REVIEW: Catalytic Mechanism and Application of Formate Dehydrogenase
V. I. Tishkov1* and V. O. Popov2
1Department of Chemical Enzymology, Faculty of Chemistry, Lomonosov Moscow State University, Moscow 119992, Russia; fax: (7-095) 959-2742; E-mail: vit@enz.chem.msu.ru2Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, Moscow 117234, Russia; fax: (7-095) 952-3441; E-mail: vpopov@inbi.ras.ru
* To whom correspondence should be addressed.
Received August 12, 2004
NAD+-dependent formate dehydrogenase (FDH) is an abundant enzyme that plays an important role in energy supply of methylotrophic microorganisms and in response to stress in plants. FDH belongs to the superfamily of D-specific 2-hydroxy acid dehydrogenases. FDH is widely accepted as a model enzyme to study the mechanism of hydride ion transfer in the active center of dehydrogenases because the reaction catalyzed by the enzyme is devoid of proton transfer steps and implies a substrate with relatively simple structure. FDH is also widely used in enzymatic syntheses of optically active compounds as a versatile biocatalyst for NAD(P)H regeneration consumed in the main reaction. This review covers the late developments in cloning genes of FDH from various sources, studies of its catalytic mechanism and physiological role, and its application for new chiral syntheses.
KEY WORDS: cloning, expression, catalytic mechanism, site directed mutagenesis, Pseudomonas sp. 101, chiral synthesis
The name formate dehydrogenase (FDH) combines several groups of enzymes strongly varied in quaternary structure, presence and type of prosthetic group, and also in substrate specificity. One of such groups is represented by NAD+-dependent formate dehydrogenases (EC 1.2.1.2), which catalyze oxidation of formate ion to carbon dioxide in the coupled reduction of NAD+ to NADH:
HCOO- + NAD+ --> NADH + CO2.
FDHs of this group consist of two identical subunits, contain no metal ions or prosthetic groups, are unable to use one-electron carriers as oxidizers, and are highly specific to both formate and NAD+.
FDHs belong to the superfamily of D-specific 2-hydroxy acid dehydrogenases [1]. The catalytic mechanism of this enzyme is specified by a direct transfer of hydride ion from the substrate onto the C4-atom of the nicotinamide moiety of NAD+ without stages of acid-base catalysis, which are present in reactions catalyzed by other related dehydrogenases. Thus, the FDH-catalyzed reaction is a suitable model for studies on the mechanism of hydride ion transfer in the active center of dehydrogenases.
FDH is widely used for regenerating NADH in enzymatic syntheses of optically active compounds with dehydrogenases [2-4]. Owing to irreversibility of the formate dehydrogenase reaction and the wide pH optimum of the activity, this enzyme is a versatile biocatalyst efficient at pH optima for many processes. The company Degussa (Germany) has developed an industrial scale process for production of tert-L-leucine with FDH as a catalyst of NADH regeneration, and this is one of the largest enzymatic processes in pharmaceutical chemistry [5].
A detailed review concerning properties, structure, and catalytic mechanism of FDH was published in 1994 [6]. Results of studies on FDH during the last decade were summarized in a review published in 2003 [7]. In the present work, the latest data on dehydrogenases will be considered, as well as the aspects of catalytic mechanism and application of FDH that have not been reviewed in the above-mentioned papers.
LOCATION AND PHYSIOLOGICAL ROLE OF FDH AND GENE CLONING
Microorganisms. Formate dehydrogenases are present in all methylotrophic microorganisms. The oxidation of formate to CO2 is one of the main energy sources in cells of these strains. NAD+-dependent FDHs have been found in all methanol-utilizing yeasts of Candida, Pichia, and Hansenula genera and isolated and characterized from various strains [6-8]. However, these enzymes are not so widely distributed in bacteria. Several types of formate-oxidizing enzymes are found in bacteria. Some of them are components of the pyruvate dehydrogenase complex and contain selenocysteine in the active center, as shown for Escherichia coli cells [9]. Other FDHs have in their active center tungsten and molybdenum ions, iron-sulfur clusters, etc. Only three such bacterial strains (Pseudomonas sp. 101, Moraxella sp. C2, and Mycobacterium vaccae N10) were known until the mid-1990s, and NAD+-dependent homogenous FDHs were isolated from them.
The yeast Hansenula polymorpha was the first microorganism from which the FDH gene was cloned [10]. Four years later, in 1990, another gene of FDH was cloned, and it was from the bacterium Pseudomonas sp. 101 [11]. Properties of FDHs isolated from yeasts and bacteria were compared in the mid-1990s, and bacterial FDHs were found to be more active and stable. FDH from Pseudomonas sp. 101 occurred to be the most stable, and up to now it is the most thermostable natural FDH known. As a result, most efforts in searches for new FDHs for application were later concentrated on the bacterial enzymes. Since 1995, FDH genes have been cloned from Moraxella sp. C2 [EMBL accession Y13245], Mycobacterium vaccae N10 [12] (it differs from the Pseudomonas sp. 101 enzyme in only two amino acids), Hyphomicrobium strain JT-17 (FERM P-16973) [13], Paracoccus sp. 12-A [14], Ancylobacter aquaticus [15], and Thiobacillus sp. KNK65MA [16]. During the same period, the genes of yeast FDHs were cloned from Candida methylica [17], Candida boidinii [18-20] (the gene of this enzyme has been cloned in a number of laboratories and differed from the gene of FDH from C. methylica in only two amino acids), and from Pichia pastoris [21].
Another and not the less important source of information about the gene structure and amino acid sequences of FDH is provided by data on sequencing whole genomes of various organisms. Highly homologous genes to those of FDH from Pseudomonas sp. 101 (>80-85%) were found in chromosomes of such bacterial pathogens as Staphylococcus aureus [22], Mycobacterium avium subsp. paratuberculosis strain k10 (GenBank accession AE017240), three strains of Bordetella including B. bronchiseptica RB50 (Alcaligenes bronchisepticus), B. parapertussis strain 12822, and B. pertussis strain Tohama I [23], of the industrial strain Streptomyces avermitilis [24], the uncultured marine alpha- and gamma-proteobacteria HOT2C01 [25] and EBAC31A08 [26], and also on the megaplasmid pSymA of symbiotic nitrogen-fixing bacterium Sinorhizobium meliloti [27]. Results of partial sequencing of genomes of 16 hemiascomycetic yeasts (Saccharomyces bayanus var. uvarum, Saccharomyces exiguus, Saccharomyces servazzii, Zygosaccaromyces rouxii, Saccharomyces kluyveri, Kluyveromyces thermotolerans, Kluyveromyces lactis var. lactis, Kluyveromyces marxianus var. marxianus, Pichia angusta, Debaryomyces hansenii var. hansenii, Pichia farinosa, Pichia sorbitophila, Candida tropicalis, and Yarrowia lipolytica) have been analyzed, and the FDH gene was found only in the strains Pichia angusta, Debaryomyces hansenii var. hansenii, Candida tropicalis, Pichia farinosa, and Saccharomyces bayanus [28]. All these strains, except S. bayanus, are of the same evolutionary group, whereas the strain S. bayanus together with the baker's yeast S. cerevisiae belongs to another group. The complete sequencing of the S. cerevisiae genome revealed the FDH gene location on the 16th chromosome (ORF SCYPL276W), and a pseudogene (ORF SCYOR388C) with a stop-codon in the middle of the gene was found on the 15th chromosome. The encoding sequence is renewed further, but with a +1 shifting of the reading frame [29]. An S. cerevisiae strain has recently been described which has the enzyme expressed from both chromosomes [30]. Analysis of the amino acid sequence of FDH from S. cerevisiae (Fig. 1) has shown rather significant differences of this enzyme from other FDHs, in particular, in the surrounding catalytically essential residues Gln313 and His332 (to facilitate the comparison of different enzymes, here and afterwards the residues will be enumerated according to the amino acid sequence of the best studied FDH from Pseudomonas sp. 101). The gene of this enzyme has been recently cloned in our laboratory [31, 32]. The genome of another important yeast strain Schizosaccharomyces pombe, which is used as a model system for studies on functioning of the cells of higher eukaryotes, lacks the FDH gene (http://www.sanger.ac.uk/Projects/S_pombe/genomic_sequences.html).
FDH genes were also cloned from fungi Aspergillus nidulans [33] and Neurospora crassa [34]. At present, databanks also contain full nucleotide sequences for other five genes from the fungi Mesembryanthemum crystallinum (Genbank Acc. BE035085), Mycosphaerella graminicola (GenBank Acc. AF123482), Magnaporthe grisea (EMBL Acc. AA415108), Ustilago maydis (GenBank Acc. XM_402785), and Gibberella zeae PH-1 (Fusarium graminearum, GenBank Acc. XM_386303).Fig. 1. Amino acid sequences of formate dehydrogenases from bacteria: Pseudomonas sp. 101 (PseFDH) [11], Thiobacillus sp. KNK65MA (TbaFDH) [16], uncultured gamma-proteobacterium EBAC31A08 (UncFDH) [26], Mycobacterium avium subsp. paratuberculosis strain k10 (MavFDH, GenBank AE017240), the N-terminal region of the enzyme from mouse Mus musculus (MmuFDH, EMBL AI505623); FDHs from higher plants: potato (PotFDH) [41], Arabidopsis thaliana (AraFDH, EMBL AF208029), barley (BarFDH, EMBL D88272), and rice Oryza sativa (RicFDH, EMBL AB019533); yeasts: Saccharomyces cerevisiae (SceFDH, EMBL Z75296), Candida boidinii (CboFDH, EMBL AF004096), and Pichia angusta (HanFDH, the former Hansenula polymorpha, EMBL P33677); fungi: Gibberella zeae PH-1 (CzeFDH, GenBank XM_386303), Magnaporthe grisea (MagFDH, EMBL AA415108), and Neurospora crassa (NeuFDH) [34]. Amino acid residues are numerated and the structural elements are placed in reference to FDH from Pseudomonas sp. 101. Catalytically essential residues are indicated with asterisks, and the gray background shows the sequence regions that form the active center of the enzyme.
