
|
REVIEW: Oxidative Stress Promotes the Regression of Fetal Liver HemopoiesisE. F. Sato1*, E. Nakagawa1, K. Hiramoto1, S. Yamamasu1, I. Moriyama-Shimamoto2, and M. Inoue11Department of Biochemistry and Molecular Pathology, Osaka City University Medical School, 1-4-3 Asahimachi, Abeno, Osaka 545-8585, Japan; fax: +81-6-6645-3721; E-mail: sato@med.osaka-cu.ac.jp2College of Nursing, Nara Medical University, Japan * To whom correspondence should be addressed.
|
Received April 30, 2003
Although apoptosis is believed to play an important role in the ontogenetic development of animals, the molecular mechanism that triggers the regression of liver hemopoiesis during the perinatal period is not known. Apoptosis is induced by many factors such as a decrease in growth factors and increased oxygen stress. Since hepatic beta-glutamyl transferase (GT) levels change markedly during the perinatal period in rodents, the metabolism of glutathione (GSH), a naturally occurring major antioxidant, might change significantly in and around liver cells. Hemopoietic cells but not hepatocytes exhibit significant apoptosis in thiol-free medium and the hemopoietic apoptosis can be inhibited by various thiols, such as L-cysteine, N-acetyl-L-cysteine, and GSH. The contribution of GSH levels in and around fetal liver cells in the triggering of apoptosis in hemopoietic cells is discussed.
KEY WORDS: apoptosis, fetal liver, beta-glutamyl transferase, glutathione, cysteine, hemopoiesis, liver, oxidative stress
The environment of the fetus changes markedly during the perinatal period. For example, the oxygen pressure of arterial blood rises from 25 to 100 mm Hg immediately after birth and the environmental temperature suddenly decreases from that in the uterus to that of atmospheric conditions outside. Furthermore, nutrients in the colostrum are still insufficient for maintaining neonatal development and maturation. The newborn must overcome severe stress caused by elevation of oxygen pressure, low temperature, dehydration, and starvation. Significant changes in fetal metabolism occur systematically to allow adaptation and survival in the extra-uterine environment.
All organs are remodeled by their constituent cells to adapt themselves to large changes in their environment during the perinatal period. Some of these adaptive changes seem to be regulated by dynamic changes in oxygen metabolism during the perinatal period. The liver is a center for hemopoiesis during the fetal period but becomes a metabolic center for detoxification and maintenance of homeostasis after birth. Hence, the liver consists mainly of hemopoietic cells during the fetal period, and of hepatocytes, sinusoidal cells, and bile ductal cells after birth. Although the regulatory mechanism for hemopoiesis has been studied extensively, the mechanism of perinatal dynamic remodeling of constituent cells in the liver is not known. This review describes the role of oxidative stress in the remodeling and conversion of the liver from a hemopoietic organ to a mature organ consisting of hepatocytes, sinusoidal cells, and bile ductal cells.
METABOLISM OF REACTIVE OXYGEN SPECIES IN THE FETUS
Defense mechanisms against oxidative stress in the fetus are significantly different from those in the adult. For example, activities of Cu/Zn-superoxide dismutase (SOD) and Mn-SOD in fetal liver are 75 and 50% of those in adult liver, respectively, and levels of both enzymes start to increase after birth. Levels of antioxidants, such as reduced glutathione (GSH) and beta-tocopherol, and activities of catalase and glutathione peroxidase are also lower in fetal liver than in adult liver [1]. Thus, lipid peroxidation in fetal liver homogenate proceeds more extensively than in that of adult liver [2]. Under strong stress caused by uterine contraction and abrupt respiration of atmospheric oxygen, oxidative stress might overwhelm the antioxidant defense mechanisms and perturb the structure and functions of fetal tissues. Thus, oxidative stress might function as a physiologically important stressor to force adaptive metabolism in the liver and other tissues during the perinatal period.
