REVIEW: Proteomic Studies of Human and Other Vertebrate Muscle Proteins
S. S. Shishkin*, L. I. Kovalyov, and M. A. Kovalyova
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, Moscow 119071, Russia; fax: (7-095) 954-2733; E-mail: shishkin@inbi.ras.ru* To whom correspondence should be addressed.
Received July 9, 2004
This review summarizes results of some systemic studies of muscle proteins of humans and some other vertebrates. The studies, started after introduction of two-dimensional gel electrophoresis of O'Farrell, were significantly extended during development of proteomics, a special branch of functional genomics. Special attention is paid to analysis of characteristic features of strategy for practical realization of the systemic approach during three main stages of these studies: pre-genomic, genomic (with organizational registration of proteomics), and post-genomic characterized by active use of structural genomics data. Proteomic technologies play an important role in detection of changes in isoforms of various muscle proteins (myosins, troponins, etc.). These changes possibly reflecting tissue specificity of gene expression may underline functional state of muscle tissues under normal and pathological conditions, and such proteomic analysis is now used in various fields of medicine.
KEY WORDS: muscle protein isoforms, proteomics, myosin and troponin families
Muscle tissues and muscle proteins are important body constituents of higher vertebrates including humans. In various species, these tissues represent up to 40% of body weight and their protein metabolism may reach 25% of total protein metabolism [1-5]. Specialized proteins determine contractility, principally important feature of these tissues that are characterized by significant morphological and biochemical differences. These differences are underlined by a characteristic spectrum of expressing genes, reflecting the existence of specific protein isoforms [4-9]. Muscle tissues also secrete proteins involved in formation of neuromuscular synapses [10] and secrete into blood circulation some specific growth factors that influence the length-mass characteristics of vertebrates [11-13].
During ontogenesis, vertebrate muscle tissues (and their proteins) undergo complex changes [1, 5, 6, 13-15] terminating in the aging period [16-18]. A wide spectrum of various diseases (from myocardial infarction and autoimmune processes to inherited neuromuscular diseases) is characterized by specific changes in muscle tissues [19-25]. All these points make vertebrate tissues interesting and perspective objects for proteomic studies. The history of these studies can be subdivided into three stages: pre-genomic, genomic (followed by formation of genomics as an independent branch of functional genomics), and post-genomic, which is successfully developed now (Fig. 1).
Fig. 1. The main stages in formation of proteomics.
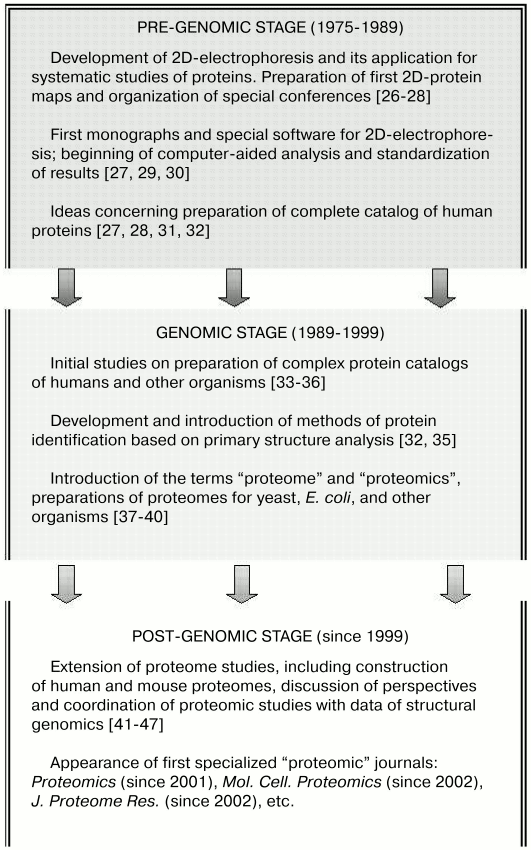
SYSTEMIC STUDIES OF MUSCLE PROTEINS IN THE PRE-GENOMIC PERIOD OF PROTEOMIC DEVELOPMENT
General features of vertebrate muscle proteins and some main families of muscle proteins. From the first half of the XX century, vertebrate muscle proteins have been intensively studied by biochemists. Usually scientists concentrated their attention on a particular representative of these proteins; they studied characteristic features of structure and function of muscle proteins (including interaction with adenine nucleotides and calcium ions), their metabolism, and some other properties [1, 2, 4, 6, 48]. In other words, scientists used a strategy of sequential study of muscle proteins. Russian biochemists made substantial contributions in contemporary knowledge of muscle proteins and an important role belonged to studies by B. F. Poglazov and his students [48-50].
In the 1980s vertebrate muscle proteins were classified into three main categories by morphological-functional criteria and extraction by various solvents: I) contractile proteins directly involved in the process of muscle contraction; II) cytoplasmic housekeeping proteins; III) proteins of various subcellular structures forming special molecular architectonics of muscle fibers [1, 2, 51-53]. An additional group (IV) would be formed by secretory muscle proteins exported by muscle tissues [10-13, 24].
From the 1980s, it was found that many muscle proteins exist as isoforms characterized by high homology of amino acid sequences and similar functions. Some isoforms are encoded by related but not identical genes, others result from alternative splicing, and a third group represents products of posttranslational modifications [5-7, 25, 54]. This promoted the concept of the origination of protein and gene families through duplication of ancestor genes followed by subsequent divergence of their initial copies [5, 6, 55, 56]. Some common properties of human muscle protein families are summarized in Tables 1-3.
Table 1. Properties of the most abundant
human muscle proteins and protein families of group I (contractile
proteins) (from www.ncbi.nlm.nih.gov and [5-7, 57])
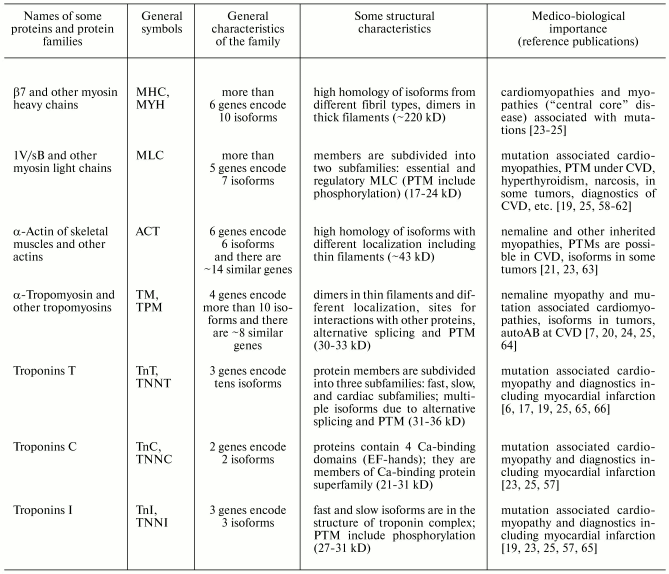
Note: autoAB) autoantibodies; CVD) cardiovascular diseases; PTM)
posttranslational modifications.
Table 2. Properties of human muscle proteins
and protein families of group II (cytoplasmic proteins) (from
www.ncbi.nlm.nih.gov and [5, 66-68])
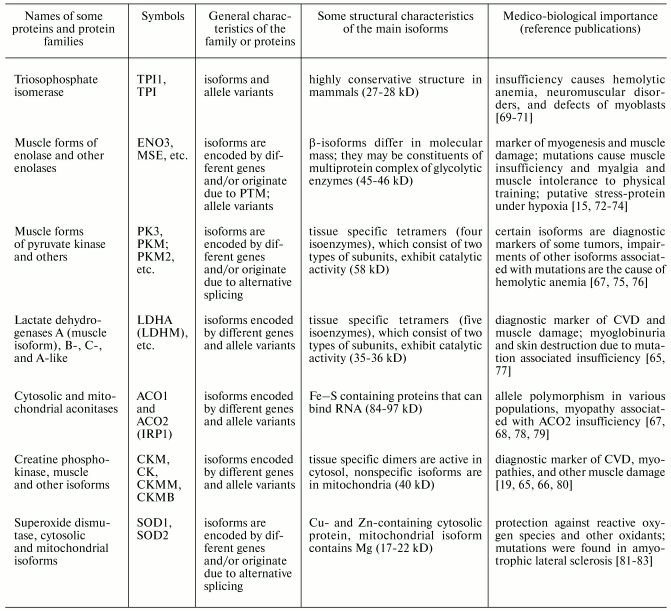
Table 3. Properties of some human muscle
proteins and protein families of group III (proteins of cytoskeleton
and various subcellular structures) (from www.ncbi.nlm.nih.gov and [5, 56, 84-86])
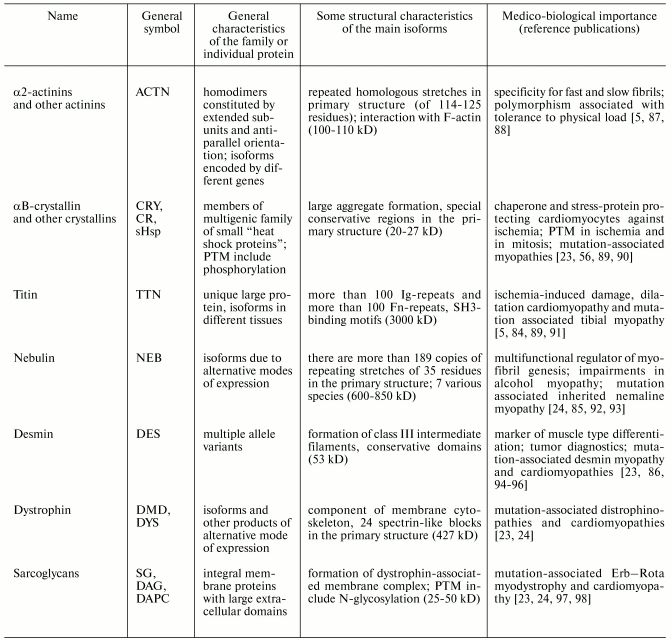
Note: Ig- or Fn-repeats, immunoglobulin-like and fibronectin-like
repeats; PTM, posttranslational modification.
