Electron Transfer in Deuterated Reaction Centers of Rhodobacter sphaeroides at 90 K According to Femtosecond Spectroscopy Data
A. G. Yakovlev1* and V. A. Shuvalov1,2
1Department of Photobiophysics, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119992, Russia; fax: (095) 939-3181; E-mail: yakov@genebee.msu.su2Institute of Basic Biological Problems, Russian Academy of Sciences, Pushchino, Moscow Region 142292, Russia; fax: (277) 90532; E-mail: shuvalov@issp.serpukhov.su
* To whom correspondence should be addressed.
Received January 4, 2003; Revision received February 26, 2003
The primary act of charge separation was studied in P+BA- and P+HA- states (P, primary electron donor; BA and HA, primary and secondary electron acceptor) of native reaction centers (RCs) of Rhodobacter sphaeroides R-26 using femtosecond absorption spectroscopy at low (90 K) and room temperature. Coherent oscillations were studied in the kinetics of the stimulated emission band of P* (935 nm), of absorption band of BA- (1020 nm) and of absorption band of HA (760 nm). It was found that in native RCs kept in heavy water (D2O) buffer the isotopic decreasing of basic oscillation frequency 32 cm-1 and its overtones takes place by the same factor ~1.3 in the 935, 1020, and 760 nm bands in comparison with the samples in ordinary water H2O. This suggests that the femtosecond oscillations in RC kinetics with 32 cm-1 frequency may be caused by rotation of hydrogen-containing groups, in particular the water molecule which may be placed between primary electron donor PB and primary electron acceptor BA. This rotation may appear also as high harmonics up to sixth in the stimulated emission of P*. The rotation of the water molecule may modulate electron transfer from P* to BA. The results allow for tracing of the possible pathway of electron transfer from P* to BA along a chain consisting of polar atoms according to the Brookhaven Protein Data Bank (1PRC): Mg(PB)-N-C-N(His M200)-HOH-O = BA. We assume that the role of 32-cm-1 modulation in electron transfer along this chain consist of a fixation of electron density at BA- during a reversible electron transfer, when populations of P* and P+BA- states are approximately equal.
KEY WORDS: photosynthesis, reaction center, electron transfer, wavepacket, femtosecond spectroscopy
Abbreviations: ÄA) absorption difference (light-minus-dark); BA and HA) monomer bacteriochlorophyll and bacteriopheophytin in the active chain, respectively; P) bacteriochlorophyll dimer; Pheo) plant pheophytin; QA) quinone; RC) reaction center.
The reaction center (RC) of photosynthesis is a hexachromophoric protein
in which light energy is converted into the energy of charge-separated
states involved further in biochemical processes in cells (see reviews
in [1, 2]). The RC of purple
bacteria consists of three protein subunits (L, M, and H), four
bacteriochlorophyll molecules, two bacteriopheophytin molecules, two
quinone molecules, and one atom of non-heme iron. The three-dimensional
structure of RC established by X-ray analysis is consistent with a
sequence of electron transfers along the chromophore chain in the
active A-branch [3-6].
The primary act of charge separation in the RC occurs between the excited primary electron donor, bacteriochlorophyll dimer P* and a monomeric bacteriochlorophyll BA within ~3 psec at 293 K (where A denotes the photoactive branch of cofactors) [7-15]. At this time an intermediate state P+BA- is formed. Then an electron is transferred from BA- to bacteriopheophytin HA within ~1 psec and further from HA- to quinone QA within ~200 psec. At low temperature (5-10 K) all of the primary electron transfer processes are accelerated by 2-3-fold. The formation of P* is accompanied by the bleaching of P absorption bands at 870 and 600 nm and by the appearance of a stimulated emission around 920 nm. Electron transfer from P* to BA is accompanied by a decrease in the stimulated emission around 920 nm, by the bleaching of the BA absorption band at 800 nm, and by the development of the BA- absorption band at 1020 nm. These processes occur simultaneously. The modified RCs in which HA is replaced by plant pheophytin a (Pheo) is an ideal object for observing the P+BA- state [12-17]. The further electron transfer in these RCs from BA-to Pheo is delayed significantly on the picosecond time scale, this helping the effective accumulation of the P+BA- state. In native RCs it is hard to register the P+BA- state due to very fast conversion of it to the P+HA- state with lower energy.