As differentiated from methylotrophs, the physiological role of FDH in other microorganisms is still insufficiently clear. According to one hypothesis, synthesis of this enzyme starts in the cell as a result of stress exposures, similarly to plants.
Higher plants. FDH from plants was first described in 1951 by D. Davison [35]. Methodical studies on properties and reaction mechanism of FDH from plants were started in the 1970s concurrently with studies on the enzymes from methylotrophic organisms [36-39]. The enzymes were isolated from pea seeds and string bean. However, the role of FDH in plants was then quite incomprehensible. A new page in studies on the physiological role of FDH was opened in 1992, when an unknown protein was found in non-photosynthetic potato tissues (stalk, roots) which constituted ~9% of the total mitochondrial protein [40]. The gene of this protein was cloned and sequenced, and the unknown polypeptide was found to have more than 55% homology with FDH from Pseudomonas sp. 101 [41]. And this polypeptide catalyzed oxidation of formate and reduction of NAD+. The amino acid sequence of FDH deduced by translation of cDNA has been compared with results of amino acid sequencing of the native protein isolated from mitochondria, and on the protein N-terminus a signal peptide has been found which is split off after the entrance into mitochondria (shown in italics in Figs. 1 and 2). The signal sequence is enriched in residues with hydroxyl or positively charged groups and can produce an amphiphilic alpha-helix. The ability of this signal peptide for transporting other proteins into mitochondria was shown on the green fluorescent protein (GFP) [42]. The structure of this sequence is very conservative: removal from the N-terminus of only two amino acids completely inhibits the transport of FDH into mitochondria [42]. Moreover, point mutations violating the amphiphility also inhibited the import of GFP into mitochondria [42].
Mitochondria of other plants also contain FDH [43]. In photosynthesizing tissues, FDH has been recently shown to be a shock protein, and its synthesis in leaves dramatically increases under various exposures (chemical reagents, low temperature, drought, hard ultraviolet, etc.), and also under unfavorable soil composition [44-46]. The main role of the enzyme under these conditions is oxidation of the generated formate. In genetically engineered potato cultivars with an artificially decreased level of FDH expression, such stress exposures resulted in accumulation of formate and increase in the content of proline [47]. Understanding of the mechanism of the response to stress seems promising for creating plants resistant to cold, drought, etc. [48]. Moreover, regulation of the energy exchange is very important when plants are used as producers of recombinant proteins [49]. Owing to the sequencing the genomes of potato, rice, and Arabidopsis thaliana, the physiological role of FDH is studied combined with analysis of expression of other genes [50-55]. The enzyme from Arabidopsis thaliana is the most popular due to the simple structure of its genome. Kinetic properties of FDH from A. thaliana were shown to be affected by heating [56].Fig. 2. Sequences of N-terminal signal peptides of plant FDHs from potato [41], tomato (GenBank AW034965), sweet potato batatas (EMBL BM878811), cotton plant (GenBank AI730437), apple Malus x domestica (EMBL CN496368), English oak (GenBank AJ577266), barley [45], bread wheat Triticum aestivum (GenBank AF479036), maize (GenBank AY111955), rice (EMBL AB019533), Arabidopsis thaliana (EMBL AF208029), Mesembryanthemum crystallinum (GenBank BE035085), and soya (EMBL AW507616 and AW317915 for isoenzymes 1 and 2, respectively). For comparison, the N-terminal region of FDH from the bacterium Pseudomonas sp. 101 is presented at the bottom. The sequence that is split off in the enzyme from potato after the entrance into mitochondria [41] is shown in italic. The amino acid sequences that are supposed to be processed in mitochondria are underlined.

Signal peptides are also present in genes of FDH from potato and other plants. Figure 2 shows N-terminal sequences of eight known sequences of FDHs from plants (potato, cotton plant, English oak, barley, rye, rice, Arabidopsis thaliana, and Mesembryanthemum crystallinum). This figure also shows N-terminal sequences for FDHs from tomato, sweet potato batatas, apple, maize, and two isoenzymes from soya deduced from corresponding cDNAs found in various data banks. In the sequence of FDH from potato, the region of the signal peptide, which is split off after the entrance into mitochondria, is shown in italic [41]. One can see that the sequences from closely related species (Solanaceae: potato and tomato; cereals: rice, barley, bread wheat, maize) display the most pronounced homology. The N-terminal sequence of the FDH gene from soya differs from other plant FDHs. First, it has two isoenzymes with different signal peptides. Second, these peptides are the longest and not homologous to others (Fig. 2). Sequences of the signal peptides were analyzed with the PSORT program [57], and they all but the sequence of the soya FDH displayed a specific sequence processed in mitochondria. All these sequences contain a conservative arginine residue by which the signal peptide is split off. With the ChloroP program, the N-terminal sequence in Arabidopsis thaliana has been shown by Olson et al. [58] to be a signal peptide for FDH transfer into chloroplasts, whereas similar sequences from rice, barley, and potato are not signal sequences for FDH transport into this cell compartment. The authors supposed that due to high reducing potential, FDH can catalyze the back reaction of CO2 reduction to formate in chloroplasts.
Plants often contain several FDH isoforms, and their presence is determined by the state of the plant. The difference in the isoenzyme composition of FDH in normal and diseased palms Pericopsis mooniana is used for selecting trees to be cut down [59]. Until recently, the mechanism of the reversible transformation of FDH isoforms was unclear. Only in 2003, phosphorylation of the enzyme was detected in mitochondria of potato [60]. FDH was phosphorylated on the residues Thr76 and Thr333 [60], and, depending on the modification degree, multiple isoforms were produced with pI varying from 6.75 to 7.19 [61]. The phosphorylation degree was inversely proportional to oxygen concentration in the cell and strongly decreased in the presence of NAD+, formate, and pyruvate. Appearance of additional isoforms was also shown to be a result of a post-translational modification caused by deamidation of the residues Asn329 and Gln330 [61].
Thus, in spite of abundant information about FDH in plants, the physiological role of this enzyme (or of its location-dependent role) remains unclear.
Higher eukaryotes. FDH genes from higher eukaryotes are not yet described. Thus, this gene has not been detected in the genome of Drosophila. For mouse, the databank presents sequences of two cDNA clones encoding an amino acid sequence with a very high homology to the N-termini of bacterial FDHs (Fig. 1). We have found in the databank of human cDNA three clones encoding the amino acid sequence region homologous to the coenzyme-binding domain of FDH. However, considering a rather high homology of the sequence in the NAD(P)+-binding domain of D-specific 2-hydroxy acid dehydrogenases, this sequence can belong to another enzyme of this superfamily. Therefore, it is premature to finally conclude that these clones belong to the gene of human FDH before finishing the genome annotation.
COMPARATIVE ANALYSIS OF AMINO ACID SEQUENCES
The databases now contain full nucleotide sequences for 33 genes of FDHs: 15 genes from bacteria, 7 genes from plants (A. thaliana, potato, rice, barley, cotton plant, English oak, Mesembryanthemum crystallinum), 5 genes from yeasts (S. cerevisiae, C. boidinii, C. methylica, Hansenula polymorpha, and Pichia pastoris), and 6 genes from fungi (A. nidulans, N. crassa, G. zeae PH-1, M. grisea, M. graminicola, U. maydis). Moreover, the set of available cDNA sequences allows us to arrange full amino acid sequences for the enzymes from tomato, apple tree, and two isoenzymes from soya. Databases also contain cDNA sequences encoding certain regions of amino acid sequences of FDHs from more than 25 sources.
Figure 1 presents a number of complete amino acid sequences of FDH from each group and also the N-terminal sequence of the enzyme from mouse. In each group, the enzymes were chosen which were the most different from one another. Thus, for bacterial FDHs, two sequences are presented of the enzymes from the methylotrophic bacteria Pseudomonas sp. 101 and Thiobacillus sp. KNK65MA and also one sequence from the gamma-proteobacterium EBAC31A08 and one sequence from the pathogen M. avium subsp. paratuberculosis strain k10.
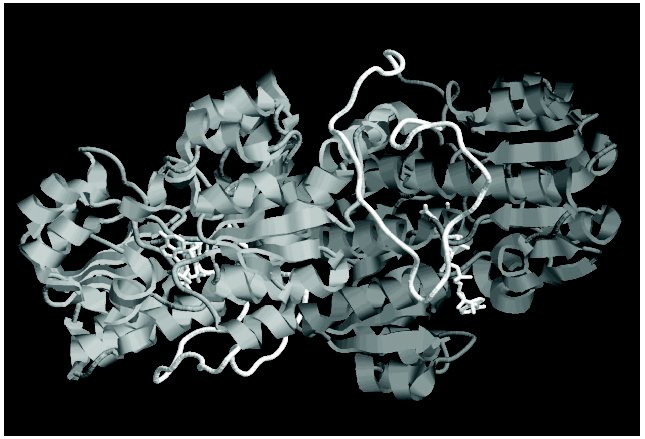
Bacterial FDHs are characterized by the longer N-terminal region (Fig. 1). This region of the polypeptide chain is an elongated loop, which covers a significant part of the enzyme subunit (in Fig. 3 (see color insert) it is shown with white color). Although this loop lacks elements of secondary structure, it is very rigid due to a high number of proline residues (seven). Interaction of this loop with other parts of the subunit is responsible for the higher thermostability of bacterial FDHs as compared to the enzymes from other sources. For example, destruction of only one ionic pair between the residue Asp43, located in the loop, and the residue Lys61 from the alpha1-helix (Fig. 1) increases sixfold the thermal inactivation rate of FDH from Pseudomonas sp. 101 [62]. In addition to multiple interactions with residues inside the subunit, this loop is involved in formation of intersubunit contacts (Fig. 3).