Isoforms of muscle proteins of the first group were originally found in muscles; initially they were considered as tissue specific isoforms, but later they were also found in non-muscle cells [1, 5-7]. Nevertheless, some muscle proteins of the second group are tissue specific muscle isoforms [72, 99]. Reliable demonstration of tissue specificity of some muscle proteins is not only of theoretical importance; this has clear practical applications for development of different diagnostic tests [6, 19, 25, 65, 73].
Almost all these human muscle proteins (listed in Tables 1-3) have been sequenced and genes encoding these proteins identified, mapped on chromosomes, and their exon-intron structure recognized (see www.ncbi.nlm.nih.gov).
Since the 1980s, the vast majority of these muscle proteins have been objects of proteomic studies [4, 8, 9, 45, 47, 58, 83, 100, 101]. Moreover, Tables 1-3 show that almost all these (and many other) proteins attract much attention of clinicians because of involvement of these proteins in pathological processes in muscle tissues and their diagnostic potential [6, 19, 23, 25, 65, 66, 102].
Huge literature has accumulated on protein-protein interactions of muscle proteins [5, 7, 48, 49]; many proteins involved in specific binding to actins, myosins, and troponins and influencing muscle contractility have been found [6, 23, 25, 57]. Many muscle proteins of groups II and III are associated with various subcellular muscle structures and influence functioning of these structures [51, 72, 85, 86, 88, 103].
Thus, the accumulation of a large amount of experimental material on the diversity of vertebrate muscle proteins and complex changes in these proteins during myogenesis and differentiation followed by formation of various types of myofibrils and changes in protein patterns under pathological conditions [6, 7, 13, 25] required the research strategy to be changed from sequential analysis of separate proteins to a strategy of parallel studies. Development of two-dimensional (2D) electrophoresis by O'Farrell, immunoblotting, microsequencing, and other highly effective technologies of protein analysis provided the methodological basis for this transition [26, 27, 29, 31, 33].
Development of a systemic approach in protein studies and its use in analysis of proteins of vertebrate muscle tissues. In 1975, P. O'Farrell [26] described a new method of highly effective protein fractionation suitable (as he believed) for systemic studies of proteins encoded in the genome of E. coli and other organisms. Soon this method of 2D-electrophoresis and this new system of protein analysis became very popular among scientists [27, 29, 31, 33]. In the beginning of the 1980s, N. Anderson and L. Anderson took into consideration some features of 2D-electrophoresis and developed a special systemic approach to studies of proteins from various organisms, cells, subcellular structures, biological fluids [27], and also muscle tissues [30, 100]. Subsequently, these ideas were supported by studies of other researchers [27, 29, 33] including Russian scientists [31].
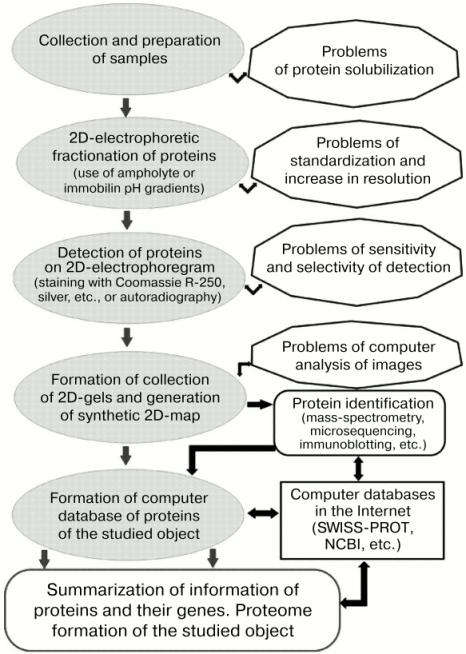
The main stages characterizing the strategy of the systemic approach in protein studies can be described schematically as follows.
1. All proteins (or the maximally possible number of proteins) contained in a particular sample are solubilized, and they represent the object for analysis. Maximally possible solubilization is achieved by complex treatments: homogenization in solution containing maximal urea concentration and a detergent (or detergents); ultrasonic treatment and (in some cases) use of hydrolases destroying non-protein macromolecules. (Addition of protease inhibitors to the lysing solutions prevents undesired proteolysis [26, 27, 29].)
2. Protein fractionation is based on two independent physicochemical properties of polypeptide chains reflecting the properties of the primary structures of the separating proteins. The first stage is isoelectrofocusing of proteins in thin polyacrylamide gel columns in the presence of urea and a nonionic detergent (Nonidet P-40 or Triton X-100). This is carried out in pH gradient created by ampholytes [34, 39, 46, 47, 104]. Separation of proteins with pI > 8.0 requires the use of a special modification of 2D-electrophoresis, which includes non-equilibrium electrophoresis in a pH gradient (NEPHGE). Many studies have also used carriers with immobilized pH gradient, which is created by special substances known as immobilins; they are bound covalently to a polyacrylamide matrix (IPG) [104, 105]. A polyacrylamide column with separated proteins is then used as a start zone from which fractionation in the second dimension begins on a polyacrylamide plate. This stage involves the widely used variant of electrophoretic protein separation in the presence of sodium dodecyl sulfate (SDS). For increase in protein resolution gel plates with concentration gradient of acrylamide are used at this stage.
3. Highly sensitive detection of protein fractions after separation. A wide range of methods from classic Coomassie Brilliant Blue R-250 to various variants of autoradiography and colored silver staining are available. Some methods can detect thousands of protein fractions on the electrophoregrams. Methods of protein detection developed in the pre-genomic stage recognized such varieties of polypeptide chains, which were comparable with evaluation of number of functioning genes in typical eukaryotic cells [29, 33, 104].
4. Systematic description of protein distribution on two-dimensional electrophoregrams using rectangular coordinates. The position of each protein fraction on a 2D-electrophoregram can be characterized by “vertical” and “horizontal” coordinates. Position on the former and the latter is a function of molecular mass and isoelectric point, respectively. Thus, elucidation of protein fraction coordinates on 2D-electrophoregrams represents the first step in systematic and objective description of protein products of gene expression in certain organs (tissues, cells). An important feature of this stage of the systematic strategy consists in formation of a “synthetic image” of protein distribution obtained on the basis of many adequate 2D-electrophoregrams (so-called “protein portrait” of the research object) or generation of standardized schemes of distribution known as “2D-maps” [27, 29, 33, 104]. Various approaches including special software for computer analysis of images have been used [29, 33-35, 106].
5. Identification of known proteins and detection of unknown proteins in the research object. At early stages of the development of the systematic approach, it was accepted that informational and implicational potentials of “2D-maps” for various applied studies significantly increased after identification of known proteins on these maps [27, 29, 30, 100]. Many various methods for protein identification after 2D-electrophoresis have been developed, and immunoblotting and microsequencing are the most important of them [29, 33, 104, 106]. Solution of identification problems was sometimes accompanied by detection of new proteins or their isoforms (see for example [107, 108]).
6. Generation of protein database of the research object. At this final stage of studies, a bulk of information on identified proteins could be systematized and summarized in the form of a specialized computer database (or databases), which can provide a basis for subsequent developments of a wide range of basic and applied problems of protein biochemistry [27, 29, 31, 33].
The properties of muscle proteins were very attractive for the use of the systematic approach (and later for proteomic studies). In the early 1980s the first publications of 2D-electrophoresis and systematic analysis for 2D-mapping of vertebrate muscle proteins appeared, and the authors were able to identify actin, myosin light chains, alpha- and beta-tropomyosins, some troponins, etc. on these maps [30, 100, 109]. In the second half of the 1980s, Russian scientists also started to use 2D-electrophoresis with the systematic approach for studies of muscle proteins [110, 111].