It was found that the excitation of primary electron donor, bacteriochlorophyll dimer P, by ultrashort (<30 fsec) light pulses with broad spectra creates a superposition of many nuclear vibrational wavefunctions known as a wavepacket [19-22]. The wavepacket is created on the P*BA potential energy surface [19, 20] and has properties of a quasi-classical particle [23]. This wavepacket moves on the potential energy surface with a frequency which is determined by the energy difference between vibronic levels. The motions of a nuclear wavepacket are visualized by femtosecond oscillations in kinetics of P* stimulated emission [19-22]. A shift of P* potential energy surface with respect to that of P leads to dependence of spectral maximum of P* stimulated emission on time. Long- and short-wavelength components of P* stimulated emission with maximums at 935 and 895 nm, respectively, are in antiphase but have the same oscillation frequencies [19, 20]. The Fourier transform spectrum of oscillations at 10 K consists of frequencies at 15, 30, 69, 92, 122, 153, 191, and 329 cm-1 [20]. Similar vibrational modes were found at 27, 73, 110, 147, 175, and 205 cm-1 in experiments of photochemical hole burning [24]. Experiments of resonance Raman scattering also gave similar frequencies at 34, 71, 95, and 128 cm-1 [25].
According to current knowledge the electron transfer between P* and BA should occur at the intersection of potential energy surfaces of the P*BA and P+BA- states [2]. In [15-18] we studied the coherent oscillations in native and pheophytin-modified RCs of Rhodobacter sphaeroides R-26 at room temperature. These studies show a coupling between primary charge separation and nuclear wavepacket motion in bacterial RCs. Femtosecond oscillations are observed in P+BA- charge separated state when P is excited by light pulses of femtosecond (<30 fsec) duration and broad spectrum [15-18]. These oscillations are observed in BA absorption band at 800 nm and BA- band at 1020 nm [15-18]. Measurements of these oscillations in the absorption band of product BA- at 1020 nm give a possibility to study an electron transfer in this band. An intensive oscillation mode at 32 cm-1 is present in the BA- absorption band at 1020 nm and in the HA bleaching band at 760 nm, while a 130-cm-1 mode dominates in P* stimulated emission band [17, 18]. A possibility of wavepacket transition from 130-140 cm-1 mode to 32 cm-1 mode was discussed in these studies. The same 32-cm-1 mode of Fourier transform spectrum of oscillations was founded around 788 nm on direct kinetics measurement [26].
In [27] the femtosecond oscillations were studied in Rh. sphaeroides RCs in the 920-1100 nm range. Using light pulses with 100-fsec duration did not allow to discern the oscillations clearly. A conclusion about the need to use pulses of 30-fsec duration or shorter in this case was made earlier in [20]. Nevertheless, the results obtained in [27] may be interpreted as an addition of out of phase oscillations of P* stimulated emission and BA- absorption bands.
In the present work the results of study of femtosecond oscillations in native RCs of Rhodobacter sphaeroides R-26 in heavy water (D2O) buffer at low (90 K) and room temperatures are presented. It is shown in this work that a 32-cm-1 mode may appear due to rotation of the water molecule which may be located between PB and BA. This is confirmed by the fact that an isotopic decreasing of fundamental oscillation frequency 32 cm-1 and its overtones by the same coefficient ~1.3 takes place in the 935-, 1020-, and 760-nm bands of native RCs placed in D2O buffer in comparison with samples placed in ordinary water. The water molecule rotation may also appear as higher harmonics up to the sixth in stimulated emission of P*. A possible pathway of electron transfer from P* to BA is suggested to be along a chain consisting of polar atoms according to the Brookhaven Protein Data Bank (file 1PRC): Mg(PB)-N-C-N(His M200)-HOH-O = BA.
MATERIALS AND METHODS
Reaction centers of Rhodobacter sphaeroides R-26 were isolated as described in [28]. RCs were suspended in 10 mM Tris-HCl (pH 8.0)/0.1% LDAO buffer. To replace H2O buffer by D2O buffer, these RCs were concentrated on a membrane and then diluted by D2O buffer. This procedure was repeated twice. It was checked that this procedure did not alter the absorption spectra of the RCs. In the 293 K measurements the optical density of the samples was 0.5 at 860 nm in the cuvette of 1-mm thickness. In the 90 K measurements the samples were mixed with a glycerol (60%) to obtain a transparent ice on freezing. The optical density of the samples at 90 K was 0.4 at 870 nm in a cuvette of 2-mm thickness. Sodium dithionite (5 mM) was added to keep the state PBAHAQA- in RCs. All measurements were carried out at 90 and 293 K.