Not considering differences in the N-terminal sequence, all FDHs can be divided into two groups. The first group includes the enzymes from bacteria and plants, and the other group includes FDHs from yeasts and fungi. It is most interesting that the second group contains an additional residue between the third glycine residue in the conservative triad (Gly/Ala)XGlyXXGly and the residue Asp221 (numeration according to the sequence of FDH from Pseudomonas sp. 101), which plays an important role in providing the specificity to NAD+ [63, 64]. Two other breaks are located between two pairs of catalytically essential residues Ile122-Asn146 and Gln313-His332 (Fig. 1).Fig. 3. Spatial structure of FDH holo-form from Pseudomonas sp. 101. Two subunits of the enzyme are shown by different tints of gray color. The elongated loop on the N-termini, which is absent in the FDH sequences from plants, yeasts, and microscopic fungi, is shown in white. The figure was obtained with the RasMol 2.71 program.
FDH is a highly conservative enzyme. The absolute homology is no less than 80-85% between enzymes of the same group and 50-55% and more between two enzymes from the different groups. At present, 36 complete and 25 partial sequences of FDH from various sources are known, and their comparison allows us to reveal 71 conservative residues, and, because the calculated mean length of the enzyme includes about 365 residues (neglecting the length of the loop in bacterial FDHs), this is nearly 20% of all residues. Analysis of spatial location of conservative residues in the structure of the FDH from Pseudomonas sp. 101 has shown that the functions of these residues are different. Some of the residues provide stability of the subunit structure (e.g., the ion pair Lys2-Asp89), others are involved in intersubunit interactions (Arg163, Asn164, Trp177, Ala180, Asp188), and only a few of them are involved in catalysis. In Fig. 1 regions of the polypeptide chain which directly form the active center of the enzyme are shown with gray color and asterisks indicate catalytically important amino acid residues: Pro97, Phe98, Ile122, Asn146, (Ala/Gly)198, Gly200, Gly203, Arg284, Gln313, and His332. Moreover, the asterisk indicates the position 255 (in the majority of FDHs it is occupied by Cys) because the residue in this position is responsible for interaction with the adenine moiety of NAD+. Among conservative residues in FDH, the residue Asp308 should be also noted because its carboxyl group forms a hydrogen bond with the amide group of the nicotinamide moiety of NAD+ [6, 65]. The mobility of some residues (Phe98, Cys255, Gln313, and His332) is strongly limited because of their neighborhood with a Pro residue. This is especially obvious in the case of residue Gln313, which in the overwhelming majority of the enzymes is fixed by two Pro residues. The picture is quite opposite for the residue in position 122 (Ile or Val). This position is occupied by different residues, because the generation of the hydrogen bond with the substrate molecule is contributed not by the side group but by the oxygen atom from the carbonyl group of the major chain [65]. And the increased flexibility of the polypeptide chain in this position provided by two neighboring Gly residues seems to be responsible for the optimal orientation of the carbonyl group of residue 122 with respect to the substrate. The residues Gly200 and Gly203 also provide maximal contact between the betaA-strand and the alphaB-helix that is required for optimal binding of NAD+ in the active center.
To reveal common residues necessary for catalysis in all D-specific 2-hydroxy acid dehydrogenases, we have designed “a portrait” of a generalized active center based on values of the Shannon's entropy [66, 67]. Such an approach is widely used in searching for catalytically essential residues of the active center based on analysis of amino acid sequences of enzymes [68]. The Shannon's entropy Hi for the i-position in the amino acid sequence is calculated using values of pij, which represents the probability of j-amino acid encounter in this position:
If the probability of one or another amino acid encounter in any position tends to unity, the probabilities for other amino acids will tend to zero. In this case the value of entropy Hi will also tend to zero, whereas for conservative invariant residues Hi = 0 for the i-position. Thus, the null value of the Shannon's entropy may be used for searching for conservative residues in families of various enzymes.
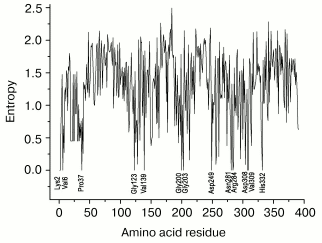
Figure 4 presents values of Shannon's entropy for various residues in sequences of 125 enzymes from the superfamily of D-specific 2-hydroxy acid dehydrogenases. The amino acid residues are enumerated for FDH from Pseudomonas sp. 101. Entropy was found to have the null value for 13 residues: Lys2, Val6, Leu37, Gly123, Val139, Gly200, Gly203, Asp249, Asn281, Arg284, Asp308, Val309, and His332. Only four residues (Gly200, Gly203, Arg284, and His332) coincide with residues of the catalytic center of FDH (Fig. 1). Moreover, 79% of the enzymes analyzed have the residues Val or Ile, and 10 and 8% have the residues Thr and Ala, respectively, in position which corresponds to the residue 122 (Ile/Val) important for the substrate binding in the active center of FDH. The residue Gln313 is absent in this list because, unlike FDH in all other D-specific 2-hydroxy acid dehydrogenases, it is replaced by the residue Glu invariant for them. This residue paired with the residue His332 forms in the active center the proton relay system, which is required for steps of acid-base catalysis. The FDH mechanism lacks such steps, and the presence of the residue Gln in this position ensures a wide pH optimum of the enzyme action. The mutation Gln313Glu in FDH from Pseudomonas sp. 101 narrows the pH optimum of the enzyme from 6.0-9.0 to 6.0-7.0 [69]. The presence of the residue Gln instead of Glu in FDHs is promising for identification of the gene encoding FDH on annotating the sequences obtained by sequencing whole genomes.
The invariance of residue Asp308 suggests its important role in orientation of the nicotinamide ring in the active center of all the enzymes. Residue Lys2 seems to support the tertiary structure of the subunit in all the enzymes, because the residues Asp and Glu in 86% of cases occupy the position 89 (Asp89 in all FDHs), whereas in 12% of cases this position is occupied by a residue Ser. The situation is similar for residue Asp249. It also forms an ion pair in FDH with residue Lys274 invariant for it, and the residue Lys is also present in this position in 91% of the other enzymes. The role of residues Val6 and Val139 is unclear because they are located beyond the active center and fail to contact with other conservative residues. Although the Shannon's entropy value of residue Pro37 is null, it is not conservative for enzymes of the superfamily under consideration because it occurs only in sequences of bacterial FDHs.Fig. 4. Values of Shannon's entropy for different residues in sequences of the superfamily of D-specific 2-hydroxy acid dehydrogenases. The plot is based on the entropy values from the HSSP database (http://hssp.ebi.ac.uk) for 125 enzymes of this family. The amino acid residues are numerated for FDH from Pseudomonas sp. 101.
Thus, comparison of the amino acid sequences of enzymes of the superfamily of D-specific 2-hydroxy acid dehydrogenases based on Shannon's entropy values reveals a similar organization of the active center of FDH and other enzymes of this family.
SPECIFIC FEATURES OF THE CATALYTIC MECHANISM OF FORMATE
DEHYDROGENASES FROM BACTERIA AND OTHER SOURCES
The molecular catalytic mechanism of FDH has been considered in detail in the reviews [6, 7]. These papers also comprehensively describe site directed mutagenesis for testing the hypothetical reaction mechanism exemplified by the enzymes from Pseudomonas sp. 101 and C. boidinii and present a summarized table with results of various amino acid substitutions [7]. We shall briefly consider specific features and differences in the catalytic mechanism of FDHs from various sources that have not been considered earlier.
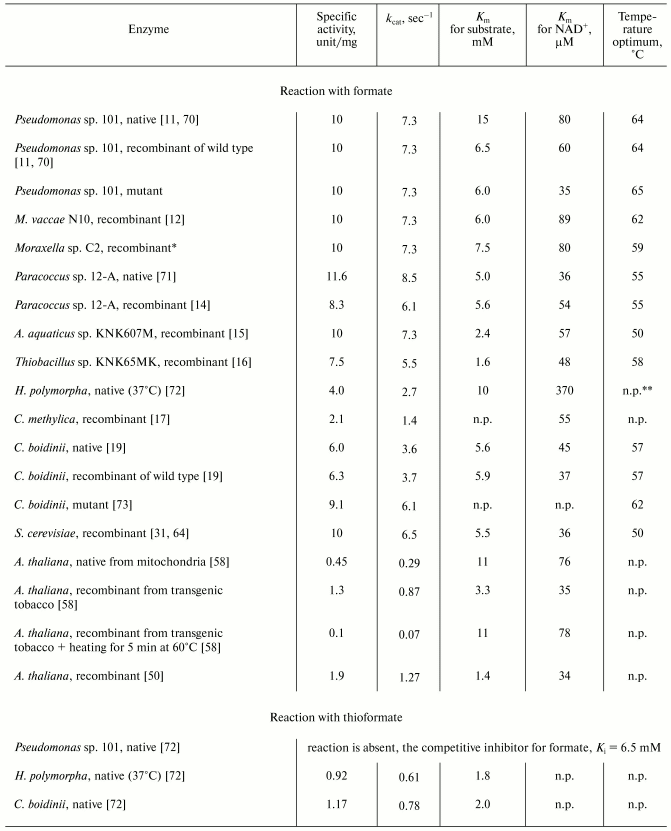
Kinetic parameters. Table 1 presents activity values and Michaelis constants for formate (thioformate) and NAD+ and also the temperature optima for native and recombinant FDHs from various sources. The data presented in the table lead to the following conclusions.