GENOMIC STAGE OF ORGANIZATION OF PROTEOMICS AND SUBSEQUENT
DEVELOPMENT OF A SYSTEMATIC APPROACH IN PROTEIN STUDIES USING MUSCLE
PROTEINS AS AN EXAMPLE
A qualitatively new stage in the development of systematic studies of proteins began in parallel with studies on decoding of genomes of various organisms and the beginning of the “Human Genome Project” [112]. Ideas concerning the use of systematic analysis of proteins for decoding the informational content of genomes were actively discussed, and this approach was included in the Russian genome project [112-114].
The term “proteome” defining the protein equivalent of the genome (“PROTEOME: entire PROTEin complement expressed by the genOME”), was originally proposed in lectures by members of the Organizing Committee of the conference “2D-Electrophoresis: from Protein Maps to Genomes” held in Siena (Italy) in 1994 [37]. Evidently, the ideology of actively developing genomic projects promoted the appearance of new terminology. Now the terms “proteome” and “proteomics” are commonly accepted and they are actively used in numerous publications and many international forums [35, 39, 42, 115].
However, the main framework of the strategy of proteome studies [34, 35, 38, 39, 106] was still within the ideology formulated in the early 1980s [27, 29]. The most important features of the genomic stage of proteomic studies included: a) qualitative changes in the problem of protein identification through the introduction of mass spectrometric methods; b) use of DNA (and genomic) technologies for protein identification and elucidation of various geno-phenotypic correlations; c) optimization of computer systems for image analyses; d) use of bioinformatic resources for solution of proteomic problems. Summarizing results of numerous studies [33, 35, 38, 39, 46], the strategy of proteomic studies of any object (e.g., muscle proteins) can be represented as a series of sequentially realized steps, where key positions are reserved for 2D-electrophoresis of proteins, protein identification by means of mass-spectrometry, and generation of a computer database (Fig. 2).
Genomic and post-genome stages were characterized by improvement and modification of the methodical arsenal for each stage of the proteomic strategy. Taking into consideration problems of solubilization of muscle proteins related to groups I and III, it is important to emphasize the significantly extended spectrum of approaches required for solution of this problem. For example, this included the use of thiourea, CHAPS, sulfobetain, Triton X-114, and other detergents and also various protease inhibitors [46, 52, 115, 116]. The proportion of studies using different variants of IPG during 2D-electrophoresis sharply increased [35, 38, 39]. New highly sensitive systems for direct detection of proteins (e.g., dyes SYPRO Orange, SYPRO Red) and improved methods for immunochemical identification (Enhanced Chemiluminescence) have been proposed [46, 115].Fig. 2. General scheme and some potential problems of the proteomic approach in protein studies (modification from [35, 38, 39, 132]).
The Danish group of Prof. J. E. Celis has demonstrated successful examples of the use of the proteomic strategy [33, 34, 46, 106]. They fractionated total protein preparations extracted from non-cultivated human keratinocyte biopsy using 2D-electrophoresis and detected protein fractions by radioautography and fluoroautography (using pre-labeling with [35S]methionine). Gel images obtained by scanning were subjected to special computer treatment using the PDQUEST II computer system, which provides smoothing of primary image, background subtraction, final smoothing of image, calculation of shape and distribution of protein spot density, etc. Use of calibrated segments containing known amount of radioactivity allowed the combining of multiple radioautographs that were obtained from the same gel by its exposure for different time intervals. This allowed pooling quantitative data about all protein fractions differing by several orders of magnitude into one synthetic computer image. For convenience of manipulations, a total image was traditionally separated into nine rectangular regions (this was accepted at the pre-genomic stage, see for example [110, 111]). Each protein fractions applied onto the map received its own number and this facilitated quantitative and qualitative analysis of information on each protein by means of protein databases. The final 2D-map reflecting the human keratinocyte proteome included 3625 proteins; 2313 of the 3625 proteins were detected using 2D-electrophoresis within the pH region 4.5-7.0; 954 proteins were found in the pH region of 7.0-11.5 (modification of NEPHGE) [46]. Among them, 1285 proteins were identified, and the resulting information formed the corresponding database (see [46] and http://biobase.dk/cgi-bin/celis). This database contained a number of non-muscle isoforms of muscle proteins and many housekeeper proteins present in muscle tissues.
Several other laboratories began to use proteomic technologies for studies of muscle proteins during the genomic period. Since 1992, M. J. Dunn's group (Harefield, England) regularly reports on progress in the creation of a database of various muscle proteins [4, 117]. In the first stage of this project more than a thousand various proteins within the pH range 4-7 were investigated; 38 of them were identified by microsequencing and immunoblotting. Summarized results placed on the Internet (HSC-2DPAGE) represented extended information on cardiac proteins of human, rat, and dog. In 1996, 220 proteins of this database were identified by Western-blotting, N-terminal and internal sequencing, and by mass-spectrometry [35].
Significant progress in the development of a human cardiac protein database was achieved by the German group of Jungblut et al. [40, 118]. These authors employed the classic method of 2D-electrophoresis with formation of ampholyte pH gradient for isoelectrofocusing. They were able to detect 3239 proteins, and 20 muscle proteins were identified by N-terminal and internal microsequencing, immunoblotting, and analysis of amino acid composition. In 1999, this database contained 120 identified proteins and the total number of proteins in this database increased to 3300 [40].
The first Russian 2D-map of human left ventricle proteins was published in 1990 [119], its second version appeared in 1995, and results obtained were summarized as a catalogue of human striated muscle proteins [8]. This catalog includes 312 muscle proteins; 33 protein fractions have been identified (including 14 fractions which were identified using microsequencing) [8, 120]. Each protein fraction is characterized by molecular mass, isoelectric point, and subcellular localization (cytosol, mitochondria). During staining with “Stains all” dye (Sigma, USA), some muscle proteins were recognized as polymorph variants demonstrating variability during embryogenesis.
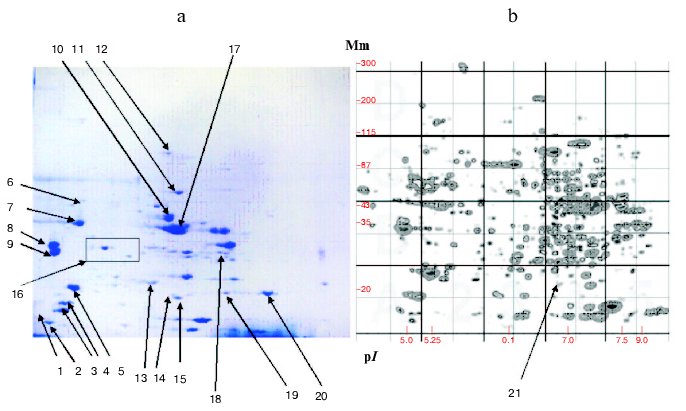
The development of the Russian catalog was accompanied by creation of an original system of uniform seven-digit numeration of protein fractions on the 2D-map. The first four numbers represent decimal logarithm of protein molecular mass, and the three last numbers are the mean of isoelectric point. Subsequently this approach and this system were used in later versions of this catalog, and each version had certain characteristic features and supplemented older versions with new information [9, 121]. For example, in the third version the total number of proteins included on the 2D-map was increased to 347 due to better resolution of proteins in acidic and alkaline zones of the pH gradient. This was achieved using a system with immobilin pH gradient during protein fractionation in the first dimension. On the final 2D-map, all fractions belonging to almost all muscle proteins listed in Tables 1-3 were identified (Fig. 3, see color insert).
Increasing attention to medical aspects of studies was typical for the genomic period. This was important for elucidation of new markers of tumor processes [35, 40, 122], neurotransmitters and neuropeptides [123], elucidation of molecular nature of cardiomyopathies [25, 40], and identification of some proteins important for diagnostics of some infections [39, 40]. This suggests the great promise of proteomics even on the background of impressive results obtained during genomic projects [124, 125].Fig. 3. 2D-electrophoresis of human muscle proteins. a) Typical electrophoregram of skeletal muscle proteins. b) 2D-map generated by the Melani software. Arrows indicate identified proteins: 1) TnC; 2) MLC type 3, fast; 3) type 3; 4) MLC type 2; 5) MLC type 1; 6) DES; 7) ACT; 8) betaTM; 9) alphaTM; 10) ENO3; 11) PK3; 12) ACO2; 13) TPI1; 14) SOD1; 15) CRYAB; 16) a region with four TnT1 fractions; 17) CKMM; 18) LDGA; 19) TnI fast type; 20) TnI isoform; 21) CRYAB (4313685) isoform. Mm, molecular mass.