Difference (light-minus-dark) absorption spectra were measured with femtosecond resolution by a laser spectrometer that was build in our laboratory on the base of a Ti:sapphire laser with amplifier, continuum generator, and optical multichannel analyzer. Detailed description of the spectrometer is in [15-18]. The frequency of measurements was 15 Hz. The duration of pump and probe pulses was about 25 fsec. Pump pulses were centered at 870 nm. The pump and probe pulses had weak (~20%) mutual perpendicular polarization while the remaining part (~80%) was depolarized. The delay between pump and probe pulses was changed with an accuracy of ~10 fsec. The temporal dispersion in the range 600-900 nm found by kinetic measurements at 600 nm (Qx transition of P) and at 900 nm (Qy transition of P and short wavelength side of stimulated emission from P*) was less than 50 fsec. In the 900-1060 nm range the temporal dispersion found by measurements of absorption band bleaching of the green filter ZS-10 (LOMO, St. Petersburg) was less than 30 fsec.
The amplitude of the spectral bands at 935, 1020, and 760 nm was measured at their maximums on the background of broadband pedestal as shown by arrows in Fig. 1. The resulting difference absorption spectra were obtained by averaging of 3000-10,000 measurements at each delay. The accuracy of spectral measurements was (1-2)*10-4 units of optical density. The kinetic curves of absorption changes (DeltaA) at different wavelengths were revealed from the difference absorption spectra measured at different delays. Then a non-oscillating quasi-exponential component was subtracted from the kinetic curves and the residual oscillatory part was Fourier transformed to obtain the spectra of oscillations. The non-oscillating part was optimized by minimum of the oscillating part. The accuracy of calculation of the frequencies from Fourier transform spectra of oscillations was 4 cm-1.
Fig. 1. Difference (light-minus-dark) absorption spectra at various femtosecond delays in native Rb. sphaeroides R-26 RCs in D2O buffer excited at 90 K by 25-fsec pulses at 870 nm: a) 990-1060 nm range (BA- absorption band); b) 735-776 nm range (HA bleaching band); c) 935-1060 nm range (long-wavelength side of P* stimulated emission band and BA- absorption band). The double-headed arrows show amplitudes of absorption changes (DeltaA) for spectral band at 1020, 760, and 935 nm, which were used further for kinetic plots (Figs. 2-5). The numbers near the curves indicate the delay in femtoseconds of the probe pulse from exciting pulse.
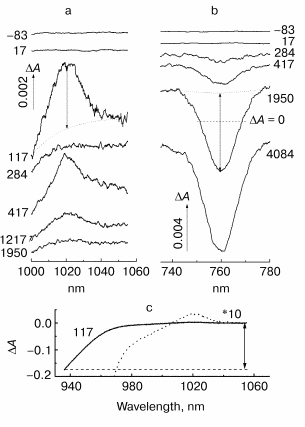
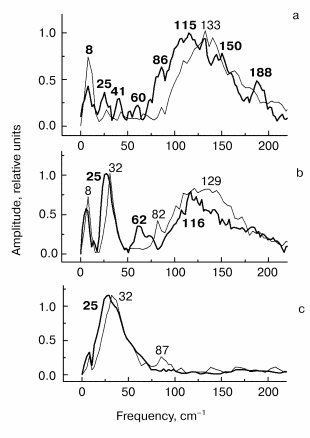
Fig. 2. Kinetic curves of DeltaA (a), its oscillatory part (b) and spectrum of Fourier transform of oscillatory part (c) at 935 nm in native Rb. sphaeroides R-26 RCs excited at 90 K by 25-fsec pulses at 870 nm. Thick curves are for RCs in D2O buffer, thin curves for RCs in H2O buffer. Numbers in part (c) show the characteristic frequencies of maximums of Fourier transform spectra.
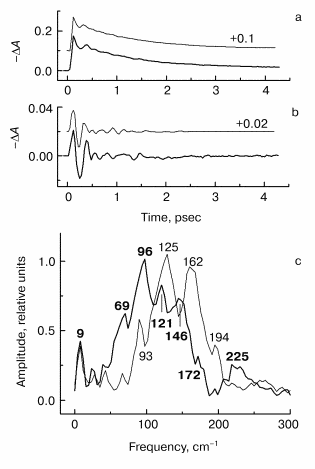
Fig. 3. Kinetic curves of DeltaA (a), its oscillatory part (b) and spectrum of Fourier transform of oscillatory part (c) at 1020 nm in native Rb. sphaeroides R-26 RCs excited at 90 K by 25-fsec pulses at 870 nm. Thick curves, RCs in D2O buffer; thin curves, RCs in H2O buffer. Numbers in part (c) show the characteristic frequencies of maximums of Fourier transform spectra.