Table 1. Kinetic properties of formate
dehydrogenases from various sources (30°C, pH 7.0-7.5)
*A. G. Galkin, personal communication.
**n.p., data not presented.
1. All the enzymes have similar Km values for formate (3-10 mM) and NAD+ (35-90 µM), and the recombinant FDHs are slightly different from the native enzymes [11-17, 19, 31, 50, 58, 64, 70-73]. Different values of the constants for the same enzyme seem to be caused by differences in conditions of their determination or in methods of purification. Thus, the higher Km value for formate of the native FDH from Pseudomonas sp. 101 than of the recombinant enzyme (Table 1) is associated with the seven amino acids shorter C-terminus of the native enzyme. In E. coli cells the subunit of the recombinant enzyme is synthesized as a full-length polypeptide containing 400 amino acids [11, 71], whereas the FDH isolated from the initial Pseudomonas sp. 101 strain was shown by amino acid sequencing to consist of 393/394 amino acids [74]. Three additional lysine residues on the C-terminus of the recombinant FDH from Pseudomonas sp. 101 also prevents its crystallization under conditions optimized for the native enzyme. The removal of six amino acids from the C-terminus of the genetically engineered FDH results in enzyme identical to the native one in both kinetic properties and ability for crystallization (Table 1, Pseudomonas sp. 101, recombinant of wild type). The recombinant FDH from A. thaliana [58] has lower values of Km for both substrates, especially for formate (Table 1); however, heating of this preparation at 60°C for 5 min is sufficient to produce enzyme identical in kinetic properties to the native FDH isolated from mitochondria.
2. Specific activities of bacterial FDHs are about 1.7-fold higher than those of the enzymes from methylotrophic yeasts. Since the molecular mass of bacterial FDHs is 10% greater, the difference in the kcat values is twofold. FDH from baker's yeast is an exception [31, 64], but because of low thermostability (Table 1) it does not seem promising for application. Methods of genetic engineering can improve both the maximal rate and the affinity of FDH for substrates. Thus, the specific activity of FDH from C. boidinii has been 1.7-fold increased by the substitution Phe285Ser [73], and a single mutation in the enzyme from Pseudomonas sp. 101 decreased the Km value for NAD+ nearly twofold and 2.4-fold increased the thermal stability [75].
Substrate specificity. Bacterial and yeast FDHs behave differently when substrates with structure similar to formate are used (Table 1). Thus, FDH from Pseudomonas sp. 101 cannot catalyze the reduction of NAD+ in the presence of thioformate. Thioformate is a competitive inhibitor of the enzyme for formate, and the Ki value is comparable to the Km value for formate [72]. Thioformate was earlier reported to be a highly effective competitive inhibitor for formate of FDH from the yeast C. boidinii (Ki = 80 µM) [76], but later this enzyme has been shown to oxidize this substrate, the ratios kcat/Km for formate and thioformate being similar [72]. Another yeast FDH, from H. polymorpha, also catalyzes the reaction with thioformate, at Km = 1.8 mM, but the kcat/Km for this substrate was fourfold lower than for formate [72].
Another significant difference in the substrate specificities of bacterial and yeast FDHs is different value of the primary kinetic isotopic effect when formate is replaced by deuteroformate. The substitution of protium by deuterium in the substrate molecule has virtually no effect on the Km value, but the ratio of maximal rates V Hmax/VDmax for FDH from the yeasts C. boidinii and C. methylica was 2.17 [77] and 2.3 [78], respectively, as compared to values of 3.0 and 3.6 at pH 7.0 and 9.0, respectively, for FDH from the bacterium Pseudomonas sp. 101 [79, 80].
Coenzyme specificity. All FDHs under consideration are highly specific to NAD+ and virtually fail to catalyze the reaction with NADP+. Among FDHs studied, the FDH from baker's yeast displayed the highest specificity to NAD+. The ratio between the kcat/Km values of this enzyme for NAD+ and NADP+ is >= 3*109 [64] (Table 2). FDH from the yeast C. methylica is also highly specific to NAD+ ((kcat/Km)NAD+/(kcat/Km)NADP+ > 250,000) [63]. The coenzyme selectivity of bacterial FDHs is much lower. For FDHs from Pseudomonas sp. 101 and Thiobacillus sp. KNK65MK the ratios (kcat/Km)NAD+/(kcat/Km)NADP+ are only 2400 [64] and >100 [16], respectively.
Table 2. Kinetic properties of mutant
formate dehydrogenases and recombinant wild type enzymes from the yeast
Saccharomyces cerevisiae (SceFDH), Candida methylica
(CmeFDH), and the bacterium Pseudomonas sp. 101
(PseFDH)*
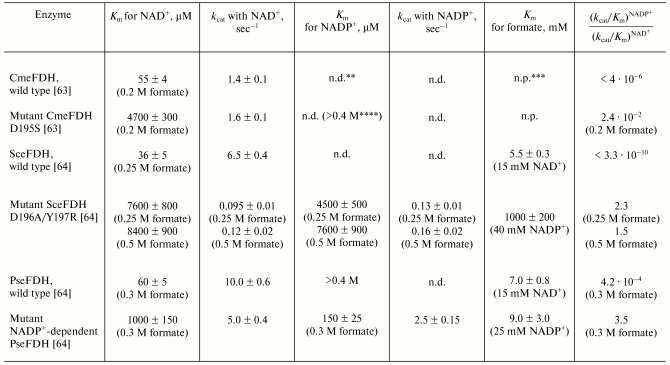
*Reproduced with some additions from the work [64].
**n.d., not detected.
***n.p., not presented.
****Evaluated from data presented in Fig. 2
[63].
There have been attempts to change the coenzyme specificity of FDHs from Pseudomonas sp. 101, C. methylica, and S. cerevisiae. Production of FDH preparations highly specific to NADP+ would promote the creation of an inexpensive and efficient system of NADPH regeneration (see below). However, at present there is no universal approach for changing the cofactor specificity of dehydrogenases, and experimental results are strongly affected by individual specific features of the active center structure of the enzyme. There are only three examples of a successful change of coenzyme specificity of dehydrogenases from NAD+ to NADP+ [81-84]. NADP+-dependent FDH was first prepared for the enzyme from Pseudomonas sp. 101 [85]. The (kcat/Km)NADP+ value of this mutant FDH was only threefold less than the (kcat/Km)NAD+ value for the wild type enzyme [64]. The Asp195Ser substitution in FDH from C. methylica increased 10,000-fold the affinity of the enzyme for NADP+ [63]; however, the enzyme was still much more specific to NAD+ than to NADP+ (Table 2). The double substitution Asp196Ala/Tyr197Arg in FDH from baker's yeast resulted in enzyme 2.4-fold more specific to NADP+ than to NAD+ [64], but the total catalytic activity of this mutant was several thousand times lower than that of the mutant NADP+-dependent FDH from Pseudomonas sp. 101 (Table 2). First of all, this is associated with the increased value of Michaelis constant of the mutant SceFDH for formate from 5.5 to 1000 mM (Table 2). Due to the so-called ordered kinetic reaction mechanism of the FDH from S. cerevisiae, formate can be bound in the active center only after generation of the double complex SceFDH-NAD+ [32]. It seems that in the case of the mutant enzyme the binding of NADP+ fails to provide an adequate conformation of the active center for effective binding of formate. In the case of FDH from Pseudomonas sp. 101, formate can be bound with the free enzyme [6, 80]. It seems that mutation in the coenzyme-binding domain introduced to improve the NADP+ binding did not affect the structure of the active center of the enzyme in the region of the formate-binding site (Table 2).
Kinetic mechanism and interaction of active centers. All eukaryotic FDHs studied catalyze oxidation of formate by the ordered kinetic mechanism [6, 37, 72, 86]. Bacterial enzymes act by an random kinetic mechanism with a rapid equilibrium between the free enzyme and double complexes FDH-formate and FDH-NAD+ [6, 80, 87]. The additional loop on the N-terminal end of bacterial FDHs (Figs. 1 and 3) is supposed to be responsible for the enzymatic reaction of this enzyme by the random kinetic mechanism. To test this hypothesis, we plan to prepare a mutant enzyme without the loop.
Active centers in FDHs from methylotrophic bacteria and yeasts function independently of one another [6, 7]. FDH from pea seeds was shown to have two types of active centers different in the affinity for formate and azide [39]. Kinetic properties of FDH from baker's yeast were studied in this laboratory, and this enzyme was found to be intermediate between FDHs with independent and dependent active centers [31]. Formate and azide are bound independently of one another in two active centers of SceFDH at pH 6.0-8.5, but at pH >= 9.0, the concentration dependence of the reaction rate did not follow the simple Michaelis-Menten equation but corresponded to the model with two types of active centers [31]. By site directed mutagenesis, mutant enzymes were prepared with mutually dependent active centers even at neutral pH values.
APPLICATION OF FORMATE DEHYDROGENASE
Optically active compounds are the most selective and efficient regulators of vital activity of the cell because of chirality of the living world. Among the 500 drug best sellers all over the world more than 58% are chiral compounds, and among new preparations, their fraction achieves 70%. Different optical isomers of the same compound can have quite opposite physiological effects, in some extreme cases resulting in disease.
According to prescriptions of the Food and Drug Administration of the United States of America, from 1999 the optical purity of all chiral compounds used as drugs has to be no less than 99%. Therefore, the number of studies concerning the application of the enzyme in pharmaceutical industry sharply increased within the last decade. Virtually all classes of enzymes can be used for synthesis of optically active compounds, but at present, various hydrolases are used most often. Unfortunately, these enzymes can be used only for separation of racemates, and 50% of the initial substance represented by the other isomer remains as a byproduct. Additional procedures are required for increasing the yield of the desired product. Thus, the remaining enantiomer can be converted into a mixture of racemates with a corresponding racemase. The rate of nonenzymatic nonspecific reaction also needs special attention when hydrolases are used for production of optically active compounds. To decrease this rate, one has sometimes to use conditions that are far from optimal for the enzyme.
