REVIEW: Biological Effects of 3,5-Diiodothyronine (T2)
F. Goglia
Dipartimento di Scienze Biologiche ed Ambientali-Universita degli Studi del Sannio-Via Port'Arsa, 11 82100 Benevento, Italy; fax: +39-082423013; E-mail: goglia@unisannio.it
Received September 15, 2004
This article is principally intended to describe the facts concerning the actions of 3,5-diiodothyronine (T2). Until recent years, T2, because of its very low affinity for thyroid hormone receptors (THR), was considered an inactive metabolite of thyroid hormones (TH) (thyroxine (T4) and triiodo-L-thyronine (T3)). Several observations, however, led to a reconsideration of this idea. Early studies dealing with the biological activities of this iodothyronine revealed its ability to stimulate cellular/mitochondrial respiration by a nuclear-independent pathway. Mitochondria and bioenergetic mechanisms seem to be major targets of T2, although outside the mitochondria T2 also has effects on carriers, ion-exchangers, and enzymes. Recent studies suggest that T2 may also affect the transcription of some genes, but again the underlying mechanisms seem to be different from those actuated by T3. The accumulated evidence permits the conclusion that the actions of T2 do not simply mimic those of T3 but instead are specific actions exerted through mechanisms that are independent of those actuated by T3 and do not involve THR.
KEY WORDS: 3,5-diiodothyronine, mitochondria, energy metabolism, cytochrome c oxidase, iodothyronine, thyroid hormones
In some people, Science and Mind
evolve like some rare “special” bottles of wine:
“they improve with age”. (Fernando Goglia)
To: my “special friend”
Vladimir Petrovich Skulachev
Energy expenditure is an important factor in the maintenance of energy homeostasis. The most accurate way to estimate energy expenditure is by measuring heat production, hence the use of the terms “thermogenesis” and/or “calorigenesis”, but in most circumstances measurement of oxygen consumption is a perfectly acceptable way of estimating the amount of energy consumed. It is universally recognized that thyroid hormones (TH) are unique in their ability to stimulate thermogenesis/calorigenesis (the well known “calorigenic effect of TH”). Their main action consists in stimulating cellular respiration while at the same time reducing metabolic efficiency. However, although this phenomenon has been known since the end of the 19th century [1] and has been the subject of a great number of reports, the mechanism by which TH exert their effects on energy metabolism is far from being firmly established. During the last half-century, a large number of hypotheses were put forward to try to explain the cellular-molecular mechanisms underlying the calorigenic effect of TH. We can distinguish three historical periods during which major hypotheses concerning the mechanism of action of TH at the cellular level were proposed and which oriented/attracted the interest of investigators. In the first of these periods, which ran from the early 1950s to the middle 1960s, the most intriguing hypothesis put forward was the “uncoupling hypothesis”. This suggested that TH stimulate metabolic rate by acting at the mitochondrial level to uncouple the electron transport chain from ATP synthesis [2, 3]. This hypothesis predicted a thyroid-dependent increase in oxygen consumption without a concomitant increase in ATP production (decreased P/O ratio). The early experiments supporting such a possibility were those performed by Lardy and Feldcott [2] and by Martius and Hess [3] who presented evidence that mitochondria prepared from T4-treated rats exhibited lower P/O ratios than those from untreated euthyroid controls. However, in the early 1960s its validity was questioned on several grounds, amongst others that uncoupling (as assessed through changes in mitochondrial respiratory parameters such as respiratory control ratio (RCR) and P/O ratio) was observed only with pharmacological doses of TH. Since some effects were also observed in vitro (in isolated mitochondria), the theory also implied that TH acted directly at the mitochondrial level. This hypothesis has never been dropped and continues to this day to be investigated using new approaches.
At the beginning of 1960s, however, results obtained by Tata and coworkers opened a new chapter in the history of our understanding of the action of TH. Those authors showed that administration of T3 to hypothyroid rats induced an increase in basal metabolic rate and, moreover, that injecting T3 in combination with actinomycin D (a well-known inhibitor of transcription) completely abolished the stimulatory effect of T3 [4, 5]. These results clearly pointed towards the involvement of transcription in the action of T3 at the cellular level. Subsequently, specific nuclear binding sites for T3 were first described by Oppenheimer's group in rat liver and kidney [6] and later in a variety of other tissues and cell cultures [7]. What is now evident from the literature is that a large number of effects attributable to T3 are indubitably initiated via thyroid hormone receptors located within the nucleus, the so-called “genomic” or “nuclear-mediated” effects of TH [8-10]. However, although transcription is considered an important target for TH, few thyroid-hormone-responsive genes have been identified and the roles played by the various nuclear-receptor isoforms in the diverse actions of TH remain unclear [10]. A new area of research in the field of thyroid hormone actions was opened up by the discovery of deiodinase enzymes, which are involved in the peripheral metabolism of iodothyronines. Indeed, three iodothyronine deiodinases (D1, D2, and D3) regulate the local and systemic availability of thyroid hormone, with D1 and D2 converting T4 to T3, while D3 converts T4 and T3 to rT3 (reverse T3) and 3,3´-diiodothyronine, respectively.
3,5-DIIODOTHYRONINE (T2): A BIOLOGICALLY ACTIVE
IODOTHYRONINE OR A DEGRADATION PRODUCT OF TH?