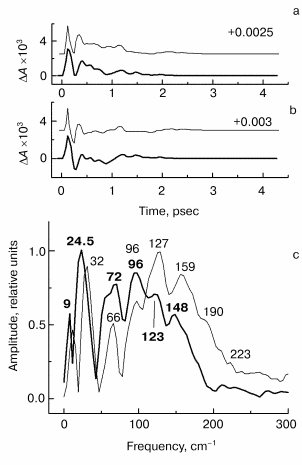
Fig. 4. Kinetic curves of DeltaA (a), its oscillatory part (b) and spectrum of Fourier transform of oscillatory part (c) at 760 nm in native Rb. sphaeroides R-26 RCs excited at 90 K by 25-fsec pulses at 870 nm. Thick curves, RCs in D2O buffer; thin curves, RCs in H2O buffer. Numbers in part (c) show the characteristic frequencies of maximums of Fourier transform spectra.
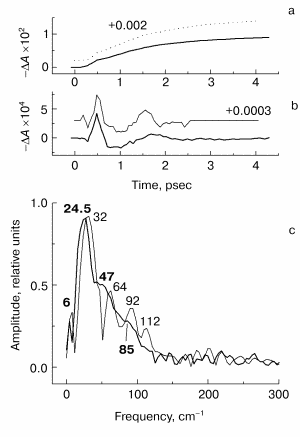
Fig. 5. Fourier transform spectra of kinetic curves DeltaA at 935 (a), 1020 (b), and 760 nm (c) in native Rb. sphaeroides R-26 RCs excited at room temperature by 25-fsec pulses at 870 nm. Thick curves, RCs in D2O buffer; thin curves, RCs in H2O buffer. Numbers in part (c) show the characteristic frequencies of maximums of Fourier transform spectra.
RESULTS
In Fig. 1 the characteristic spectra DeltaA of native Rb. sphaeroides R-26 RCs placed in D2O buffer at 90 K are shown at various delays. The shape of the BA- absorption band at 1020 nm (Fig. 1a) and position of its maximum remain invariable over the whole range of delays. This means that femtosecond oscillations observed in kinetics of this band (Figs. 3 and 5) are caused by coherent nuclear motions between different electronic states (probably between P* and P+BA-) rather than by motions inside one P+BA- state. The same is true for pheophytin HA absorption band at 760 nm (Fig. 1b) in which the oscillations are caused by coherent nuclear motions between different electronic states (most probably between P+HA- and P+BA-).
Amplitudes of the bands at 935 nm (stimulated emission of P*), 1020 nm (development of BA-), and 760 nm (bleaching of HA) were measured on the background of the broadband pedestal as shown in Fig. 1 by double arrows. This method of measurements separates the changes of the studied band from the background of other bands (for example, P* stimulated emission band, P* absorption band). The band amplitudes obtained in this way were used to plot the kinetics.
In Fig. 2 the results of kinetic measurements in the 935-nm band (P* stimulated emission band) at 90 K are shown. The kinetics of RCs in D2O buffer (Fig. 2a, thick curves here and further in Figs. 3-5) shows the decay of the stimulated emission within the same characteristic time ~1.5 psec as the analogous kinetics of RCs in H2O buffer show (Fig. 2a, thin curves here and further in Figs. 3-5). This fact indicates an absence of changes in RC structure after the procedure of buffer change. A small increasing of a period of the initial intensive oscillations and a small increasing of oscillation decay time are observed in oscillations of RCs in D2O buffer (Fig. 2b). A broad band with a center at 125 cm-1 containing the peaks at 93, 125, 162, and 194 cm-1 dominates in Fourier transform spectrum of oscillations of RCs in H2O buffer (Fig. 2c, thin curve). The distances between neighboring peaks are 32, 37, and 32 cm-1 that is close to fundamental frequency 32 cm-1 which is well observed in the Fourier transform spectra of the oscillations at 1020 nm (Fig. 3c) and 760 nm (Fig. 4c). The center of this band is shifted down to 96 cm-1 (coefficient ~1.3) under substitution of H2O buffer by D2O buffer (Fig. 2c, thick curve). Probably, all peaks with frequencies 69, 96, 121, and 146 cm-1 are shifted by the same value. The distances between neighboring peaks are decreased also to the new values 27, 25, and 25 cm-1 after this substitution.