BEGINNING OF THE POST-GENOMIC STAGE: SOME ACHIEVEMENTS OF
STRUCTURAL GENOMICS AND PROTEOMIC STUDIES OF VERTEBRATE MUSCLE
PROTEINS
The turn of the century was characterized by intensive studies of genomes of various organisms (especially of the human genome). This resulted in the accumulation of principally important information for many branches of general biology and medicine [42, 43, 124, 125]. The identification of special research objects (genomes), specificity of solving problems, and the development and use of a whole complex of special technologies emphasize the fact that a new branch of science has been formed--genomics, or more precisely structural genomics [36, 39]. The creation of GenBank, a giant and constantly developing computer database on decoded nucleotide sequences [36, 39, 126] and availability of some other important computer resources (e.g., www.ncbi.nlm.nih.gov; http://cn.expasy.org, etc.) were the most important events determining development of this field. Structural genomics has summarized data on genomes of rather complex organisms, both prokaryotes and eukaryotes [36, 124-126] (including two mammalian species, human and mouse) [124-127]. Structural genomics also operates with materials on more than a thousand relatively short genomes related to cellular organelles, natural plasmids, viruses, etc. (cited by www.ncbi.nlm.nih.gov). Undoubtedly, results of complete sequencing of the human genome are the most important information for medicine (and human biology in general) [124, 125, 127]. One of the key reports in this series contained results of an international consortium that included about 20 research groups from the USA, United Kingdom, Japan, France, Germany, and China [124]. The other report was published by C. J. Venter et al; his group working for Celera Genomics developed their own strategy of genomic analysis [125].
Analyzing 26,383 genes in the human genome, scientists from Celera Genomics characterized putative functions for more than 60% of these genes [125]. Many of the characterized genes should function in muscle tissues; 376 genes are responsible for muscle contractility (motor); 296 genes encode structural muscle proteins, 876 genes encode structural proteins of cytoskeleton, etc. Since the existence of multiple products of gene expression is firmly recognized for well-studied genes encoding muscle proteins, the information available in the human genome indicates the possibility for formation of many thousands of proteins in human muscle tissues.
In general, the achievements of structural genomics provided a good background for the transition to so-called functional genomics [43, 128]. The research task of functional genomics is often defined as study of gene expression and its results; however, the main emphases are made on various aspects of such study [39, 40, 122, 123, 129]. Nevertheless, gene functions represent the main object of functional genomics. Consequently, various studies related to genome operation which extend our knowledge on theoretical considerations on phenotype formation under normal and pathological conditions can be referred to the sphere of functional genomics.
Considering the existence of several levels of formation of functional characteristics (direct gene control of amino acid sequences, alternative promoters and alternative splicing responsible for multiple products of gene expression, posttranslational modifications, protein-protein interactions, etc.), we obtain in the first approximation the sphere of research interests of functional genomics. These research interests may be subdivided into four actively developing directions [39, 40, 42, 128, 129]: study of coordinated gene expression followed by formation of primary transcripts, their splicing, and formation of mature mRNAs (transcriptomics); study of protein products of gene expression including posttranslational modification of the protein products (proteomics); study of genetic mechanisms and genetic control of formation of subcellular structures, cell differentiation, and histogenesis (cytomics); study of genetic mechanisms and genetic control of various phenotype formation under normal and pathological conditions.
Considering the main research tasks of functional genomics, the principal goals of proteomics of the post-genomic period consist of recognition and identification (with known proteins) of the maximally possible number of protein fractions obtained from a particular object. On “protein portraits”, these fractions are usually characterized by certain coordinates. Identification of certain fractions with known proteins (including various isoforms) can be used to compare genomic, transcriptomic, and proteomic information; this significantly increases the value of protein databases [38, 43, 106]. Thus, the major role of proteomics can be defined as integral analysis of the functional state of the genome at the level of protein products of gene expression [46, 47, 128].
Considering the major research tasks, the proteomics and systematic approach in protein studies naturally fit to the central problem of molecular biology of genomic and post-genomic stages [4, 40, 45, 128, 130]. Analysis of publications that appeared in the literature from 1995 to 2002 by a common set of key words--“human”, “cardiac”, “protein”--revealed about 150-250 papers per year and 10-20 papers of them contained information on amino acid sequence of myocardial muscle proteins [45]. Muscle proteins attract much interest from other tissues [4, 130-132].
A report on muscle proteins published by a British group in 2001 [4] and a report on myocardial proteins published by a German group in 2002 [133] represent typical examples of results of studies carried out in the post-genomic stage. Both groups used laboratory animals as experimental models that the authors suggest may model certain pathological conditions in humans and approaches for their treatments. In both studies, proteins were fractionated by 2D-electrophoresis and identified by mass-spectrometry; the resulting images were treated using the Melani computer program. Both groups detected about 500-600 protein fractions, and several tens of muscle proteins belonging to three main groups were identified. Our database also contains about 500 muscle proteins, and more than 50 of these proteins have been identified [9].
Results reported by Gajendran et al. [10] demonstrated another aspect of proteomic analysis of muscle proteins. These authors characterized 1200 proteins form cultivated muscle cells So18; 140 of the 1200 proteins were identified by mass-spectrometry. Comparison of proteomic maps and results of co-electrophoresis of protein preparations of cultivated cells, culture medium, and proteins from muscle extracts revealed some proteins secreted by muscle cells.
Proteomic studies of muscle proteins typical for the post-genomic period have some characteristic features. Beside wide use of mass-spectrometry for protein identification, use of genomic information and transcriptome analysis, these include special attention to certain groups of muscles and even to some subcellular components of muscles (sub-proteomics) [45, 52, 53, 58, 130]. A study by Li et al. [130] is a typical example of such investigations. These authors analyzed proteins of human m. thyreoarytenoideus. These muscles are of particular interest because of their unique functions providing speech facility and protection of the respiratory tracts from aspiration. Six autopsies were investigated. Their proteins were fractionated by 2D-electrophoresis using IPG with broad (variant I) and narrow (variant II) pH values (pH ranges of 3-10 and 5-8, respectively). Using silver staining the variant revealed 547 protein fractions, and 75% of them were located within the pH zone of 5-8. Although variant II detected 1087 proteins, 26 fractions from pH region of 3-5 and 110 fractions of pH region of 8-10 were absent. The authors tried to identify 150 protein fractions, which were cut from 2D-electrophoregrams, but only 75 proteins could be identified by mass-spectrometry. The identified proteins were referred to the following functional classes: a) membrane proteins, membrane receptors and proteins involved in signaling (8.5%); b) proteins of cytoskeleton and myofibrils (14.6%); c) proteins associated with mitochondria and energy production of cells (28%); d) proteins involved into stress response (8.5%); e) proteins bound to DNA or RNA (10.9%); f) non-classified proteins (29.2%). These results confirmed the notion that many muscle proteins exist as a set of isoforms and isoform composition (e.g., composition of myosin heavy and light chains and also composition of troponin complex) of muscle proteins determines functional capacities of muscles including contractility. Li et al. [130] generated their own database of human muscle proteins. They believe that subsequent development of such studies will help to create an effective resource for molecular analysis of normal and pathological process in striated muscles.
In the post-genomic period, possibilities and approaches to the solution of proteomic tasks without the use of 2D-electrophoresis are actively discussed in the literature [44, 115, 134]. For example, it was suggested that reversed-phase HPLC [135], capillary isoelectrofocusing and capillary electrophoresis [136], design of special protein chips [134, 137], and also combinations of various techniques for protein fractionation including modern variants of affinity and ion-exchange chromatography [135, 137] might provide an alternative to 2D-electrophoresis. A practical example of such approaches (called gel-free approaches) was demonstrated by Ruse et al. [45]. They studied human myocardial proteome. Myocardial samples obtained from five patients without cardiac pathology were used for maximally possible solubilization of proteins. The solubilized preparations were treated by trypsin and proteolytic peptides were analyzed by chromatographic methods in combination with mass-spectrometry. Such proteomic analysis consisted of 11 sequential stages; 267 proteins including 47 phosphoproteins were identified. For protein identification, these authors used the broad capacities of bioinformatics and special databases (http://www.harefield.nthames.nhs.uk/nhli/protein/index.html and http://www.mdc-berlin.de/~emu/heart/). The identified proteins were determined as mitochondrial (98), nuclear (75), and cytoplasmic (65) proteins and 22 proteins belonged to various structures of cytoskeleton.
Although several attempts have been made to use gel-free approaches in proteomics, many researchers believe that 2D-electrophoresis remains the main tool for protein (including muscle protein) fractionation [46, 47, 58, 102, 115].
Characterizing proteomics of the post-genomic period as the branch of functional genomics, we should emphasize that it still actively uses the systematic approach for studies of protein products of gene expression for elucidation of principles of genome functioning. Now the main “proteomic pathway” to selected targets is related to creation of computer databases based on various protein catalogs corresponding to proteomes of the studied objects including whole vertebrate organisms (and the human body as well) [39, 41, 45-47, 102, 131]. It should be noted that this viewpoint completely corresponds to the notions that appeared in the pre-genomic period [27, 29, 31, 33]. The construction of proteomes (as well as genomes) should become (and they have actually become) new important species characteristics and data obtained begin to form a new theoretical basis for the development of many general biological and medical problems [39, 43, 115, 127, 129].