Unlike hydrolases, oxidoreductases, and in particular dehydrogenases can be used to produce optically active compounds from nonchiral ones (thus, L- and D-lactate can be prepared from pyruvate using L- and D-lactate dehydrogenases). All dehydrogenases are extremely stereospecific in the transfer of hydride ion between the substrate and coenzyme. Thus, L-lactate dehydrogenase from porcine muscles [88] and alcohol dehydrogenase from yeast [89] commit only one “stereochemical” error per 1*107 and 7*109 catalytic cycles, respectively. Moreover, as differentiated from hydrolases, the rate of nonenzymatic process in reactions catalyzed by dehydrogenases virtually equals zero. Therefore, these enzymes are promising for production of optically active compounds with very high optical purity (99.9-99.99%).
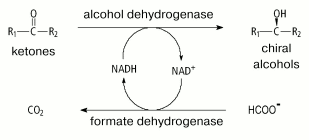
Because of the high price of NADH and especially of NADPH (>US$ 12,000/kg), processes involving only dehydrogenases is economically unprofitable. To decrease the contribution of the cost of reduced coenzymes to the cost of the final product, another reaction has been proposed be used which converts NAD(P)+ back to NAD(P)H [2, 3, 90]. This reaction can be catalyzed by both the major enzyme (e.g., addition into the system of excess isopropanol in processes with involvement of alcohol dehydrogenases) and other additional enzyme. Figure 5 presents the general scheme of such a process, with FDH as the second enzyme. The reaction catalyzed by FDH fits the best requirements for the system of NADH regeneration [75].
1. The reaction of formate oxidation to carbon dioxide is irreversible, and this allows us to use thermodynamic pressure on the major process and obtain 99-100% yield of the desired product.
2. The FDH activity does not change in the pH range from 5.5 to 11.0 and values of Michaelis constants for NAD+ and formate are unchanged in the pH range from 6.0 to 9.5 [91], thus FDH can be used in combination with any dehydrogenase that has activity optimum in this range.
3. Formate salts are very low-cost and available substrates. Formate ion does not influence activities of enzymes used in the proposed process. At present, only one enzyme is known (xylitol reductase [92]) that is inhibited by formate, and the inhibition constant (182 mM) is comparable with formate concentrations used in practice.
4. Carbon dioxide, which is the product of the FDH-catalyzed reaction, also does not inhibit the majority of dehydrogenases, does not interfere with purification of the desired compound, and can be easily removed from the reaction mixture at decreased pressure.
5. FDHs from bacteria and yeasts are highly stable enzymes and can work in systems for weeks and months.
6. FDHs are inexpensive and available, and the price for the production of the enzyme is low. The content of FDH in the starting strains of methylotrophic bacteria and yeasts can be up to 10-18% of the total cell protein [93, 94], and technology of recombinant DNA provides for obtaining of still higher expression of the desired product.
Shortcomings of FDHs are their rather low specific activities: 6-7 and 10 units/mg protein for homogenous FDHs from yeasts [18, 95] and bacteria [71], respectively (Table 1). Moreover, NADP+-specific FDHs have not yet been found in nature, and this prevents using this enzyme for regeneration of NADPH. Such systems were first elaborated after a mutant FDH from Pseudomonas sp. 101 with coenzyme specificity changed from NAD+ to NADP+ was prepared in 1993 in this laboratory [85, 96].Fig. 5. A scheme for synthesis of optically active compounds using dehydrogenases and formate dehydrogenase for regeneration of NADH.
The use of FDHs for regeneration of NADH was proposed even 25 years ago [2, 3, 90], but the high price cost of the enzyme production from natural strains retarded their active application. In fact, of native enzymes only leucine dehydrogenase and FDH from C. boidinii [5] were used for industrial production of tert-L-leucine, and this still remains the largest-scale process using dehydrogenases even after turning to recombinant enzymes.
The price of FDHs as biocatalysts for regeneration of NADH might be decreased by some approaches: increasing the operational stability of FDH, decreasing the price of production and storage, and improvement of kinetic properties.
These three factors are closely interrelated. Thus, increase in the thermal stability will allow us to increase the temperature of the process, which will also increase the specific activity of the enzyme (the improvement of kinetic properties). Increased thermal stability can be also used for purification of the enzyme isolated from the recombinant strain from admixtures of E. coli proteins by heat treatment of the cell-free extract, and this stage needs no expensive equipment and additional reagents and carriers (decreasing the price of production). Increase in chemical and thermal stability will also prolong the storage life of the enzyme without loss of its activity. Thus, FDH from wild type C. boidinii must be stored in 50% glycerol at -20°C [97], whereas the more thermostable FDH from Pseudomonas sp. 101 (and especially its genetically engineered modification) can be stored at 4°C for several years without loss of the activity [75].
Increasing operational stability. Loss of enzyme activity during the process is associated with both chemical modification of catalytically essential residues and thermal denaturation. In the case of FDH, the enzyme is chemically inactivated because of oxidation of cysteine residues with oxygen of the air or their modification with microimpurities in the initial reagents [19, 98]. The content of cysteine residues in the enzyme varies depending on the source. FDH from C. boidinii has the lowest number of cysteine residues (two residues per subunit, Cys23 and Cys262). The number of cysteine residues is maximal (seven per subunit) in FDHs from the methylotrophic bacteria (Pseudomonas sp. 101, M. vaccae N10, Moraxella sp. C-2, Hyphomicrobium strain JT-17 (FERM P-16973), Paracoccus sp. 12-A, A. aquaticus sp. KNK607M). Other bacterial FDHs have three to five cysteine residues per subunit. Most yeast, plant, and fungal FDHs contain three Cys residues per subunit.
Chemical inactivation of FDH from Pseudomonas sp. 101 is mainly caused by oxidation or modification of the Cys255 residue [74]. This residue is located in the coenzyme-binding domain of the enzyme, interacts with the adenine moiety of NAD+ molecule, and in the apo form of FDH is accessible to the solvent. The residue in position 255 plays a key role in maintaining chemical stability of bacterial FDHs, and this is well exemplified by the enzyme from Thiobacillus sp. NK65MA [16]. This enzyme is the most homologous to FDH from Pseudomonas sp. 101 and is significantly superior to the latter in chemical stability due to Val residue in position 255 instead of Cys (Fig. 1). It also has an Ala residue in position 288 (Cys288 in FDH from Pseudomonas sp. 101). Of 15 bacterial FDHs with known sequences, ten have Cys in position 255, two have Ala, and the remaining three FDHs have Val, Thr, or Ser. None of 16 known sequences of plant FDHs has Cys in this position (13 FDHs have Thr and three FDHs have Met). Four of seven yeast FDHs (~70%) and seven of seven fungal FDHs (100%) have Cys in this position.
In 1993 in this laboratory (Faculty of Chemistry, Lomonosov Moscow State University) mutant FDHs were prepared from Pseudomonas sp. 101 with substitutions Cys255Ser and Cys255Met [99]. These mutants displayed at least two order of magnitude higher chemical stability than the wild type enzyme. On inactivation with bivalent mercury ions, the stabilization effect was no less than 1000-fold increased. Unfortunately, both mutations impaired the binding of NAD+. Later, the mutant Cys255Ala was obtained [100] with kinetic properties identical to those of the wild type recombinant FDH. The FDH molecule also contains other Cys residues, which are much less reactive than Cys255, but their modification also resulted in the loss of activity [99]. Analysis of protein structure has shown that two other cysteine residues, Cys145 and Cys354, are accessible to modifying agents. The Cys354 residue is located on the surface of the protein globule, while the Cys145 residue is found in the enzyme active site near the Asn146 residue, which is involved in the binding of formate ion (Fig. 1). To elucidate the role of these residues in stability and reaction mechanism of FDH from Pseudomonas sp. 101, point mutants have been obtained with substitutions of Cys145 with Ala and Ser and of Cys354 with Ala, Ser, and Arg [100]. The best mutations were combined with the substitution Cys255Ala into double mutants, which were found to be 1000-fold more stable against chemical inactivation.
Similar studies were performed on FDH from Mycobacterium vaccae N10 [101]. The Cys255 residue was substituted with residues Ala, Ser, and Val, and the Cys145 residue was substituted with Ala and Ser. The Cys5 residue was also substituted with Ala, Ser, and Val. Altogether 13 mutants were obtained including point mutants C145S, C255S, C255V, double mutants C6A/C255S, C6V/C255S, C6S/C255A, C6S/C255V, C6A/C255V, C145S/C255S, C145S/C255V, and triple mutants C6S/C145S/C255S, C6A/C145S/C255V, and C6A/C145A/C255V. Unfortunately, these results cannot be compared with data on FDH from Pseudomonas sp. 101 because kinetic properties and stability have not been studied in detail, and the resistance was assessed by values of residual activity of the mutants after incubation in aqueous solutions of some chloro-organic compounds. Based on similar values of residual activity of the mutants C6S/C145S/C255S and C145S/C255S, one can conclude that the Cys5 residue is not essential in providing chemical stability of FDH.
FDH from C. boidinii has only two cysteine residues per subunit: Cys23 and Cys262. The latter is equivalent in its position to residue Cys288 in FDH from Pseudomonas sp. 101 (Fig. 1). The model of this enzyme structure shows that the Cys23 residue is exposed to the solvent [19]. Mutants of C. boidinii FDH have been obtained: C23S, C262V, C23S/C262V, and C23S/C262A. These substitutions did not influence the kinetic properties but significantly increased the stability against inactivation by Cu2+, and under conditions of the technological process, the half-life of the mutants C23S and C23S/C262A increased to 500 from 60-70 h of the wild type enzyme [19].