The results of the measurements in the BA- absorption band at 1020 nm are shown in Fig. 3. The kinetics of RCs in D2O buffer and in H2O buffer are the same (Fig. 3a) that shows the absence of destructive consequences of deuteration. The kinetics show very fast initial development of absorption (within ~100 fsec) and further more slow decay (within ~1 psec). Thus, the BA- band exists only during the first 1.5-2 psec after excitation because of fast electron transfer further to pheophytin HA. The oscillatory part of the kinetics of RCs in D2O buffer (Fig. 3b) shows an increasing of oscillation period in comparison with kinetics in H2O buffer. The Fourier transform spectrum of oscillations of RCs in H2O buffer (Fig. 3c, thin curve) contains a narrow intense band at 32 cm-1 and broad band centered at 127 cm-1 which contains the peaks at 66, 96, 127, 159, and 190 cm-1. Note that the distances between the neighboring peaks 30, 31, 32, and 31 cm-1 are close to the fundamental frequency 32 cm-1 in this case too. The 32 cm-1 band is shifted to 24.5 cm-1 (coefficient ~1.3) and the center of the broad band is shifted from 127 to 96 cm-1 with the same coefficient ~1.3 in deuterated RCs. The other peaks also are shifted to the new values 72, 96, 123, and 148 cm-1, and the new distances between the neighboring peaks are equal to 24, 27, and 25 cm-1 that is close to the value 24.5 cm-1 of fundamental frequency of RCs in D2O buffer.
The results of the measurements in the HA absorption band are shown in Fig. 4. The kinetics of both kinds of RCs (Fig. 4a) show a small initial delay ~0.3 psec followed by quasi-exponential increase of the bleaching within approximately equal time of ~1.5 psec. A similarity of kinetics proves that RCs were not damaged when the buffer was substituted. The oscillatory parts of the kinetics (Fig. 4b) shows an integration over time of the corresponding peaks of the 1020-nm oscillations, that means the efficiency of electron transfer to HA when the wavepacket appears on the P+BA- surface. The main oscillation period of RCs in D2O buffer (Fig. 4b, thick curve) is bigger compared with that of RCs in H2O buffer (Fig. 4b, thin curve). The Fourier transform spectrum of RCs in H2O buffer at 760 nm (Fig. 4c, thin curve) contains an intense band at 32 cm-1 and weak bands at 64, 92, and 112 cm-1. The frequencies of these bands decreases by the factor of ~1.3 when H2O buffer is substituted by D2O buffer (Fig. 4c, thick curve). The new frequency values are 24.5 (fundamental frequency), 47, and 85 cm-1 (small shoulders) after deuteration. Note that the Fourier transform spectrum of the kinetics at 760 nm does not contain the band centered at 125-130 cm-1 in contrast to Fourier transform spectra of the kinetics at 935 and 1020 nm. This means that the 130 cm-1 mode and its overtones are not active when the electron is transferred to the P+HA- surface.
At room temperature the deuteration of RCs also leads to frequency shifts of the Fourier transform spectra of the kinetics (Fig. 5), but this effect is less pronounced compared with the measurements at 90 K. At 293 K clear peaks in the 130-cm-1 band at 935 and 1020 nm are absent. A comparison of the Fourier transform spectra of the kinetics at 935 nm (Fig. 5a) shows the shift of the 130-cm-1 band to lower frequencies when H2O buffer is substituted by D2O buffer. The decreasing of the 32-cm-1 fundamental frequency caused by buffer substitution is most pronounced in the 1020- and 760-nm bands (Figs. 5b and 5c).
DISCUSSION
The main result of this work is that all characteristic frequencies of the Fourier transform spectra of the kinetics at 935, 1020, and 760 nm are shifted to the lower frequencies by the same factor ~1.3 when RCs are deuterated in the measurements at 90 K. This fact suggests that all observed modes with frequencies approximately equal to the fundamental frequency are overtones of this frequency. Indeed, the 32-cm-1 fundamental frequency has calculated overtones at 64, 96, 128, 160, 192, and 224 cm-1. After decreasing by the factor of 1.3 this fundamental frequency has a new value of 24.5 cm-1 and forms the following series of calculated overtones: 49, 73.5, 98, 122.5, 147, and 171.5 cm-1. The overtone frequencies observed in experiment are very close to the calculated ones, which proves their origin from one and the same fundamental frequency which is shifted by the same factor when RCs are deuterated. The shift of the fundamental frequency is mostly pronounced in the measurements at 1020 and 760 nm, while the shift of its overtones is mostly pronounced in the 935-nm measurements.
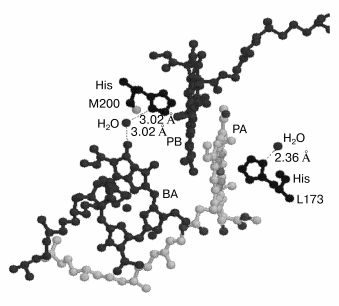
The appearance of the higher harmonics up to the sixth is characteristic of rotational modes rather than vibrational modes. Probably, this is the case of a rotation of small molecules like OH- or H2O connected by hydrogen bond to the photochemically active chromophores. For example, in Rhodopseudomonas viridis RCs H2O molecules are known (Brookhaven protein databank) to be bound by a hydrogen bond to the N atom of His M200 and L173 (axial ligands of PB and PA, respectively) on one side and to the keto carbonyl group of ring V of BA (and BB) on the other side (Fig. 6). This is true for RCs of Rhodobacter sphaeroides too, the structure of which is very similar to that of Rhodopseudomonas viridis [6].