It should also be noted that many researchers are concentrating their attention again on recognition and studies of separate proteins (including those which represent a combination of several isoforms) by proteomic and other technologies [7, 75, 102]. (This often reflects changes in functional properties of muscle tissues [23, 25, 138].) Other priorities include studies of muscle proteins with certain tissues specificity and/or studies of proteins that are characterized by clear variability during ontogenesis and also under different treatments [10, 18, 132, 133, 139].
PROTEOMIC STUDIES OF TISSUE SPECIFICITY AND POLYMORPHISM OF SOME
MUSCLE PROTEINS
Results of numerous studies suggest that contractile function of myofibrils under normal and pathological conditions depends on the presence of certain forms of muscle proteins and also on their posttranslational modifications [5, 60, 61, 139]. (We cannot analyze this bulk of information because the accumulated material significantly exceeds all possible limits reserved for this review.) So all these problems studied by means of proteomic technologies will be considered using certain muscle proteins as examples (myosin light chains, troponins T, enolases, and crystallins). These proteins were selected because of the availability of rather comprehensive information in our group [8, 9, 108, 111, 114, 119, 140].
Since polymorphism of proteins related to group I muscle proteins and directly involved in muscle contraction is of special interest, the proteomic technologies have been actively used for investigation of polymorphism of almost all proteins of this group. Myosin heavy chains (MHC) are the only exception due to the remaining problems of fractionation of poorly soluble proteins with molecular mass of 100 kD and above by the method of 2D-electrophoresis. Although there are some reports on detection of MHC fractions during 2D-electrophoresis [30], all the information on tissue specificity of MHC isoforms has been obtained in studies using technologies other than proteomic ones [5, 141].
In contrast to MHC, myosin light chains (MLC) can be successfully analyzed by the proteomic technologies. These proteins are reliably detected and identified within the region of 2D-electrophoregram limited by the coordinates from 28 to 15 kD and pI from 4.8 to 5.5 [8, 9, 108]. Besides major MLC proteins, this region also contains more than twenty other protein fractions that still require better identification (Fig. 3). Special analysis of one of these proteins [108] specific for atrium revealed that its amino acid sequence contains at least two sites that share high homology with the corresponding sequences of other MLC. For some MLC polymorphic variants are known; for example, 2D-electrophoretic analysis of human autopsies revealed the variant known as MLC-1V/sB, which differed from the normal pI value; this was due to a single amino acid substitution 144(N-->H) [140]. Some MLCs are subjected to phosphorylation and their phosphorylated forms are reliably registered by the proteomic technologies; these forms have been listed in the proteome databases [4, 117], especially in HSC-2DPAGE and other English computer resources (http://www.harefield.nthames.nhs.uk/nhli/protein/index.html).
There is increasing evidence for medical importance of various proteins of the MLC family. Some hypertrophic cardiomyopathies are associated with mutations of MLC-1V/sB and MLC-2s/v genes encoding essential and regulatory myosin light chains, respectively [25], and MLC-2 phosphorylation/dephosphorylation processes are significantly altered in cardiovascular diseases [58], hyperthyroidism [60], during narcosis [61], and in some tumors [62]. This emphasizes the importance of further studies of MLC. Tests on MLC are already used in clinical practice because these proteins are specific markers of cardiac damage [19]. Thus, certain experimental evidence exists that skeletal muscles and myocardial ventricles and atria contain specific spectra of MLC proteins that are changed during ontogenesis and in pathology [5, 8, 58, 111].
The family of troponin T (TnT) proteins attracts much attention due to the problem of tissues specificity of proteins; this family is further subdivided into fast skeletal, slow skeletal, and cardiac isoforms of troponins [6, 57]. Separate members of this family are studied using proteomic technologies for solution of various medico-biological problems [6, 25, 57, 132].
Human (and other vertebrate) muscle tissues of various types contain specific sets of TnT isoforms; fast skeletal TnTs are the most abundant isoforms. Their formation involves alternative mechanisms of expression of one gene, and the calculated number of such isoforms can reach 128 [6, 54, 57]. One gene encoding slow skeletal TnT is responsible for formation of four isoforms of particular species due to alternative splicing [142]. Interestingly, 2D-electrophoresis of human skeletal muscles with subsequent mass-spectrometry also revealed four fractions of slow skeletal TnT (Fig. 3).
TnT together with troponins C and I form a troponin complex in thin filaments; this complex interacts with tropomyosin molecules and plays an important role in the regulation of muscle contraction. Consequently, the presence of certain forms of TnT should emphasize some specificity in the functioning of certain muscles [5, 6, 57]. Moreover, convincing evidence exists that mutations in genes encoding TnT cause certain types of muscle pathology [6, 23, 25]. Comparative 2D-electrophoretic study of skeletal muscle samples (n = 8) obtained from patients with amyotrophic lateral sclerosis and corresponding controls revealed that a group of closely positioned proteins clearly and specifically detected in muscles under normal conditions is absent in patients with this disease [143]. Mass-spectrometric analysis identified the missed proteins as protein products of expression of slow skeletal muscle TnT gene (TNNT1) [144]. Mass-spectrometry analysis of the other protein fraction, which might be related to the considered group by its electrophoretic mobility and which has been detected in muscle samples of healthy individuals and patients with amyotrophic lateral sclerosis, identified this proteins as the protein product of slow skeletal muscle TnT gene; this product is apparently truncated at the C-terminus because it lacked the C-terminal part. Although appearance of amyotrophic lateral sclerosis is often associated with mutations in superoxide dismutase gene (SOD1) [82], disappearance of isoforms of TnT suggests their involvement in pathogenesis of this disease.
Proteomic technologies are successfully used for studies of numerous proteins of group II. For example, interesting results have been obtained during studies of an isoform of enolase specific for muscle tissues (ENO3) [4, 72, 103]. This glycolytic enzyme attracts much attention as a putative marker of early myogenesis and muscle impairments under increased physical load (e.g., during very intensive sport trainings) [15, 73].
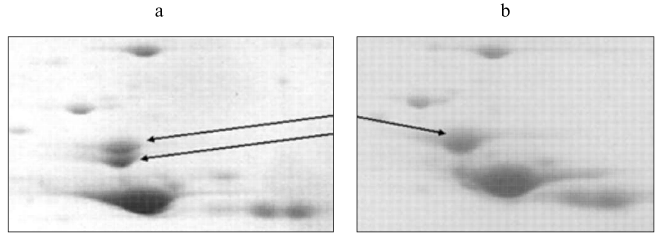
Using 2D-electrophoresis, Merkulova et al. [72] demonstrated that human fast muscles (type II) contain two isoforms of beta-enolase that differ in molecular mass: the H isoform is of 46 kD, whereas the L isoform is of 45 kD. In different people, beta-enolase forms three isoform patterns: the enzyme may be present as H isoform, L isoform, or mixture of equal quantities of H and L isoforms. This suggests the genetic nature of human beta-enolase polymorphism.
Recently, we also detected the position of beta-enolase (together with some other proteins) on the 2D-map of human muscle proteins (Fig. 3) using mass-spectrometry. This study was carried out in the Proteomic Center at the Institute of Biomedical Chemistry, Russian Academy of Medical Sciences [144]. Subsequent investigation of representative set of biopsies (n = 20) and autopsies (n = 34) of skeletal muscles also revealed the existence of two isoforms of beta-enolase (Fig. 4). Their frequencies corresponded to expected values using the Hardy-Weinberg equation. This indirectly supports the suggestion of di-allele state of ENO3 gene. It is possible that polymorphism of beta-enolase may influence energy supply to muscle tissues. This is indirectly supported by the fact that some missense mutations in ENO3 gene cause enzymatic insufficiency, manifesting by specific myopathy characterized by myalgia and inability for muscle training [74].
In some proteomic studies alphaB-crystallin, a protein of group III, has been intensively investigated [8, 34, 45, 130, 133]. In 1995, our group determined its position on the 2D-map of muscle proteins using microsequencing (Fig. 3) [8]. Later mass-spectrometry revealed that fraction 4313685 is also a crystallin. This fraction is presumably a phosphorylated form of this protein [56, 90]. Similar results were obtained by Z-B. Li et al. [130]. Analyzing proteins of m. thyreoarytenoideus, these authors identified two fractions as alphaB-crystallins and one fraction as alpha-crystallin (polypeptide 2). Study of changes in protein composition of human heart transplant (n = 3) during its rejection revealed sharp reduction of alphaB-crystallin fraction and marked increase of two neighboring fractions, 4312685 (kD/pI 20.6/6.85) and 4262672 (kD/pI 18.3/6.72) [121]. This observation possibly reflects changes in the set of small heat shock proteins or degradation of alphaB-crystallins; it is also possible that these changes are associated with the reaction of heart transplant rejection.Fig. 4. Isoforms of human muscle beta-enolase: a) heterozygote (H and L isoforms are present); b) homozygote (H form is present).
