuftsin on phagocytosis of S. typhimurium virulent 415 strain bacteria by mouse peritoneal macrophages in vitroFig. 2. Scatchard analysis of specific 125I-labeled pentarphin binding to mouse peritoneal macrophages. B and F are molar concentrations of bound and free labeled peptide, respectively.
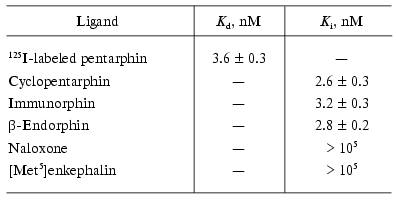
To characterize the specificity of 125I-labeled pentarphin specific binding to macrophages, competition experiments were performed using unlabeled cyclopentarphin, immunorphin, beta-endorphin, naloxone, and [Met5]enkephalin as potential competitors. The results are presented in Table 4. It is obvious that cyclopentarphin, immunorphin, and beta-endorphin actively inhibited 125I-labeled pentarphin specific binding (Ki values are 2.6 ± 0.3, 3.2 ± 0.3, and 2.8 ± 0.2 nM, respectively). Naloxone and [Met5]enkephalin were inactive (Ki > 100 µM). Thus, the receptors revealed using 125I-labeled pentarphin bind cyclopentarphin, immunorphin, and beta-endorphin with high affinity, and do not interact with naloxone and [Met5]enkephalin.
Table 4. Inhibition of
125I-labeled pentarphin binding to mouse peritoneal
macrophages by unlabeled peptides and naloxone
DISCUSSION
Phagocytosis of both opsonized and non-opsonized bacteria includes their binding, capture, and digestion by macrophages and neutrophils [20]. It was shown that opsonized bacteria bind to the following receptors on macrophages: IgG Fc-receptors, receptors for C3b-component of complement and, probably, IgM receptors. Nonspecific capture of non-opsonized bacteria is also preceded by their binding to phagocyte surface, but the receptors participating in this binding are not identified yet. A bound microorganism is embraced by pseudopodia which are formed near the binding site. The intracellular system of contractile proteins becomes activated and provides pseudopodium elongation until the bacterium is inside the phagosome. The phagosome fuses with granules and lysosomes containing proteolytic enzymes to form a phagolysosome, in which the microorganism is digested.
The most important consequence of phagocytosis is increased macrophage secretory activity. Activated macrophages produce and secrete into the milieu a wide spectrum of bioactive compounds and in this respect they have no equal among other cell types. For macrophage activity regulation, synthesis and secretion of active oxygen forms, such as superoxide anion (O2-), hydrogen peroxide (H2O2), and hydroxyl radical (OH*), nitrogen oxide (NO), and prostaglandins (PGE1, PGE2, PGD2, PGF2, 6-keto-PGF1alpha) are of most interest [20].
At present there is incomplete understanding of the mechanisms of macrophage phagocytic activity initiation and regulation. The results of numerous investigations indicate that at least three interrelated regulatory systems participate in these processes: Ca2+-dependent, cAMP-dependent, and cytoskeletal systems. There is much speculation that the system controlling Ca2+ concentration in macrophage cytoplasm plays the central role in initiation and regulation of phagocytosis [20-28]. Actually, the ionophore A23187 that transports Ca2+ inside the cell, initiates the production of active oxygen forms, an increase in cytoplasmic cAMP concentration, and synthesis and secretion of prostaglandins [29-31]. Changes in Ca2+ concentration within physiological range modulate the activity of NAD(P)H-oxidase localized in phagosome membranes and plasmatic membrane vesicles [32]. It seems that superoxide production is a calmodulin-dependent reaction, as it is inhibited by trifluoperazine--an inhibitor of calmodulin-stimulated processes [32]. The prostaglandin synthesis key enzyme--phospholipase A2--is Ca2+-dependent [33]. Aggregation/disaggregation of microtubules is also a calmodulin-dependent process [34].
The phagocytosis of Salmonella typhimurium virulent strain 415 by mouse peritoneal macrophages in vitro was used as a model for selective influence of peptides on phagocytic activity of macrophages. The tetrapeptide tuftsin (Thr-Lys-Pro-Arg, 289-292 fragment of IgG CH2 domain) served as a positive control. It has been reported to stimulate the bactericidal activity of macrophages against Staphylococcus aureus, St. londres, Listeria monocytogenesis, Leishmania parasites, Escherichia coli, and Serratia marcescens [4]. The phagocytosis characteristics of mouse peritoneal macrophages against S. typhimurium 415 in the absence and presence of tuftsin are presented in Table 3. A comparison of the characteristics obtained in the presence of 100 nM tuftsin and control values indicates that this peptide enhances the macrophage ability to digest captured microbes. By 12 h of phagocytosis in the presence of tuftsin there were approximately 10% of phagocytes with one or two non-digested bacteria, and the number of destroyed phagocytes did not exceed 8%.