Thus, site directed mutagenesis of cysteine residues resulted in production of bacterial and yeast FDHs with increased chemical stability.
Increasing thermal stability. Substitution of Cys residues decreases the thermal stability of FDH. In the case of FDA from Pseudomonas sp. 101, the rate of thermal inactivation of the mutants Cys255Ala and Cys354Ser increased four- and twofold, respectively, as compared to the wild type FDA. Combining these mutations displayed an additive effect on the thermal stability, and the rate of thermal inactivation of the double mutant increased eightfold. The effect was still stronger in the case of FDH from C. boidinii. The thermal inactivation rate constants for the mutants Cys23Ser and Cys262Val increased 6- and 19-fold, and for the double mutants C23S/C262V and C23S/C262A they increased 100- and 33-fold, respectively [19].
Such a decrease in thermal stability was not critical for the FDH from Pseudomonas sp. 101 because even the double mutant C255A/C354S of this enzyme had higher thermal stability than the wild type FDH from C. boidinii (the activities were 50% decreased within 20 min at 59 and 57°C, respectively). For the double mutants C23S/C262V and C23S/C262A these temperatures decreased to 44 and 48°C, respectively [19]. The thermal stability of mutants from C. boidinii was increased by random mutagenesis using a “directed evolution” technique [73]. The authors found 11 mutations increasing the enzyme thermal stability, but the substitutions Glu151Asp and Arg178Ser were the most important because they determined more than 90% of the stabilization effect. The Tm value of the best mutant C23S/E151D/R178S/K206R/T315N was 62°C [73], which was equal to that of FDH from the wild type Pseudomonas sp. 101. Increasing the thermal stability of FDH from C. boidinii allowed the process temperature of production of tret-L-leucine to be increased from 30 to 40°C (K. Drauz, personal communication).
To increase the thermal stability of FDH from Pseudomonas sp. 101, site directed mutagenesis was used. Positions for the mutagenesis were chosen using general principles of protein stabilization based on hydrophobization of alpha-helices [102], optimization of electrostatic interactions [62] and conformation of the polypeptide chain [103, 104], etc. As a result, 14 substitutions were found which increased the enzyme thermal stability. Seven of them, which at least did not affect the kinetic properties of FDH, were chosen for producing a multiple-point mutant. The resulting mutant which combined the substitutions to increase both chemical and temperature stability had the tenfold higher thermal stability than the wild type enzyme. This mutant did not lose the activity after incubation for 3 months at 45°C, whereas the wild type FDH completely lost its activity after 60 days.
Decreasing the price of FDH production. Optimization of the culture. The most important advantage of recombinant strains is the possibility of obtaining the expression level of the desired protein several times higher than of the initial strains and cultivating recombinant strains under conditions of high-density cultures. Technology of recombinant DNA allowed us to significantly increase the FDH yield on culturing. The creation of a highly efficient recombinant strain as a producer of the desired enzyme is a multiparametric process. For FDH from Pseudomonas sp. 101 it is described in [75, 105]. This work resulted in creation of producer strain expressing FDH up to 50-55% of E. coli proteins as an active soluble enzyme, and this expression system operates equally well with all mutants prepared. At present, the time-space yield reaches 25,000 units of FDH activity per liter per 24 h in fermenters with volume of 400 liters. In the case of small culture volumes, the system productivity reaches 40,000 units (or 4 g enzyme) per liter per 24 h. Preliminary calculations show that this parameter can still be increased two-threefold.
In the case of FDH from C. boidinii, the expression level is not more than 25% of the total protein of E. coli [17-20]. High-density cultures are reported in the literature [106], but no data are presented on the productivity of this process.
Purification of recombinant FDHs. The increase in the enzyme content in recombinant strains resulted in a significant decrease in volumes of solutions to be treated and in expenditure of reagents required for purification. The increase in thermal stability of FDH allowed us to use heat treatment as a separate effective stage of purification. On isolation of recombinant mutant FDHs from Pseudomonas sp. 101, the heating of the cell-free extract at 58-60°C for 15-20 min increases the enzyme purity from 50 to 80-85%. Such purity is quite sufficient for application of FDH, and the subsequent purification of the enzyme includes removal of DNA, oligosaccharides, and other cell components. In particular, this can be achieved by extraction in biphasic water-polyethylene glycol-salt systems [97, 105] or by hydrophobic chromatography.
Genetic engineering methods significantly increased the FDH stability and developed highly effective processes of culture and purification. At present, the cost of the enzyme production is more than tenfold decreased and is not more than US $ 0.010-0.015 per activity unit. For production of tert-L-leucine, the cost of enzymes in the total cost of production is now only 5-8%.
Regeneration of NAD(P)H during enzymatic chiral syntheses. FDHs are so often used for regeneration of reduced cofactors that these situations are analyzed in a number of reviews [3, 4, 107-113]. Several approaches are used in technological processes.
1. Various types of flow membrane reactor with two enzymes and with free or immobilized on polyethylene glycol NAD+ [114, 115]. This principle underlies the synthesis of tert-L-leucine [5].
2. Flow membrane reactor with two enzymes and free NAD(P)+ in the recirculation regime. Such a reactor is used for saturating the solution with oxygen [116].
3. Periodic reactor of ideal mixing with free NAD(P)+. The reactor works in the case of both homogenous solutions and microemulsions or unmixed aqueous-organic phases [117-119].
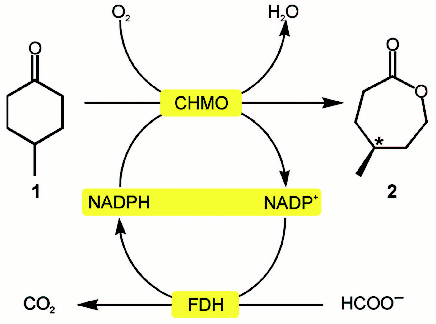
Both free enzymes and intact cells with all necessary enzymes expressed (sometimes up to four enzymes concurrently) are used as biocatalysts [120-122]. As an interesting example, let us consider the production of chiral lactones by the Baeyer-Villiger reaction catalyzed by cyclohexanone monooxygenase where NADPH is regenerated with the mutant NADP+-specific FDH from Pseudomonas sp. 101 [123] (Fig. 6, see color insert). This reaction is characterized by a cycle increased by one atom with a concurrent generation of the chiral center.
In conclusion, note that although NAD+-dependent FDH has been studied for more than a decade and the protein itself is one of the most completely and comprehensively characterized enzymes, its great potential in biotechnology is the reason for extending studies for its application.Fig. 6. Baeyer-Villiger reaction using cyclohexanone monooxygenase and mutant NADP+-dependent formate dehydrogenase [116].
In the future data will be obtained on the structure of FDHs from various sources and of different mutant FDHs, and this will allow us to explain how fine differences in the structure of closely related enzymes influence their stability and kinetic and regulatory properties.
This work was supported by the Russian Foundation for Basic Research (project No. 02-04-49415) and NATO (projects No. LST.CLG 979818 and No. LST.CLG 977839).
REFERENCES
1.Vinals, C., Depiereux, E., and Feytmans, E. (1993)
Biochem. Biophys. Res. Commun., 192, 182-188.
2.Wichmann, R., Wandrey, C., Buckmann, A. F., and
Kula, M. R. (1981) Biotechnol. Bioeng., 23,
2789-2802.
3.Hummel, W., and Kula, M. R. (1989) Eur. J.
Biochem., 184, 1-13.
4.Zhao, H., and van der Donk, W. A. (2003) Curr.
Opin. Biotechnol., 14, 583-589.
5.Bommarius, A. S., Schwarm, M., Stingl, K.,
Kottenhahn, M., Huthmacher, K., and Drauz, K. (1995) Tetrahedron:
Asymmetry, 6, 2851-2888.
6.Popov, V. O., and Lamzin, V. S. (1994) Biochem.
J., 301, 625-643.
7.Popov, V. O., and Tishkov, V. I. (2003) in
Protein Structures: Kaleidoscope of Structural Properties and
Functions (Uversky, V. N., ed.) Research Signpost, Kerala, India,
pp. 441-473.
8.Rodionov, Yu. V. (1981) Usp. Mikrobiol., No.
16, 104-138.
9.Khangulov, S. V., Gladyshev, V. N., Dismukes, G.
C., and Stadtman, T. C. (1998) Biochemistry, 37,
3518-3528.
10.Hollenberg, C. P., and Janowicz, Z. (1989)
Eur. Patent EP1987000110417, Bulletin 89/03.
11.Tishkov, V. I., Galkin, A. G., and Egorov, A. M.
(1991) Dokl. Akad. Nauk SSSR, 317, 345-348.
12.Galkin, A., Kulakova, L., Tishkov, V., Esaki, N.,
and Soda, K. (1995) Appl. Microbiol. Biotechnol., 44,
479-483.
13.Mitsunaga, T., Tanaka, Y., Yoshida, T., and
Watanabe, K. (2000) Jap. Patent JP245471A2 of 12.09.2000.
14.Shinoda, T., Satoh, T., Mineki, S., Iida, M., and
Taguchi, H. (2002) Biosci. Biotechnol. Biochem., 66,
271-276.
15.Nanba, H., Takaoka, Y., and Hasegawa, J. (2003)
Biosci. Biotechnol. Biochem., 67, 720-728.
16.Nanba, H., Takaoka, Y., and Hasegawa, J. (2003)
Biosci. Biotechnol. Biochem., 67, 2145-2153.