Thus, proteomic studies of muscle proteins represent one of the intensively developing regions of modern proteomics, which has good perspectives and which may substantially contribute to new achievements in muscle protein research.
This work was supported within agreement 01-03/04 with Institute of Medico-Biological Problems, Russian Academy of Sciences, and also by the Russian Foundation for Basic Research (grant 03-04-49376).
REFERENCES
1.Zengbush, P. (1982) Molecular and Cellular
Biochemistry [Russian translation], Vol. 2, Mir, Moscow, pp.
148-199.
2.Berezov, T. T., and Korovkin, B. F. (1998)
Biological Chemistry [in Russian], Meditsina, Moscow, pp.
645-660.
3.Volkov, N. I., Nesen, E. N., Osipenko, A. A., and
Korsun, S. N. (2000) Biochemistry of Muscular Activity [in
Russian], Olimpiiskaya Literatura, Kiev, pp. 286-485.
4.Yan, J. X., Harry, R. A., Wait, R., Welson, S. Y.,
Emery, P. W., Preedy, V. R., and Dunn, M. J. (2001) Proteomics,
1, 424-434.
5.Schiaffino, S., and Reggiani, C. (1996) Physiol.
Rev., 76, 371-423.
6.Perry, S. V. (1998) J. Muscle Res. Cell.
Motil., 19, 575-602.
7.Perry, S. V. (2001) J. Muscle Res. Cell.
Motil., 22, 5-49.
8.Kovalyov, L. I., Shishkin, S. S., Efimochkin, A.
S., Kovalyova, M. A., Ershova, E. S., Egorov, T. A., and Musalyamov, A.
K. (1995) Electrophoresis, 16, 1160-1169.
9.Shishkin, S. S., Kovalyov, L. I., and Gromov, P. S.
(2000) in Diversity of Modern Human Genetics (Shishkin, S. S.,
ed.) [in Russian], Gilem, Moscow-Ufa, pp. 17-50.
10.Gajendran, N., Frey, J. R., Lefkovits, I., Kuhn,
L., Fountoulakis, M., Krapfenbauer, K., and Brenner, H. R. (2002)
Proteomics, 2, 1601-1615.
11.McPherron, A. C., Lawler, A. M., and Lee, S.-J.
(1997) Nature, 387, 83-90.
12.Yang, S. Y., and Goldspink, G. (2002) FEBS
Lett., 522, 156-160.
13.Shishkin, S. S. (2004) Usp. Biol. Khim.,
44, 209-262.
14.Kovalyov, L. I. (1995) Systemic approach in
study of tissue-specific gene expression in human striated muscle
tissue: Abstract of doctoral dissertation [in Russian], Russian
Academy of Medical Sciences, Moscow.
15.Fougerousse, F., Edom-Vovard, F., Merkulova, T.,
Ott, M. O., Durand, M., Butler-Browne, G., and Keller, A. (2001) J.
Muscle Res. Cell. Motil., 22, 535-544.
16.Yarasheski, K. E., Bhasin, S., Sinha-Hikim, I.,
Pak-Loduca, J., and Gonzalez-Cadavid, N. F. (2002) J. Nutr. Health
Aging, 6, 343-348.
17.Roth, S. M., Martel, G. F., Ivey, F. M., Lemmer,
J. T., Tracy, B. L., Hurlbut, D. E., Metter, E. J., Hurley, B. F., and
Rogers, M. A. (1999) J. Appl. Physiol., 86,
1833-1840.
18.Cobon, G. S., Verrills, N., Papakostopoulos, P.,
Eastwood, H., and Linnane, A. W. (2002) Biogerontology,
3, 133-136.
19.Goto, T., Takase, H., Toriyama, T., Sugiura, T.,
Sato, K., Ueda, R., and Dohi, Y. (2003) Heart, 89,
1303-1307.
20.Fujita, A., Kuroda, S., Tada, H., Hidakka, Y.,
Kimura, M., Takeoka, K., Nagata, S., Sato, H., and Amino, N. (2000)
Clin. Chem. Acta, 299, 179-192.
21.Arrell, D. K., Neverova, I., and van Eyk, J. E.
(2001) Circ. Res., 88, 763-773.
22.Tontsch, D., Pankuweit, S., and Maisch, B. (2000)
Clin. Exp. Immunol., 121, 270-274.
23.Gorbunova, V. N., Saveleva-Vasileva, E. A., and
Krasilnikov, V. V. (2000) Molecular Neurology. Part 1. Diseases of
Neuromuscular System [in Russian], Intermedika, St. Petersburg, pp.
19-190.
24.Vainzof, M., and Zatz, M. (2003) Braz. J. Med.
Biol. Res., 36, 543-555.
25.Bonne, G., Carrier, L., Richard, P., Hainque, B.,
and Schwartz, K. (1998) Circ. Res., 83, 580-593.
26.O'Farrell, P. H. (1975) J. Biol. Chem.,
250, 4007-4021.
27.Anderson, N. G., and Anderson, L. (1982) Clin.
Chem., 28, 739-748.
28.Clark, B. F. C. (1981) Nature, 292,
491-492.
29. Celis, J. E., and Bravo, R. (eds.) (1984) Two-Dimensional
Gel Electrophoresis of Proteins: Methods and Applications, Academic
Press, New York.
30.Giometti, C. S., Anderson, N. G., and Anderson,
N. L. (1979) Clin. Chem., 25, 1877-1884.
31.Shishkin, S. S. (1985) Vestnik AMN SSSR,
No. 1, 78-84.
32.Klose, J. (1989) Electrophoresis,
10, 140-152.
33.Celis, J. E., Madsen, P., Rasmussen, H. H.,
Leffers, H., Honore, B., Gesser, B., Dejgaard, K., Olsen, E.,
Magnusson, N., Kiil, J., et al. (1991) Electrophoresis,
12, 802-872.
34.Celis, J. E., Rasmussen, H. H., Gromov, P. S.,
Olsen, E., Madsen, P., Leffers, H., Honore, B., Dejgaard, K., Vorum,
H., Kirstensen, D. B., et al. (1995) Electrophoresis, 16,
2177-2240.
35.Abst. II Siena 2D-Electrophoresis Meet.
“From Genome to Proteome”, Siena, Italy, September 16-18,
1996.
36.Baxevanis, A., and Ouellette, B. F. F. (eds.)
(1998) Bioinformatics: A Practical Guide to Analysis of Genes and
Proteins, John Wiley and Sons Inc., NY.
37.Dunn, M. J., Hochstrasser, D., Pallini, V., and
Bini, L. (1995) Electrophoresis, 16, Editoral.
38.Wilkins, M. R., Williams, K. L., Appel, R. D.,
and Hochstrasser, D. F. (1997) Proteomic Research: New Frontiers in
Functional Genomics (Principle and Practice), Springer Verlag.
39. Abst. Human Genome Meet. '99, Brisbane, Australia, March
27-30, 1999.
40.Jungblut, P. R., Zimny-Arndt, U.,
Zeindl-Eberhart, E., Stulik, J., Koupilova, K., Pleissner, K. P., Otto,
A., Muller, E. C., Sokolowska-Kohler, W., Grabher, G., and Stoffler, G.
(1999) Electrophoresis, 20, 2100-2110.
41.Anderson, N. G., Matheson, A., and Anderson, L.
(2001) Proteomics, 1, 3-12.
42.Archakov, A. I. (2000) Vopr. Med. Khim.,
46, 335-343.
43.Peltonen, L., and McKusick, V. A. (2001)
Science, 291, 1224-1229.
44.Apweiler, R., Biswas, M., Fleischmann, W.,
Kanapin, A., Karavidopoulou, Y., Kersey, P., Kriventseva, E. V.,
Mittard, V., Mulder, N., Phan, I., and Zdobnov, E. (2001) Nucleic
Acids Res., 29, 44-48.
45.Ruse, C. I., Tan, F.-L., Kinter, M., and Bond, M.
(2004) Proteomics, 4, 1505-1516.
46.Gromov, P. S., and Tselis, Kh. E. (2000) Mol.
Biol. (Moscow), 34, 597-611.
47.Sanchez, J. C., Chiappe, D., Converset, V.,
Hoogland, C., Binz, P. A., Paesano, S., Appel, R. D., Wang, S.,
Sennitt, M., Nolan, A., Cawthorne, M. A., and Hochstrasser, D. F.
(2001) Proteomics, 1, 136-163.
48.Poglazov, B. F., and Levitsky, D. I. (1982)
Myosin and Biological Activity [in Russian], Nauka, Moscow.
49.Podlubnaya, Z. A., Levitsky, D. I., Shuvalova, L.
A., and Poglazov, B. F. (1986) Dokl. Akad. Nauk SSSR,
290, 1015-1017.
50.Levitsky, D. I., Shnyrov, V. L., Khvorov, N. V.,
Bukatina, A. E., Vedenkina, N. S., Permyakov, E. A., Nikolaeva, O. P.,
and Poglazov, B. F. (1992) Eur. J. Biochem., 209,
829-835.