According to a classical monograph [29], the rotational spectra of small two- or many-atom molecules are determined by dipole electric moment and include a set of lines reflecting the population of harmonics in the gas phase. Taking into account the selection rules DeltaJ = ±1 and DeltaK = 0 (here J and K are quantum numbers) the infrared frequencies are determined as follows [29]:Fig. 6. Three-dimensional structure (Brookhaven Protein Data Bank, file 1PRC) of special pair of bacteriochlorophylls PA and PB and monomeric chlorophyll BA. His M200 liganding Mg of PB is connected by hydrogen bond via H2O to oxygen of the keto carbonyl group of ring V of BA. Thus PB is connected to BA via the sequence of following polar atoms: Mg(PB)-N-C-N(His)-H-O-H(water)-O = BA which may represent a pathway for the electron transfer from P* to BA. The rotation of the water molecule in this system with 32 cm-1 frequency induced by the electron flow from P* may serves as a mechanism fixing separated charges in the state P+BA-. This mechanism allows the surrounding molecules to stabilize the charge separated state (see text for details).
nu = 2B(J + 1) - 4Dj(J + 1)3, (1)
where B and Dj are rotational and centrifugal constants, respectively (B>>Dj). The distance between infrared lines is ~2B if 4Dj(J + 1)3 << 2B(J + 1) in expression (1) (this is true for small J). The constant B is determined as follows [29]:
B = h/(8pi2cµr2), (2)
where h is Plank's constant, c speed of light, µ equivalent mass, r distance from rotating atom to rotation axis. For OH B ~19 cm-1 and nu ~ 38 cm-1 (µ = 0.941, r = 0.9707 Å). The expression (1) can be used for asymmetrical three-atom molecules like H2O too. For H2O three types of rotation of two protons around oxygen are possible. To find nu values for H2O one should simplify this molecule down to a two-atom molecule in which two protons are connected in one point according to [29]. As a result one can find the following values of rotation frequencies nu for H2O: ~32, ~52, and ~20 cm-1 for three types of rotation (µ = 1.778, the angle between hydrogen bonds is 104.5°). The experimental values of H2O rotational frequencies in gas phase are close to theoretical ones with good accuracy [29].
Note that the 32-cm-1 frequency is exactly consistent with the fundamental frequency observed in the oscillations of the RCs kinetics. It is interesting that the difference between two other frequencies (52 and 20 cm-1) is equal to 32 cm-1 too, and the ~52-cm-1 frequency is observed in RCs in which QA is doubly reduced [30].
Therefore the appearance of the peaks at 32, 66, 93-96, 125-127, 159-162, 190-194, and 223 cm-1 in the Fourier transform spectra of the native RCs kinetics at 935 and 1020 nm at 90 K (Figs. 2-4) can be explained in terms of modulation of the kinetics by the rotation of H2O (or OH-) molecules. The same is probably true at room temperature (Fig. 5). One can suggest that this modulation is along the reaction coordinate of the primary charge separation P* --> P+BA- since all these frequencies are observed in the oscillations of the product BA- band at 1020 nm. This modulation may be important for electron transfer from neutral P* to BA with formation of a charge separated state P+BA-.
The isotopic exchange of H2O by D2O shows (Figs. 2-4) that the frequencies observed in the Fourier transform spectra of the kinetics may belong to the higher harmonics of the 32-cm-1 mode, since all these frequencies are shifted by the same factor ~1.3 after H2O-D2O exchange. This result also shows that all these frequencies are related to the vibration or rotation of the H containing groups. The lowest mode observed at 9 cm-1 is shifted by the smaller factor in the oscillations at 1020 and 760 nm and nearly is not shifted in the oscillations at 935 nm after H2O-D2O exchange. The nature of this mode is unclear. According to expression (2) the coefficient of the frequency isotopic shift is ~1.9 that is different from its experimental value ~1.3. This discrepancy can probably be explained by some isotopic fractionation that does not allow exchanging both protons by deuterium in water molecules in RCs at the mentioned positions (Fig. 6) [31]. According to expression (2) the isotopic shift coefficient is ~1.4 for the DOH molecule, that is close to the experimental value ~1.3. In 90-K experiments the samples were mixed with glycerol containing OH groups, that increase the probability to obtain DOH molecules instead of DOD. An absence of deuteration in some fraction of RCs may be another reason for discrepancy between calculations and experiment. In this case the Fourier transform spectrum of oscillations should be a superposition of corresponding spectra of the RCs in H2O and D2O buffers. Probably, this is an explanation for the more complicated structure of the oscillation spectrum and of changing of relative oscillation amplitudes of the RCs in D2O buffer (Figs. 2-5). Note that H2O-D2O exchange may influence the protons which are not included in the water molecules. Recent experiments show that in dry films of RCs, where water is almost absent, the isotopic shift of femtosecond oscillation frequencies is much smaller than the shift in the water buffer (A. G. Yakovlev, V. A. Shuvalov, unpublished results).