The pentapeptides tested in parallel with tuftsin also stimulated the macrophage ability to digest virulent microorganisms, but at lower concentrations compared to tuftsin. In the presence of 1 nM cyclopentarphin or pentarphin, macrophages virtually completely digested all captured microorganisms by 7 or 12 h of phagocytosis, respectively (Table 2). Thus, cyclopentarphin demonstrated higher activity than pentarphin, and both were about two orders more potent than tuftsin.
Previously, we showed that the decapeptide immunorphin, which encompasses the pentarphin sequence, binds with high affinity to non-opioid beta-endorphin receptors on mouse peritoneal macrophages and thymocytes [7], human T-lymphocytes [8-11], and rat brain synaptic membranes [35]. The results of the present study demonstrate that 125I-labeled pentarphin binds with high affinity to murine peritoneal macrophages (Kd = 3.6 ± 0.3 nM). The analysis of binding specificity revealed that this binding was not inhibited by naloxone or [Met5]enkephalin, but was completely inhibited by cyclopentarphin (Ki = 2.6 ± 0.3 nM), immunorphin (Ki = 3.2 ± 0.3 nM), and beta-endorphin (Ki = 2.8 ± 0.2 nM). Thus, the effects of pentarphin and cyclopentarphin on macrophages are mediated by naloxone-insensitive receptors which also bind immunorphin and beta-endorphin with high affinity. beta-Endorphin is known to interact with both µ (morphine) and delta (enkephalin) opioid receptors [36] as well as with non-opioid receptors. In contrast, immunorphin, pentarphin, and cyclopentarphin inhibit beta-endorphin binding only to non-opioid receptors and are unlikely to interact with other receptors, thus they may be considered as selective agonists of non-opioid receptors for beta-endorphin.
The most effective currently available approach to the treatment of infectious diseases is the use of antibiotics. The last generation antibiotics are highly potent and have relatively low toxic and allergenic properties. Nevertheless, their use (especially prolonged) may cause a number of serious complications. The microorganisms may acquire resistance even to “strong” antibiotics. Thus, antibiotic therapy inevitably leads to the decrease or loss of sensitivity to them. This continuous “arms race” results in constant need for novel, more potent drugs.
The encounter of the immune system with antigens is a determining factor for its maturation and proper functioning. The persistence of bacteria in the body is tonic for the immune system. Therefore, the intestinal dysbacteriosis inevitable in oral administration of antibiotics leads to the weakening of immunity that opens the way for viral infections and allergies.
The results of this study demonstrate that pentarphin and cyclopentarphin are potent stimulators of phagocytosis. Our previous studies have shown that in the physiological concentration range both peptides are non-immunogenic and non-toxic (LD50 > 2500 mg/kg administered intraperitoneally to mice) [37], and catabolism of the peptides yields amino acids. Thus, their use in combination with antibiotics is likely to provide the possibility to decrease the therapeutic doses and, hence, the negative side effects of the latter.
This work was supported by the Russian Foundation for Basic Research (grant No. 02-04-48424) and the International Science and Technology Center (grant No. 1462).
REFERENCES
1.Najiar, V. A., and Nashioka, K. (1970)
Nature, 228, 672-673.
2.Herman, Z. S., Stachura, Z., Opielka, L., Siemion,
I. Z., and Nawrocha, E. (1981) Experientia, 37,76-77.
3.Herman, Z. S., Stachura, Z., Kraemifski, T., Plech,
A., Siemion, I. Z., and Nawrocha, E. (1983) Ann. N. Y. Acad.
Sci., 419, 156-163.
4.Val'dman, A. V., Bondarenko, N. A., Kamysheva, V.
A., Kozlovskaia, M. M., and Kalikhevich, V. N. (1982) Byul. Eksp.
Biol. Med., 93, 57-60.
5.Julliard, J. H., Shibasaki, T., Ling, N., and
Guilemin, R. (1980) Science, 208, 183-185.
6.Houck, J. C., Kimbull, C., Chang, C., Pedigo, N.
W., and Yamamura, H. J. (1980) Science, 207, 78-80.
7.Zav'yalov, V. P., Zaitseva, O. R., Navolotskaya, E.
V., Abramov, V. M., Volodina, E. Yu., and Mitin, Y. V. (1996)
Immunol. Lett., 49, 21-26.
8.Navolotskaya, E. V., Malkova, N. V., Lepikhova, T.
N., Krasnova, S. B., Zargarova, T. A., Zav'yalov, V. P., and Lipkin, V.