LIVER HEMOPOIESIS AND GSH METABOLISM
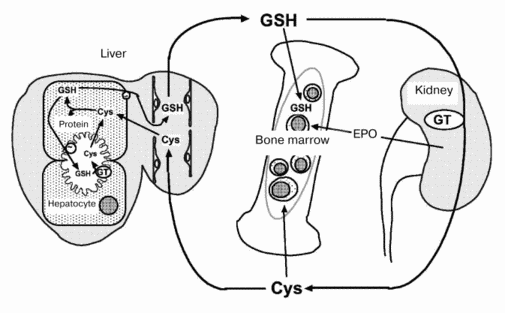
Metabolic profiles of GSH, an important and naturally occurring antioxidant, also differ between fetus and adult [3]. In adult animals, GSH metabolism occurs predominantly via intra- and inter-organ cycles (Fig. 1). GSH in hepatocytes is secreted into Disse's space by an active transport system (20 nmol/min per g liver). In rodents, the secreted GSH enters systemic circulation because the hepatic activity of beta-glutamyl transferase (GT), a membrane-associated enzyme that degrades extracellular GSH, is low. Plasma GSH is degraded to its constituent amino acids (Glu, Cys, Gly) by GT and peptidases in the kidney and other tissues. The cyst(e)ine thus released is supplied to extrahepatic tissues and is used for the synthesis of GSH and proteins. The inter-organ cycle of GSH maintains the plasma GSH level at 5 to 25 µM and regulates oxidative stress in the circulation and in various tissues [4]. However, a GSH inter-organ cycle such as this has not been established in perinatal animals. During late gestation, hepatic GT activity is elevated transiently but significantly to levels (3 to 6 U/g tissue) that would be sufficient for the degradation of GSH secreted by fetal liver cells (7 nmol/min per g tissue). Thus, metabolism of hepatic GSH occurs principally via intra-organ cycles during the perinatal period. The histological structure of fetal liver also differs from that of adult liver. The former consists of hemopoietic foci, parenchymal hepatocytes, and blood vessels while the latter lacks hemopoietic cells.
Levels of GSH, GT, glutathione peroxidase, and glutathione reductase in hepatocytes and hemopoietic cells isolated from rat liver were analyzed on days 18, 20, and 21 of gestation and on day 1 after birth (table). The GSH level in hepatocytes decreased by 50% from day 18 to 20 of gestation and recovered thereafter. GSH levels in hemopoietic cells were significantly lower than those in hepatocytes and remained unchanged during the perinatal period. GT activity in hepatocytes increased markedly from day 18 to 20 of gestation and remained high until day 1 after birth, and GT activity in hemopoietic cells was significantly lower than that in hepatocytes. The marked decrease in GSH levels with a concomitant increase in GT activity in hepatocytes suggests that both secretion and degradation of GSH by hepatocytes are enhanced during the perinatal period. The released amino acids are absorbed and used for the synthesis of GSH and other proteins by liver constituent cells. Tateishi et al. [5] reported that in fetal liver, cysteine is used to synthesize proteins other than GSH. From day 18 to 20 of gestation, protein content in hepatocytes increased 1.5-fold to a level significantly higher than that in hemopoietic cells. Moreover, electron microscopic observation revealed that during this period hepatocytes proliferate rapidly and possess well-developed intracellular organelles, such as rough endoplasmic reticulum. These observations indicate that the reabsorption of constituent amino acids and protein synthesis are also enhanced in hepatocytes relative to hemopoietic cells. Although intracellular levels of GSH decrease in hepatocytes, levels of glutathione peroxidase and reductase increase significantly. Because the metabolism of GSH is enhanced during late gestation, oxidative stress and thiol status in the liver might change significantly both in and around hepatic constituent cells.Fig. 1. Hemopoiesis and inter-organ cycle of GSH in adult animals. GT, beta-glutamyl transferase; EPO, erythropoietin.
Levels of GSH and related enzymes in liver constituent cells during
perinatal period (HPC, hemopoietic cells)

To elucidate the effect of oxidative stress and thiol status on the survival of fetal liver cells, hepatocytes and hemopoietic cells were isolated from fetal livers and cultured for 4 h in the presence or absence of various antioxidants. Cellular DNA then was analyzed by agarose gel electrophoresis. Hemopoietic cells but not hepatocytes exhibited marked fragmentation of nuclear DNA and this fragmentation was inhibited by adding either L-cysteine, N-acetyl-L-cysteine (NAC), or GSH to the culture medium. L-Cystine, SOD, and catalase did not exhibit such an inhibitory effect [6]. These results indicate that both extracellular and intracellular thiols are important for the survival of hemopoietic cells in fetal liver.
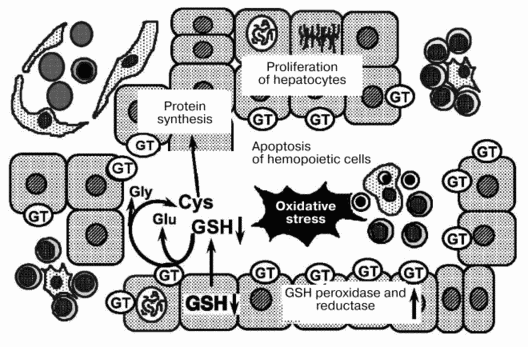
On the basis of such observations, we suggest a mechanism for the regression of fetal liver hemopoiesis during the perinatal period (Fig. 2). During late gestation, extracellular levels of GSH are affected by GT expressed on the outer surface of hepatocytes. The decrease in extracellular GSH seems to trigger apoptosis of hemopoietic cells. Moreover, oxidative stress during the perinatal period might accelerate the regression of liver hemopoiesis.