17.Allen, S. J., and Holbrook, J. J. (1995)
Gene, 162, 99-104.
18.Sakai, Y., Murdanoto, A. P., Konishi, T.,
Iwamatsu, A., and Kato, N. (1997) J. Bacteriol., 179,
4480-4485.
19.Slusarczyk, H., Felber, S., Kula, M. R., and
Pohl, M. (2000) Eur. J. Biochem., 267, 1280-1289.
20.Labrou, N. E., and Rigden, D. J. (2001)
Biochem. J., 354, 455-463.
21.Goldberg, S. L., Cino, P. M., Patel, R. N.,
Nanduri, V. B., Johnston, R. M., and Johnston, R. (2004) USA Patent US
2004/0038237 Al of 26.02.2004.
22.Baba, T., Takeuchi, F., Kuroda, M., Yuzawa, H.,
Aoki, K.-I., Oguchi, A., Nagai, Y., Iwama, N., Asano, K., Naimi, T.,
Kuroda, H., Cui, L., Yamamoto, K., and Hiramatsu, K. (2002)
Lancet, 359, 1819-1827.
23.Parkhill, J., Sebaihia, M., Preston, A., Murphy,
L. D., Thomson, N., Harris, D. E., Holden, M. T. G., Churcher, C. M.,
Bentley, S. D., Mungall, K. L., Cerdeno-Tarraga, A. M., Temple, L.,
James, K., Harris, B., Quail, M. A., Achtman, M., Atkin, R., Baker, S.,
Basham, D., Bason, N., Cherevach, I., Chillingworth, T., Collins, M.,
Cronin, A., Davis, P., Doggett, J., Feltwell, T., Goble, A., Hamlin,
N., Hauser, H., Holroyd, S., Jagels, K., Leather, S., Moule, S.,
Norberczak, H., O'Neil, S., Ormond, D., Price, C., Rabbinowitsch, E.,
Rutter, S., Sanders, M., Saunders, D., Seeger, K., Sharp, S., Simmonds,
M., Skelton, J., Squares, R., Squares, S., Stevens, K., Unwin, L.,
Whitehead, S., Barrell, B. G., and Maskell, D. J. (2003) Nat.
Genet., 35, 32-40.
24.Omura, S., Ikeda, H., Ishikawa, J., Hanamoto, A.,
Takahashi, C., Shinose, M., Takahashi, Y., Horikawa, H., Nakazawa, H.,
Osonoe, T., Kikuchi, H., Shiba, T., Sakaki, Y., and Hattori, M. (2001)
Proc. Natl. Acad. Sci. USA, 98, 12215-12220.
25.De la Torre, J. R., Christianson, L. M., Beja,
O., Suzuki, M. T., Karl, D. M., Heidelberg, J., and De Long, E. F.
(2003) Proc. Natl. Acad. Sci. USA, 100, 12830-12835.
26.Beja, O., Aravind, L., Koonin, E. V., Suzuki, M.
T., Hadd, A., Nguyen, L. P., Jovanovich, S. B., Gates, C. M., Feldman,
R. A., Spudich, J. L., Spudich, E. N., and De Long, E. F. (2000)
Science, 289, 1902-1906.
27.Barnett, M. J., Fisher, R. F., Jones, T., Komp,
C., Abola, A. P., Barloy-Hubler, F., Bowser, L., Capela, D., Galibert,
F., Gouzy, J., Gurjal, M., Hong, A., Huizar, L., Hyman, R. W., Kahn,
D., Kahn, M. L., Kalman, S., Keating, D. H., Palm, C., Peck, M. C.,
Surzycki, R., Wells, D. H., Yeh, K. C., Davis, R. W., Federspiel, N.
A., and Long, S. R. (2001) Proc. Natl. Acad. Sci. USA,
98, 9883-9888.
28.Souciet, J. L., Aigle, M., Artiguenave, F.,
Blandin, G., Bolotin-Fukuhara, M., Bon, E., Brottier, P., Casaregola,
S., de-Montigny, J., Dujon, B., Durrens, P., Lepingle, A., Llorente,
B., Malpertuy, A., Neuveglise, C., Ozier-Kalogeropoulos, O., Potier,
S., Saurin, W., Tekaia, F., Toffano-Nioche, C., Wesolowski-Louvel, M.,
Wincker, P., and Weissenbach, J. (2000) FEBS Lett., 487,
3-12.
29.Ostergaard, S., Olsson, L., and Nielsen, J.
(2000) Microbiol. Rev., 64, 34-50.
30.Overkamp, K. M., Kotter, P., van Der, H. R.,
Schoondermark-Stolk, S., Luttik, M. A. H., van Dijken, J. P., and
Pronk, J. T. (2002) Yeast, 19, 509-520.
31.Serov, A. E. (2002) Structure-Function
Relationships in Recombinant Formate Dehydrogenases from Baker's Yeast
and Methylotrophic Bacteria: Ph.D. Thesis (Chemistry) [in Russian],
Lomonosov Moscow State University, Moscow.
32.Serov, A. E., Popova, A. S., and Tishkov, V. I.
(2002) Doklady RAN, 382, 401-405.
33.Saleeba, J. A., Cobbett, C. S., and Hynes, M. J.
(1992) Mol. Gen. Genet., 235, 349-358.
34.Chow, C. M., and Raj Bhandary, U. L. (1993) J.
Bacteriol., 175, 3703-3709.
35.Davison, D. C. (1951) Biochem. J.,
49, 520-526.
36.Kanamori, T., and Suzuki, Y. (1968)
Enzymologia, 35, 185-197.
37.Peacock, D., and Boulter, D. (1970) Biochem.
J., 120, 763-769.
38.Ohyama, T., and Yamazaki, I. (1974) J.
Biochem. (Tokyo), 75, 1257-1263.
39.Ohyama, T., and Yamazaki, I. (1975) J.
Biochem. (Tokyo), 77, 845-852.
40.Colas des Francs-Small, C., Ambard-Bretteville,
F., Darpas, A., Sallantin, M., Huet, J. C., Pernollet, J. C., and Remy,
R. (1992) Plant Physiol., 98, 273-278.
41.Colas des Francs-Small, C., Ambard-Bretteville,
F., Small, I. D., and Remy, R. (1993) Plant Physiol.,
102, 1171-1177.
42.Ambard-Bretteville, F., Small, I., Grandjean, O.,
and Colas des Francs-Small, C. (2003) Biochem. Biophys. Res.
Commun., 311, 966-971.
43.Jansch, L., Kruft, V., Schmitz, U. K., and Braun,
H. P. (1996) Plant J., 9, 357-368.
44.Hourton-Cabassa, C., Ambard-Bretteville, F.,
Moreau, F., de Virville, J. D., Remy, R., and Colas des Francs-Small,
C. (1998) Plant Physiol., 116, 627-635.
45.Suzuki, K., Itai, R., Suzuki, K., Nakanishi, H.,
Nishizawa, N. K., Yoshimura, E., and Mori, S. (1998) Plant
Physiol., 116, 725-732.
46.Thompson, P., Bowsher, C. G., and Tobin, A. K.
(1998) Plant Physiol., 118, 1089-1099.
47.Ambard-Bretteville, F., Sorin, C., Rebeille, F.,
Hourton-Cabassa, C., and Colas des Francs-Small, C. (2003) Plant
Mol. Biol., 52, 1153-1168.
48.Hanson, A. D., Gage, D. A., and Shachar-Hill, Y.
(2000) Trends Plant Sci., 5, 206-213.
49.Kreuzwieser, J., Schnitzler, J. P., and
Steinbrecher, R. (1999) Plant Biol., 1, 149-159.
50.Li, R., Ziola, B., and King, J. (2000) J.
Plant Physiol., 157, 161-167.
51.Olson, B. J., Skavdahl, M., Ramberg, H.,
Osterman, J. C., and Markwell, J. (2000) Plant Sci., 159,
205-212.
52.Shiraishi, T., Fukusaki, E., and Kobayashi, A.
(2000) J. Biosci. Bioeng., 89, 241-246.
53.Li, R., Bonham-Smith, P. C., and King, J. (2001)
Canad. J. Botany-Revue Canadienne de Botanique, 79,
796-804.
54.Herman, P. L., Ramberg, H., Baack, R. D.,
Markwell, J., and Osterman, J. C. (2002) Plant Sci., 163,
1137-1145.
55.Li, R., Moore, M., and King, J. (2003) Plant
Cell Physiol., 44, 233-241.
56.Nakai, K., and Kanehisa, M. (1992)
Genomics, 14, 897-911.
57.Emannuelsson, O., Nielsen, H., and von Heijne, G.
(1999) Protein Sci., 8, 978-984.
58.Baack, R. D., Markwell, J., Herman, P. L., and
Osterman, J. C. (2003) J. Plant Physiol., 160,
445-450.
59.Weerasinghe, P. A., Weerasekera, M. L. M. C., and
van Holm, L. H. J. (1999) Biologia Plantarum, 42,
541-547.
60.Bykova, N. V., Egsgaard, H., and Moller, I. M.
(2003) FEBS Lett., 540, 141-146.
61.Bykova, N. V., Stensballe, A., Egsgaard, H.,
Jensen, O. N., and Moller, I. M. (2003) J. Biol. Chem.,
278, 26021-26030.
62.Fedorchuk, V. V., Galkin, A. G., Yasnyi, I. E.,
Kulakova, L. B., Rojkova, A. M., Filippova, A. A., and Tishkov, V. I.
(2002) Biochemistry (Moscow), 67, 1145-1151.
63.Gul-Karaguler, N., Sessions, R. B., Clarke, A.
R., and Holbrook, J. (2001) Biotechnol. Lett., 23,
283-287.