M. (2001) Bioorg. Chem. (Moscow),27, 359-363.
9.Navolotskaya, E. V., Zargarova, T. A., Malkova, N.
V., Lepikhova, T. N., Zav'yalov, V. P., and Lipkin, V. M. (2001)
Peptides, 22, 2009-2013.
10.Navolotskaya, E. V., Malkova, N. V., Zargarova,
T. A., Krasnova, S. B., and Lipkin, V. M. (2002) Biochemistry
(Moscow),67,357-363.
11.Navolotskaya, E. V., Zargarova, T. A., Malkova,
N. V., Krasnova, S. B., Zav'yalov, V. P., and Lipkin, V. M. (2002)
Biochem. Biophys. Res. Commun., 292, 799-804.
12.Gershkovitch, A. A., and Kibirev, V. K. (1992)
Chemical Peptide Synthesis [in Russian], Naukova Dumka, Kiev,
pp. 95-283.
13.Schnolzer, M., Alewood, P., Jones, A., Alewood,
D., and Kent, S. B. H. (1992) Int. J. Peptide Protein Res.,
40, 180-193.
14.Nakagawa, S. H., and Kaiser, E. T. (1983) J.
Org. Chem., 48, 678-685.
15.Uchitel, I. Ya. (1978) Macrophages in the
Immunity [in Russian], Meditsina, Moscow, pp. 168-180.
16.Navolotskaya, E. V. (1994)
Structure-functional studies of human
alpha2-interferon, interleukin 2 and
immunoglobulin G using synthetic peptides: Doctoral dissertation
[in Russian], Institute of Immunology, Moscow.
17.Salacinski, P. R. P., Lean, C. M., Sykes, J. E.
C., Climent-Jones, V. V., and Lowry, P. J. (1981) Analyt.
Biochem., 117, 136-146.
18.Chang, K.-J., Jacobs, S., and Cuatrecasas, P.
(1975) Biochim. Biophys. Acta, 406, 294-303.
19.Chang, Y. C., and Prusoff, W. H. (1973)
Biochem. Pharmacol., 22, 3099-3108.
20.Prishchepov, E. D., Erin, A. N., Perelygin, V.
V., and Vorob'ev, A. A. (1984) J. Microbiol. Epidemiol. Immunobiol.
(Moscow), 6, 3-9.
21.Bromberg, Y., and Pick, E. (1981) Cell.
Immunol., 61,90-103.
22.Lew, P. D. (1989) Eur. J. Clin. Invest.,
19, 338-346.
23.Stendahl, O., Krause, K. H., Kricher, J.,
Jerstrom, P., Theler, J. M., Clark, R. A., Carpentier, J. L., and Leu,
D. P. (1994) Science, 265, 1439-1441.
24.Theler, J. M., Lew, D. P., Jaconi, M. E., Krause,
K. H., Wollheim, C. B., and Schlegel, W. (1995) Blood,
85, 2194-2201.
25.Clapham, D. (1995) Cell,80,
259-268.
26.Helberg, C., Molony, L., Zheng, L., and
Andersson, T. (1996) Biochem. J.,317, 403-409.
27.Berridge, M. (1997) J.
Physiol.,499, 291-306.
28.Malik, Z. A., Denning, G. M., and Kusner, D. J.
(2000) J. Exp. Med., 191 (2), 287-302.
29.Gemza, D., Seitz, M., Kramer, W., Grimm, W.,
Till, G., and Resch, K. (1979) Exp. Cell. Res., 118,
55-62.
30.Pick, E., and Keisari, Y. (1981) Cell.
Immunol., 59, 301-308.
31.Weidemann, M. J., Peskar, B. A., Wrogemann, K.,
Rietschel, E. T., Staudinder, H., and Fisher, H. (1978) FEBS
Lett., 89,136-140.
32.Leu, P. D., and Stassel, T. P. (1981) J. Clin.
Invest., 67, 1-9.
33.Wong, P. Y. K., and Cheung, W. Y. (1979)
Biochem. Biophys. Res. Commun., 90, 473-480.
34.Welsh, M. J., Dedman, J. R., Brinkley, B. R., and
Means, A. R. (1979) J. Cell. Biol.,81, 624-634.
35.Navolotskaya, E. V., Zargarova, T. A., Malkova,
N. V., Krasnova, S. B., Zav'yalov, V. P., and Lipkin, V. M. (2002)
Peptides, 23, 1115-1119.
36.Li, C. H. (1982) Cell, 31,
504-505.
37.Korpela, T., Navolotskaya, E. V., Zargarova, T.
A., and Zav'yalov, V. P. (2002)Patent F120020121AO. Finland. Pub. Date
23.01.2002.