After birth, GT activity decreases rapidly in the liver and increases in the kidney [7]. After such significant changes, the inter-organ cycles for GSH are established. The sites for hemopoiesis and erythropoietin production are transferred from the liver to the bone marrow and the kidney, respectively. In the bone marrow, hemopoietic cells might receive sufficient amounts of cysteine and related thiols partly because of the absence of cells competing for these substances. Additionally, oxygen tension and energy metabolism in the bone marrow are fairly stable. The bone marrow might be the ideal site for hemopoiesis in adult animals because of these environmental conditions.Fig. 2. Mechanism for the regression of liver hemopoiesis during the perinatal period. GT, beta-glutamyl transferase.
REGULATION OF GT GENE IN FETAL LIVER
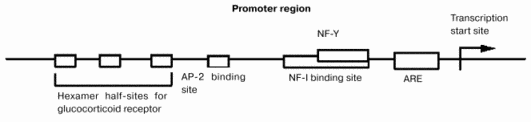
The mechanism by which the gene expression of GT is regulated in fetal liver is not known. Six different promoters are localized upstream of the GT gene in rats [8]. From these promoters, six different mRNAs are synthesized. Although the 5beta-untranslated regions of these mRNAs differ, proteins derived from these mRNAs are the same because their coding regions have identical sequences. Different organs and cells express different mRNAs for GT. For example, type I and II GT mRNAs are expressed in rat kidney while type VI is predominant in mouse small intestine [9, 10] and type III GT mRNA is expressed in fetal liver and hepatoma cells. Binding sites for glucocorticoid receptor, AP-2, nuclear factors I (NF-I) and III (NF-Y), and antioxidant responsive element are localized in the type III promoter region (Fig. 3) [11].
Gene transfection experiments using the promoter region and hepatoma cell lines have revealed that the NF-I and NF-Y families are critically important for the regulation of gene expression. These protein families regulate the differentiation of hepatocytes, albumin synthesis, gluconeogenesis, and GT expression. These findings suggest that these proteins might regulate the expression of GT in fetal liver during late gestation. The glucocorticoid concentration in maternal blood increases transiently on day 19 of gestation, which might also stimulate the gene expression of GT. The presence of the antioxidant responsive element in the promoter region suggests the possibility that oxidative stress or metabolism of reactive oxygen species or both might also regulate the expression of GT. Although the mechanism by which the gene expression of GT is regulated in fetal liver is not known, GT might trigger the regression of liver hemopoiesis and the growth of parenchymal hepatocytes during the perinatal period. The molecular mechanisms by which GT expression is triggered should be studied further.Fig. 3. Type III promoter region of the GT gene. ARE, antioxidant-responsive element.
REFERENCES
1.Yoshioka, T., Takehara, Y., Shimatani, M.,
Abe, K., and Utsumi, K. (1982) Tohoku J. Exp. Med., 137,
391-400.
2.Utsumi, K., Yoshioka, T., Yamanaka, N., and
Nakazawa, T. (1977) FEBS Lett., 79, 1-3.
3.Taniguchi, M., Hirayama, K., Yamaguchi, K.,
Tateishi, N., and Suzuki, M. (1989) in Coenzymes and Cofactors.
Vol. 3. Glutathione. Pt. B (Dolphin, D., Poulson, R., and
Avramovic, O., eds.) John Wiley & Sons Inc, New York, pp.
645-727.
4.Inoue, M., Minamiyama, Y., Yu, H., Yamamasu,
S., Inoue, K., and Sato, E. F. (1995) in Biothiols in Health and
Disease (Packer, L., and Cadenas, E., eds.) Marcel Decker Inc, New
York, pp. 287-304.
5.Higashi, T., and Tateishi, N. (1978) in
Functions of Glutathione in Liver and Kidney (Sies, H., and
Wendel, A., eds.) Springer-Verlag, Berlin, pp. 22-26.
6.Yamamasu, S., Sato, E. F., Ogita, S., and Inoue, M.
(1997) Free Rad. Biol. Med., 23, 100-109.
7.Taniguchi, M., and Inoue, M. (1986) J.
Biochem., 100, 1457-1463.
8.Habib, G. M., Carter, B. Z., Sepulveda, A. R., Shi,
Z. Z., Wan, D. F., Lebovitz, R. M., and Liebermann, M. W. (1995)
J. Biol. Chem., 270, 13711-13715.
9.Kurauchi, O., Lahuna, O., Darbouy, M.,
Aggerbeck, M., Chobert, M. N., and Laperche, Y. (1991)
Biochemistry, 30, 1618-1623.
10.Carter, B. Z., Habib, G. M., Sepulveda, A. R.,
Barrios, R., Wan, D. F., Lebovits, R. M., and Liebermann, M. W. (1994)
J. Biol. Chem., 269, 24581-24585.
11.Brouillet, A., Darbouy, M., Okamoto, T.,
Chobert, M. N., Lahuna, O., Garlatti, M., Goodspeed, D., and
Laperche, Y. (1994) J. Biol. Chem., 269, 14878-14884.