64.Serov, A. E., Popova, A. S., Fedorchuk, V. V.,
and Tishkov, V. I. (2002) Biochem. J., 367, 841-847.
65.Lamzin, V. S., Dauter, Z., Popov, V. O.,
Harutyunyan, E. H., and Wilson, K. S. (1994) J. Mol. Biol.,
236, 759-785.
66.Shannon, C. E. (1997) MD Comput.,
14, 306-317 (reprinting of the paper The Mathematical Theory
of Communication (1963)).
67.Wilson, A. G. (1970) Entropy in Urban and
Regional Modelling, Pion Limited, London.
68.Sander, C., and Schneider, R. (1991)
Proteins, 9, 56-68.
69.Tishkov, V. I., Matorin, A. D., Rojkova, A. M.,
Fedorchuk, V. V., Savitsky, P. A., Dementieva, L. A., Lamzin, V. S.,
Mezentzev, A. V., and Popov, V. O. (1996) FEBS Lett.,
390, 104-108.
70.Iida, M., Kitamura-Kimura, K., Maeda, H., and
Mineki, S. (1992) Biosci. Biotechnol. Biochem., 56,
1966-1970.
71.Tishkov, V. I., Galkin, A. G., Gladyshev, V. N.,
Karzanov, V. V., and Egorov, A. M. (1992) Biotekhnologiya, No.
5, 52-59.
72.Mezentsev, A. V., Ustinnikova, T. B., Tikhonova,
T. V., and Popov, V. O. (1996) Prikl. Biokhim. Mikrobiol.,
32, 589-595.
73.Slusarczyk, H., Felber, S., Kula, M.-R., and
Pohl, M. (2003) Eur. Patent EP1295937-A2N of 26.03.2003.
74.Popov, V. O., Shumilin, I. A., Ustinnikova, T.
B., Lamzin, V. S., and Egorov, Ts. A. (1990) Bioorg. Khim.,
16, 324-335.
75.Tishkov, V. I. (2002) Bull. Moscow Univ., Ser.
2, Chemistry, 43, 380-387.
76.Blanchard, J. S., and Cleland, W. W. (1980)
Biochemistry, 19, 3543-3550.
77.Hermes, J. D., Morrical, S. W., O'Leary, M. H.,
and Cleland, W. W. (1984) Biochemistry, 23,
5479-5488.
78.Tishkov, V. I., Galkin, A. G., and Egorov, A. M.
(1989) Biokhimiya, 54, 299-305.
79.Popov, V. O., Egorov, A. M., and Berezin, I. V.
(1978) Dokl. Akad. Nauk SSSR, 240, 217-220.
80.Tishkov, V. I., Galkin, A. G., and Egorov, A. M.
(1989) Biochimie, 71, 551-557.
81.Bocanegra, J. A., Scrutton, N. S., and Perham, R.
N. (1993) Biochemistry, 32, 2737-2740.
82.Chen, Z., Lee, W. R., and Chang, S. H. (1991)
Eur. J. Biochem., 202, 263-267.
83.Chen, Z., Tsigelny, I., Lee, W. R., Baker, M. E.,
and Chang, S. H. (1994) FEBS Lett., 356, 81-85.
84.Chen, R., Greer, A., and Dean, A. M. (1996)
Proc. Natl. Acad. Sci. USA, 93, 12171-12176.
85.Tishkov, V. I., Galkin, A. G., and Egorov, A. M.
(1993) Proc. Int. Conf. “Enzyme Engineering XII”, Deu
Ville, France, p. 54.
86.Shutte, H., Flossdorf, J., Sahm, H., and Kula,
M.-R. (1976) Eur. J. Biochem., 62, 151-160.
87.Popov, V. O., Rodionov, Yu. V., Egorov, A. M.,
and Berezin, I. V. (1978) Bioorg. Khim., 4, 117-129.
88.La Reau, R. D., and Anderson, V. E. (1989) J.
Biol. Chem., 264, 15338-15343.
89.Weinhold, E. G., Glasfeld, A., Ellington, A. D.,
and Benner, S. A. (1991) Proc. Natl. Acad. Sci. USA, 88,
8420-8424.
90.Shaked, Z., and Whitesides, G. M. (1980) J.
Am. Chem. Soc., 102, 7104-7105.
91.Mesentsev, A. V., Lamzin, V. S., Tishkov, V. I.,
Ustinnikova, T. B., and Popov, V. O. (1997) Biochem. J.,
321, 475-480.
92.Neuhauser, W., Steininger, M., Haltrich, D.,
Kulbe, K. D., and Nidetzky, B. (1998) Biotechnol. Bioeng.,
60, 277-282.
93.Berezin, I. V., Tishkov, V. I., Karzanov,
V. V., Avilova, T. V., Egorov, A. M., Petkyavichene, R. I.,
Vaitkyavichus, R.-K., and Glemzha, A. A. (1989) USSR Patent, No.
1479513, Byul. Izobret. (1989) No. 18.
94.Weusterbotz, D., and Wandrey, C. (1995) Proc.
Biochem., 30, 563-571.
95.Labrou, N. E., Rigden, D. J., and Clonis, Y. D.
(2000) Eur. J. Biochem., 267, 6657-6664.
96.Seelbach, K., Riebel, B., Hummel, W., Kula, M.
R., Tishkov, V. I., Egorov, A. M., Wandrey, C., and Kragl, U. (1996)
Tetrahedron Lett., 37, 1377-1380.
97.Weuster-Botz, D., Paschold, H., Striegel, B.,
Gieren, H., Kula, M.-R., and Wandrey, C. (1994) Chem. Eng.
Technol., 17, 131-137.
98.Rodionov, Yu. V., Avilova, T. V., and Popov, V.
O. (1977) Biokhimiya, 42, 2020-2026.
99.Tishkov, V. I., Galkin, A. G., Marchenko, G. N.,
Egorova, O. A., Sheluho, D. V., Kulakova, L. B., Dementieva, L. A., and
Egorov, A. M. (1993) Biochem. Biophys. Res. Commun., 192,
976-981.
100.Odintseva, E. R., Popova, A. S., Rojkova, A.
M., and Tishkov, V. I. (2002) Bull. Moscow Univ., Ser. 2,
Chemistry, 43, 356-359.
101.Mitsuhashi, K., Yamamoto, H., and Kimoto, N.
(2001) Eur. Patent EP1211316-A of 27.11.2001.
102.Rojkova, A. M., Galkin, A. G., Kulakova, L. B.,
Serov, A. E., Savitsky, P. A., Fedorchuk, V. V., and Tishkov, V. I.
(1999) FEBS Lett., 445, 183-188.
103.Serov, A. E., and Tishkov, V. I. (2002)
Bull. Moscow Univ., Ser. 2, Chemistry, 43, 345-349.
104.Serov, A. E., Odintseva, E. R., Uporov, I. V.,
and Tishkov, V. I. (2005) Biochemistry (Moscow), in press.
105.Tishkov, V. I., Galkin, A. G., Fedorchuk, V.
V., Savitsky, P. A., Rojkova, A. M., Gieren, H., and Kula, M. R. (1999)
Biotechnol. Bioeng., 64, 187-193.
106.Reichert, U., Knieps, E., Slusarczyk, H., Kula,
M. R., and Thommes, J. (2001) J. Biochem. Biophys. Meth.,
49, 533-552.
107.Leonida, M. (2001) Curr. Med. Chem.,
8, 345-369.
108.Bornscheuer, U. T., and Pohl, M. (2001)
Curr. Opin. Chem. Biol., 5, 137-143.
109.Hummel, W. (1999) Trends Biotechnol.,
17, 487-492.
110.Kragl, U., Kruse, W., Hummel, W., and Wandrey,
C. (1996) Biotechnol. Bioeng., 52, 309-319.
111.Liese, A., and Filho, M. V. (1999) Curr.
Opin. Biotechnol., 10, 595-603.
112.Patel, R. N. (2000) Adv. Appl.
Microbiol., 47, 33-78.
113.Carrea, G., and Ottolina, G. (2002)
Biocatal. Biotransf., 18, 119-132.
114.Kragl, U., VasicRacki, D., and Wandrey, C.
(1996) Bioproc. Eng., 14, 291-297.
115.Seelbach, K., and Kragl, U. (1997) Enzyme
Microb. Technol., 20, 389-392.
116.Rissom, S., Schwarz-Linek, U., Vogel, M.,
Tishkov, V. I., and Kragl, U. (1997) Tetrahedron: Asymmetry,
8, 2523-2526.
117.Liese, A., Zelinski, T., Kula, M. R., Kierkels,
H., Karutz, M., Kragl, U., and Wandrey, C. (1998) J. Mol. Catal.
B-Enzymatic, 4, 91-99.
118.Orlich, B., Berger, H., Lade, M., and
Schomacker, R. (2000) Biotechnol. Bioeng., 70,
638-646.
119.Zelinski, T., Liese, A., Wandrey, C., and Kula,
M. R. (1999) Tetrahedron: Asymmetry, 10, 1681-1687.
120.Galkin, A., Kulakova, L., Yoshimura, T., Soda,
K., and Esaki, N. (1997) Appl. Environ. Microbiol., 63,
4651-4656.
121.Takeshita, K., Ishida, Y., Takada, G., and
Izumori, K. (2000) J. Biosci. Bioeng., 90, 545-548.
122.Galkin, A., Kulakova, L., Yamamoto, H.,
Tanizawa, K., Tanaka, Y., Esaki, N., and Soda, K. (1997) J. Ferment.
Bioeng., 83, 299-300.
123.Schwarz-Linek, U., Krodel, A., Ludwig, F. A.,
Schulze, A., Rissom, S., Kragl, U., Tishkov, V. I., and Vogel, M.
(2001) Synthesis-Stuttgart, 33, 947-951.