Figure 6 shows that in Rhodopseudomonas viridis RCs [5] (the structure of which is very similar to that of Rhodobacter sphaeroides [6]) there is a polar group chain connecting PB and BA as follows (Brookhaven protein databank, file 1PRC): a) coordination bond between Mg atom of PB and N atom of His M200 molecule; b) hydrogen bond between another N atom of His M200 molecule and H-O-H; c) hydrogen bond between H-O-H and keto carbonyl group of ring V of BA with distances indicated by numbers in Fig. 6. This chain is suggested to be used for electron transfer from P* to BA (at least in part of the RCs). Then the sequence of electron transfers may be as follows. Before the exciting light arrives the chain is in the form presented in Fig. 6 and is completely ready for electron transfer. The femtosecond excitation of P dimer forms the nuclear vibration wavepacket with frequency of 130-140 cm-1 inside P. The appearance of the stimulated emission of P* at 935 nm with period of 260 fsec can be considered as a result of a decrease in the distance between porphyrin rings of PA and PB. This can create strong exciton coupling between PA and PB and possible charge transfer exciplex PB-PA+. This is consistent with molecular orbital calculations which show the electron density shift from PA to PB (ratio 0.24 to 0.76, respectively) in the excited state of P* [32]. It is known that the electron density in PB is mostly located near atoms N coordinating Mg, that is very close to His M200 nitrogen. Perhaps the electron density is reversibly shifted via the mentioned chain from PB- to an iso-energetic vacant orbital of BA with the same time period (260 fsec) observing in the P* band at 935 nm. If the 32 cm-1 modulation is connected to the H2O rotation it means that the mentioned chain for electron transfer is destroyed and created again with period of the H2O rotation. The start of the H2O rotation at ~100 fsec is consistent with first shift of the electron density from P* to BA and a change of the electrostatic interaction between groups involved in mentioned hydrogen bonds. This seems to give an initial impulse for H2O rotation. It is worth note that the 32-cm-1 mode is absent in the P* stimulated emission at 935 nm (Fig. 2). The ~260-fsec oscillation (~130 cm-1) seems to be created in P* and then propagated to BA with the formation of the P+BA- state. The modulation with a frequency of 32 cm-1 itself is most probably due to the fact that the electron density shift from PA to PB is stabilized in time. One can imagine that the rotation of the water molecule causes a periodic transfer of an electron from PB to BA and back with a frequency of the rotation of this molecule. The role of the 32-cm-1 modulation can be considered as a fixation of the electron density on BA- during reversible electron transfer with approximately equal population of P* and P+BA-. This fixation may additionally stabilize the BA- state faster than the relatively slow relaxation of the surrounding molecules according to the new electronic configuration. The main stabilizing molecule is most probably Tyr M210 located between P and BA.
The authors are grateful to Dr. A. V. Sharkov for technical assistance and V. A. Shkuropatova for preparation of the RCs. This study was supported in part by the Russian Foundation for Basic Research (grant No. 02-04-48650) and an NWO (The Netherlands) grant.
REFERENCES
1.Shuvalov, V. A. (1990) Primary Conversion of
Light Energy at Photosynthesis [in Russian], Nauka, Moscow.
2.Shuvalov, V. A. (2000) Conversion of Light
Energy in Primary Act of Charge Separation in Reaction Centers of
Photosynthesis [in Russian], Nauka, Moscow.
3.Deisenhofer, J., Epp, O., Miki, K., Huber, R., and
Michel, H. (1984) Mol. Biol., 180, 385-398.
4.Deisenhofer, J., Epp, O., Miki, K., Huber, R., and
Michel, H. (1985) Nature, 318, 618-624.
5.Michel, H., Epp, O., and Deisenhofer, J. (1986)
EMBO J., 5, 2445-2451.
6.Komiya, H., Yeates, T. O., Rees, D. C., Allen, J.
P., and Feher, G. (1988) Proc. Natl. Acad. Sci. USA,
85, 9012-9016.
7.Shuvalov, V. A., Klevanik, A. V., Sharkov, A. V.,
Matveetz, Yu. A., and Krukov, P. G. (1978) FEBS Lett.,
91, 135-139.