51.Clark, K. A., McElhinny, A. S., Beckerle, M. C.,
and Gregorio, C. C. (2002) Annu. Rev. Cell. Dev. Biol.,
18, P.637-706.
52.Marci, J., McGee, B., Thomas, J. N., Du, P.,
Stevenson, T. I., Kilby, G. W., and Rapundalo, S. T. (2000)
Electrophoresis, 21, 1685-1693.
53.Taylor, S. W., Warnock, D. E., Glenn, G. M.,
Zhang, B., Fahy, E., Gaucher, S. P., Capaldi, R. A., Gibson, B. W., and
Ghosh, S. S. (2002) J. Proteome Res., 1, 451-458.
54.Breitbart, R. E., Andreadis, A., and
Nadal-Ginard, B. (1987) Ann. Rev. Biochem., 56,
467-495.
55.Stoltzfus, A., Spencer, D. F., Zuker, M.,
Logsdon, J. M., and Doolittle, W. F. (1994) Science, 265,
202-207.
56.Gusev, N. B., Bogatcheva, N. V., and Marston, S.
B. (2002) Biochemistry (Moscow), 67, 511-519.
57.Filatov, I. L., Katrukha, A. G., Bulargina, T.
V., and Gusev, N. B. (1999) Biochemistry (Moscow), 64,
969-985.
58.Szczesna-Cordary, D., Guzman, G., Ng, S. S., and
Zhao, J. (2004) J. Biol. Chem., 279, 3535-3542.
59.Roopnarine, O. (2003) Biophys. J.,
84, 2440-2449.
60.Yamada, T., Inashima, S., Matsunaga, S., Nara,
I., Kajihara, H., and Wada, M. (2004) Acta Physiol. Scand.,
180, 79-87.
61.Eleveld, D. J., Kopman, A. F., Proost, J. H., and
Wierda, J. M. (2004) Br. J. Anaesth., 92, 373-380.
62.Suzuki, E., Ota, T., Tsukuda, K., Okita, A.,
Matsuoka, K., Murakami, M., Doihara, H., and Shimizu, N. (2004) Int.
J. Cancer, 108, 207-211.
63.Clement, S., Orlandi, A., Bocchi, L., Pizzolato,
G., Foschini, M. P., Eusebi, V., and Gabbiani, G. (2003) Virchows
Arch., 442, 31-38.
64.Pawlak, G., McGarvey, T. W., Nguyen, T. B.,
Tomaszewski, J. E., Puthiyaveettil, R., Malkowicz, S. B., and Helfman,
D. M. (2004) Int. J. Cancer, 110, 368-373.
65.Saprygin, D. B., and Romanov, M. Yu. (1999)
Lab. Med., No. 2, 16-23.
66.Penttila, K., Koukkunen, H., Halinen, M.,
Rantanen, T., Pyorala, K., Punnonen, K., and Penttila, I. (2002)
Clin. Biochem., 35, 647-653.
67.Roychoudhury, A. K., and Nei, M. (1988) Human
Polymorphic Genes: World Distribution, Oxford University Press, New
York.
68.Eisenstein, R. S. (2000) Annu. Rev. Nutr.,
20, 627-662.
69.Pekrun, A., Neubauer, B. A., Eber, S. W.,
Lakomek, M., Seidel, H., and Schroter, W. (1995) Clin. Genet.,
47, 175-179.
70.Orosz, F., Olah, J., Alvarez, M., Keseru, G. M.,
Szabo, B., Wagner, G., Kovari, Z., Horanyi, M., Baroti, K., Martial, J.
A., Hollan, S., and Ovadi, J. (2001) Blood, 98,
3106-3112.
71.Ationu, A., Humphries, A., Wild, B., Carr, T.,
Will, A., Arya, R., and Layton, D. M. (1999) Lancet, 353,
1155-1156.
72.Merkulova, T., Thornell, L. E., Butler-Browne,
G., Oberlin, C., Lucas, M., Lamande, N., Lazar, M., and Keller, A.
(1999) J. Muscle Res. Cell. Motil., 20, 55-63.
73.Chosa, E., Sekimoto, T., Sonoda, N., Yamamoto,
K., Matsuda, H., Takahama, K., and Tajima, N. (2003) Clin. J. Sport
Med., 13, 209-212.
74.Comi, G. P., Fortunato, F., Lucchiari, S.,
Bordoni, A., Prelle, A., Jann, S., Keller, A., Ciscato, P., Galbiati,
S., Chiveri, L., Torrente, Y., Scarlato, G., and Bresolin, N. (2001)
Ann. Neurol., 50, 202-207.
75.Zhang, B., Chen, J. Y., Chen, D. D., Wang, G. B.,
and Shen, P. (2004) World J. Gastroenterol., 10,
1643-1646.
76.Van Wijk, R., van Solinge, W. W., Nerlov, C.,
Beutler, E., Gelbart, T., Rijksen, G., and Nielsen, F. C. (2003)
Blood, 101, 1596-1602.
77.Maekawa, M., Sudo, K., Li, S. S.-L., and Kanno,
T. (1991) Hum. Genet., 88, 34-38.
78.Rouault, T. A., Tang, C. K., Kaptain, S.,
Burgess, W. H., Haile, D. J., Samaniego, F., McBride, O. W., Harford,
J. B., and Klausner, R. D. (1990) Proc. Natl. Acad. Sci. USA,
87, 7958-7962.
79.Drugge, U., Holmberg, M., Holmgren, G., Almay, B.
G. L., and Linderholm, H. (1995) J. Med. Genet., 32,
344-347.
80.Ventura-Clapier, R., Kaasik, A., and Veksler, V.
(2004) Mol. Cell. Biochem., 256/257, 29-41.
81.Hirano, M., Huang, W.-Y., Cole, N., Azim, A. C.,
Deng, H.-X., and Siddique, T. (2000) Biochem. Biophys. Res.
Commun., 276, 52-56.
82.Stathopulos, P. B., Rumfeldt, J. A. O., Scholz,
G. A., Irani, R. A., Frey, H. E., Hallewell, R. A., Lepock, J. R., and
Meiering, E. M. (2003) Proc. Natl. Acad. Sci. USA, 100,
7021-7026.
83.Allen, S., Heath, P. R., Kirby, J., Wharton, S.
B., Cookson, M. R., Menzies, F. M., Banks, R. E., and Shaw, P. J.
(2003) J. Biol. Chem., 278, 6371-6383.
84.Tskhovrebova, L., and Trinick, J. (2003) Nat.
Rev. Mol. Cell. Biol., 4, 679-689.
85.McElhinny, A. S., Kazmierski, S. T., Labeit, S.,
and Gregorio, C. C. (2003) Trends Cardiovasc. Med., 13,
195-201.
86.Goldfarb, L. G., Vicart, P., Goebel, H. H., and
Dalakas, M. C. (2004) Brain, 127 (Pt. 4), 723-734.
87.North, K. N., Yang, N., Wattanasirichaigoon, D.,
Mills, M., Easteal, S., and Beggs, A. H. (1999) Nat. Genet.,
21, 353-354.
88.Yang, N., MacArthur, D. G., Gulbin, J. P., Hahn,
A. G., Alan, H., Beggs, A. H., Easteal, S., and North, K. (2003) Am.
J. Hum. Genet., 73, 627-631.
89.Golenhofen, N., Arbeiter, A., Koob, R., and
Drenckhahn, D. (2002) J. Mol. Cell. Cardiol., 34,
309-319.
90.Inaguma, Y., Ito, H., Iwamoto, I., Saga, S., and
Kato, K. (2001) Eur. J. Cell. Biol., 80, 741-748.
91.Hackman, P., Vihola, A., Haravuori, H., Marchand,
S., Sarparanta, J., de Seze, J., Labeit, S., Witt, C., Peltonen, L.,
Richard, I., and Udd, B. (2002) Am. J. Hum. Genet., 71,
492-500.
92.Siebrands, C. C., Sanger, J. M., and Sanger, J.
W. (2004) Cell Motil. Cytoskeleton, 58, 39-52.
93.Hunter, R. J., Neagoe, C., Jarvelainen, H. A.,
Martin, C. R., Lindros, K. O., Linke, W. A., and Preedy, V. R. (2003)
J. Nutr., 133, 1154-1157.
94.Carlsson, L., and Thornell, L. E. (2001) Acta
Physiol. Scand., 171, 341-348.
95.Goudeau, B., Dagvadorj, A., Rodrigues-Lima, F.,
Nedellec, P., Casteras-Simon, M., Perret, E., Langlois, S., Goldfarb,
L., and Vicart, P. (2001) Hum. Mutat., 18, 388-396.
96.Abbott, J. J., Amirkhan, R. H., and Hoang, M. P.
(2004) Arch. Pathol. Lab. Med., 128, 686-688.
97.Brown, R. H. (1997) Annu. Rev. Med.,
48, 457-466.