8.Shuvalov, V. A., and Duysens, L. N. M. (1986)
Proc. Natl. Acad. Sci. USA, 83, 1690-1694.
9.Arlt, T., Schmidt, S., Kaiser, W., Lanterwasser,
C., Meyer, M., Scheer, H., and Zinth, W. (1993) Proc. Natl. Acad.
Sci. USA, 90, 11757-11761.
10.Chekalin, S. V., Matveetz, Yu. A., Shkuropatov,
A. Ya., Shuvalov, V. A., and Yartzev, A. P. (1987) FEBS Lett.,
216, 245-248.
11.Schmidt, S., Arlt, T., Hamm, P., Huber, H.,
Nagele, T., Wachtveitl, J., Meyer, M., Scheer, H., and Zinth, W. (1994)
Chem. Phys. Lett., 223, 116-120.
12.Schmidt, S., Arlt, T., Hamm, P., Huber, H.,
Nagele, T., Wachtveitl, J., Zinth, W., Meyer, M., and Scheer, H. (1995)
Spectrochim. Acta. Part A, 51, 1565-1578.
13.Shkuropatov, A. Ya., and Shuvalov, V. A. (1993)
FEBS Lett., 322, 168-172.
14.Kennis, J. T. M., Shkuropatov, A. Ya., van
Stokkum, I. H. M., Gast, P., Hoff, A. J., Shuvalov, V. A., and Aartsma,
T. J. (1997) Biochemistry, 36, 16231-16238.
15.Yakovlev, A. G., and Shuvalov, V. A. (2000) J.
Chin. Chem. Soc., 47, 709-714.
16.Yakovlev, A. G., Shkuropatov, A. Ya., and
Shuvalov, V. A. (2000) FEBS Lett., 466, 209-212.
17.Yakovlev, A. G., and Shuvalov, V. A. (2001)
Biochemistry (Moscow), 66, 211-220.
18.Yakovlev, A. G., Shkuropatov, A. Ya., and
Shuvalov, V. A. (2002) Biochemistry, 41, 2667-2674.
19.Vos, M., Rappaport, F., Lambry, J.-C., Breton,
J., and Martin, J.-L. (1993) Nature, 363, 320-325.
20.Vos, M., Jones, M. R., Hunter, C. N., Breton, J.,
Lambry, J.-C., and Martin, J.-L. (1994) Biochemistry, 33,
6750-6757.
21.Vos, M. H., Jones, M. R., McGlynn, P., Hunter, C.
N., Breton, J., and Martin, J.-L. (1994) Biochim. Biophys. Acta,
1186, 117-122.
22.Vos, M. H., Jones, M. R., Hunter, C. N., Breton,
J., and Martin, J.-L. (1994) Proc. Natl. Acad. Sci. USA,
91, 12701-12705.
23.Sokolov, A. A., Loskutov, Yu. M., and Ternov, I.
M. (1962) Quantum Mechanics [in Russian], Moscow.
24.Shuvalov, V. A., Klevanik, A. V., Ganago, A. O.,
Shkuropatov, A. Ya., and Gubanov, V. S. (1988) FEBS Lett.,
237, 57-60.
25.Cherepy, N. J., Shreve, A. P., Moore, L. J.,
Franzen, S., Boxer, S. G., and Mathies, R. A. (1994) J. Phys.
Chem., 98, 6023-6029.
26.Vos, H. M., Rischel, C., Jones, M. R., and
Martin, J.-L. (2000) Biochemistry, 39, 8353-8361.
27.Spörlein, S., Zinth, W., and Watchtveitl, J.
(1998) J. Phys. Chem. B, 102, 7492-7496.
28.Shuvalov, V. A., Shkuropatov, A. Y., Kulakova, S.
M., Ismailov, M. A., and Shkuropatova, V. A. (1986) Biochim.
Biophys. Acta, 849, 337-348.
29.Herzberg, G. (1949) Vibrational and Rotational
Spectra of Polyatomic Molecules [Russian translation], Izd-vo
Inostr. Literatury, Moscow.
30.Streltsov, A. M., Vulto, S. I. E., Shkuropatov,
A. Ya., Hoff, A. J., Aartsma, T. J., and Shuvalov, V. A. (1998) J.
Phys. Chem. B, 102, 7293-7298.
31.Gardner, K. H., and Kay, L. E. (1998) Annu.
Rev. Biophys. Biomol. Struct., 27, 357-406.
32.Plato, M., Lendzian, F., Lubitz, W., Trankle, E.,
and Mobius, K. (1988) The Photosynthetic Bacterial Reaction
Center: Structure and Dynamics (Breton, J., and Vermeglio, A.,
eds.) Plenum Press, N.Y.-London, pp. 379-388.