98.Tsubata, S., Bowles, K. R., Vatta, M., Zintz, C.,
Titus, J., Muhonen, L., Bowles, N. E., and Towbin, J. A. (2000) J.
Clin. Invest., 106, 655-662.
99.Freemont, P. S., Dunbar, B., and
Fothergill-Gilmore, L. A. (1988) Biochem. J., 249,
779-788.
100.Giometti, C. S., and Anderson, N. G. (1981)
Clin. Chem., 27, 1918-1921.
101.Shishkin, S. S. (1986) Vopr. Med. Khim.,
No. 5, 139-140.
102.Van den Heuvel, L. P., Wevers, M. H., van
Engelen, B. G. M., and Smeitink, J. A. M. (2003) Ann. Clin.
Biochem., 40, 9-15.
103.Foucault, G., Vacher, M., Merkulova, T.,
Keller, A., and Arrio-Dupont, M. (1999) Biochem. J., 338
(Pt. 1), 115-121.
104.Anderson, N. G., and Anderson, L. (1996)
Electrophoresis, 17, 443-453.
105.Gorg, A., Boguth, G., Obermaier, C., Posch, A.,
and Weiss, W. (1995) Electrophoresis, 16, 1079-1086.
106.Celis, J. E., Gesser, B., Rasmussen, H. H.,
Madsen, P., Leffers, H., Dejgaard, K., Honore, B., Olsen, E., Ratz, G.,
Larodsen, J., Basse, B., Mouridsen, S., Hellerup, M., Andersen, A.,
Walbum, E., Celis, A., Bauw, G., Puype, M., van Damme, J., and
Vandekerckhove, J. (1990) Electrophoresis, 11,
989-1071.
107.O'Mahoney, J. V., Guven, K. L., Lin, J., Joya,
J. J., Robinson, C. S., Wade, R. P., and Hardeman, E. C. (1998) Mol.
Cell. Biol., 18, 6641-6652.
108.Kovalyova, M. A., Kovalyov, L. I., Shishkin, S.
S., Egorov, T. A., and Musalyamov, A. K. (1999) Biochemistry
(Moscow), 64, 1108-1110.
109.Mikawa, T., Takeda, S., Shimizu, T., and
Kitaura, T. (1981) J. Biochem., 89, 1951-1962.
110.Kovalyov, L. I., Shishkin, S. S., Ivolgina, G.
L., Gromov, P. S., and Shandala, A. M. (1986) Biokhimiya,
51, 896-908.
111.Shishkin, S. S., and Kovalyov, L. I. (1987)
Biokhimiya, 52, 423-429.
112.Baev, A. A. (1990) The Project “Human
Genome”: Its Creation, Content, and Development, in
Advances in Science and Technology, Human Genome Series,
Vol. 1, VINITI, Moscow, pp. 4-33.
113.Shishkin, S. S. (1990) Abst. First All-Union
Conf. “Human Genome”, October 8-12, 1990,
Pereslavl-Zalessky, pp. 33-34.
114.Shishkin, S. S., Kovalyov, L. I., Lucashova, I.
V., Ershova, E. S., Chudaidatov, A. I., Musolyamov, A. Kh., Egorov, Ts.
A., Shilov, A. G., and Kucharenko, V. I. (1994) Abst. “2D
Electrophoresis: from Protein Maps to Genomes”, Italy, Siena,
September 5-7, 1994, p. 65.
115.Govorun, V. M., and Archakov, A. I. (2002)
Biochemistry (Moscow), 67, 1109-1123.
116.Labugger, R., McDonough, G. L., Neverova, I.,
and van Eyk, J. E. (2002) Proteomics, 2, 673-678.
117.Baker, C. S., Corbett, J. M., May, A. J.,
Yacoub, M. H., and Dunn, M. J. (1992) Electrophoresis,
13, 723-726.
118.Jungblut, P., Otto, A., Zeindl-Eberhart, E.,
Pleibner, K. P., Kreckt, M., Regitz-Zagrosek, V., Fleck, E., and
Wittman-Liebold, B. (1994) Electrophoresis, 15,
685-707.
119.Pulyaeva, E. V., Kovalyov, L. I., Tsvetkova, M.
N., Shishkin, S. S., and Boldyrev, N. I. (1990) Biokhimiya,
55, 489-498.
120.Shishkin, S. S., Egorov, Ts. A., Laptev, A. V.,
Tsvetkova, M. N., Pulyaeva, E. V., and Barbashov, S. F. (1991)
Biokhimiya, 56, 136-140.
121.Ershova, E. S. (1998) Structure-functional
study of protein products of gene expression in striated human muscle
tissue: Abstract of Candidate's dissertation [in Russian], Russian
Academy of Medical Sciences, Moscow.
122.Kaiser, C., von Stein, O., Laux, G., and
Hoffmann, M. (1999) Electrophoresis, 20, 261-268.
123.Civelli, O. (1998) FEBS Lett,
430, 55-58.
124.International Human Genome Sequencing
Consortium (2001) Nature, 409, 860-921.
125.Venter, C. J., Adams, M. D., Myers, E. W., Li,
P. W., Mural, R. J., Sutton, G. G., Smith, H. O., Yandell, M., Evans,
C. A., Holt, R. A., Gocayne, J. D., Amanatides, P., Ballew, R. M.,
Huson, D. H., Wortman, J. R., Zhang, Q., Korida, C. D., Zheng, X. H.,
Chen, L., Skupski, M., et al. (2001) Science, 291,
1304-1351.
126.Benson, D. A., Boguski, M. S., Lipman, D. J.,
Ostell, J., Ouellette, B. F. F., Rapp, B. A., and Wheeler, D. L. (1999)
Nucleic Acids Res., 27, 12-17.
127.Bork, P., and Copley, R. (2001) Nature,
409, 818-820.
128.Fields, S., Kohara, Y., and Lockhart, D. J.
(1999) Proc. Natl. Acad. Sci. USA, 96, 8825-8826.
129.Shishkin, S. S. (2002) Vestn. RAMN, No,
4, 11-16.
130.Li, Z.-B., Lehar, M., Braga, N., Westra, W.,
Liu, L.-H., and Flint, P. W. (2003) Proteomics, 3,
1325-1334.
131.Talamo, F., D'Ambrosio, C., Arena, S., Del
Vecchio, P., Ledda, L., Zehender, G., Ferrara, L., and Scaloni, A.
(2003) Proteomics, 3, 440-460.
132.Pinet, F., Poirier, F., Fuchs, S., Tharaux, P.
L., Caron, M., Corvol, P., Michel, J. B., and Joubert-Caron, R. (2004)
FASEB J., 18, 585-586.
133.Schwertz, H., Langin, T., Platsch, H., Richert,
J., Bomm, S., Schmidt, M., Hillen, H., Blaschke, G., Meyer, J., Darius,
H., and Buerke, M. (2002) Proteomics, 2, 988-995.
134.Jenkins, R. E., and Pennington, S. R. (2001)
Proteomics, 1, 13-29.
135.Patterson, S. D. (2000) Curr. Opin.
Biotechnol., 11, 413-418.
136.Manabe, T. (2000) Electrophoresis,
21, 1116-1122.
137.Nelson, R. W., Nedelkov, D., and Tubbbs, K. A.
(2000) Electrophoresis, 21, 1155-1163.
138.Ogut, O., and Jin, J.-P. (2000) J. Biol.
Chem., 275, 26089-26095.
139.Formigli, L., Meacci, E., Vassalli, M., Nosi,
D., Quercioli, F., Tiribilli, B., Tani, A., Squecco, R., Francini, F.,
Bruni, P., and Zecchi Orlandini, S. (2004) J. Cell Physiol.,
198, 1-11.
140.Laptev, A. V., Shishkin, S. S., Kovalyov, L.
I., Galyuk, M. A., Musalyamov, A. Ch., and Egorov, Ts. A. (1993)
Biochem. Genet., 31, 253-258.
141.Tikunov, B. A., Sweeney, H. L., and Rome, L. C.
(2001) J. Appl. Physiol., 90, 1927-1935.
142.Samson, F., Mesnard, L., Minovilovic, T. G.,
Potter, J. J. M., Roses, A. D., and Gilbert, J. R. (1994) Biochem.
Biophys. Res. Commun., 199, 841-847.
143.Kovalyov, L. I., Khudaitov, A. I., Galyuk, M.
A., Shishkin, S. S., Niyazbekova, A. S., Gelashvili, N. N., and
Zavalishin, I. A. (1994) Vopr. Med. Khim., No. 5, 42-45.
144.Kovalyov, L. I., Kovalyova, M. A., Gerasimov,
E. V., Budina, A. A., Tikhonova, O. V., Moshkovsky, S. A.,
Serebryakova, M. V., and Shishkin, S. S. (2004) Proc. III Congr.
Rus. Soc. Genetics and Selection “Genetics in XXI Century: State
of Art and Future Perspectives”, Vol. 2, URSS Publishers,
Moscow, p. 27.