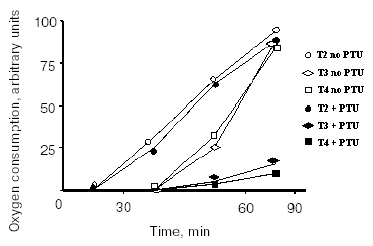
Between the mid-1980s and the beginning of the 1990s, it became evident that some TH effects are undoubtedly non-genomic in origin [11-14], although the sites of origin of some of these effects remain unknown to this day. Similar evidence exists for other hormones, such as reproductive hormones [15] and other steroid hormones [16], as well as for glucocorticoids [17, 18]. The non-nuclear actions of TH are those actions in which the involvement of the nucleus can be excluded. Extranuclear effects of TH have been described at different cellular levels (plasma membrane, mitochondria, cytoplasm, cytoskeleton, etc.). As these extranuclear effects are independent of a direct interaction with nuclear receptors, it is not surprising that they have hormone structure-activity relationships that are distinct from those of the nuclear-mediated actions. In fact, it became evident some years ago that iodothyronines other than T3 possess biological activity. Because of the presence of the above-mentioned nuclear receptors and because these receptors have their greatest affinity for T3, the belief developed that T3 is the only active iodothyronine. However, this idea has been under reconsideration for some time and now, to judge from the literature, at least four iodothyronines possess significant biological activities. These four are: thyroxine (T4), 3,3´,5-triiodo-L-thyronine (T3), reverse T3 (rT3), and, of particular interest for the present purposes, 3,5-diiodothyronine (T2). Concerning the effect of TH on energy metabolism, a decade and a half ago surprising results were published showing that (among a lot of iodothyronines tested) T2, like T3, at a concentration as low as 1 pM induced a rapid (few minutes) stimulation of oxygen consumption in perfused livers isolated from hypothyroid rats. In the same study, it was shown that the effect of T3 was largely abolished by the addition of an inhibitor of D1 deiodinase, while the effect of T2 was not. Moreover, T2 was as potent as T3 but exerted its effect more rapidly (Fig. 1) [19].
Fig. 1. Effect of iodothyronines on the O2 consumption of isolated perfused livers from hypothyroid rats. Experiments were performed in the absence or presence of propylthiouracil (PTU) (adapted from Horst et al., 1989).
STUDIES in vivo
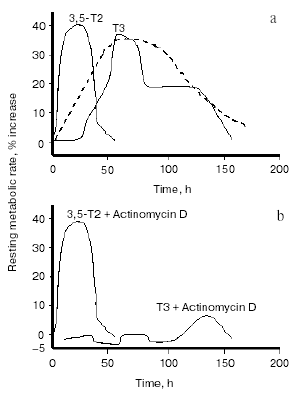
Stimulated by that report, and other paper showing an interaction of 3,3´-T2 with mitochondria [20], other investigators began to focus more sharply on a putative physiological role for T2. Energy metabolism, cellular respiration, and mitochondria were the major areas of interest. Several reports issuing from various laboratories at the beginning of the last decade showed that acute or chronic administration of T2 to rats resulted in significant changes in mitochondrial activities. When either T3 or T2 were acutely injected, T2 had a more rapid effect on mitochondrial respiration than T3, that of T2 being evident as soon as 1 h after its injection, while that of T3 became evident only after 24 h. Moreover, the rapid effect of T2 was shown to be independent of protein synthesis (since it was evident even in the presence of cycloheximide) [21-23]. The interpretation of these results was that the effects of T2 are mediated by a direct interaction with mitochondria, whilst those of T3 are initiated via the nucleus. Similar observations on oxygen consumption were also made in human mononuclear blood cells [24]. The realization that there were effects of T2 on mitochondrial activities stimulated researchers into trying both to uncover a physiological role for T2 and to find evidence for a possible effect of T2 on the energy metabolism of the whole animal. The experimental design used to test the latter possibility was basically the same as that employed by J. R. Tata in the early 1960s [25, 26]. It consisted in acutely injecting a single dose of T2 in hypothyroid rats, then following the time course of the induced changes in the resting metabolic rate (RMR) of the animals and comparing those results with the ones obtained by injecting T3. One problem encountered when injecting various iodothyronines is the need to exclude the possibility that some of the effects seen after their administration might be due to their deiodinated products: for this reason, the deiodinase enzymes need to be inhibited. This objective, particularly important when injecting T3, can be achieved by inducing hypothyroidism by the simultaneous administration of propylthiouracil (PTU) and iopanoic acid (IOP). This yields animals with severe hypothyroidism and at the same time, with a powerful inhibition of all three types of deiodinase enzymes. Indeed: i) PTU inhibits TH production by the thyroid gland (via an inhibition of thyroid peroxidase activity) and at the same time induces a strong inhibition of type I deiodinase enzyme (D1), while ii) iopanoic acid has no influence over TH production but exerts an inhibitory action over all three deiodinase enzymes, including type II (D2) and type III (D3) [27]. In studies employing this method, it was observed some years ago that RMR was considerably lower in hypothyroid rats than in the euthyroid controls and that in hypothyroid animals, injection of either T3 or T2 significantly increased the RMR [28, 29]. In fact, injection of T3 caused an increase of about 35% in RMR: the increase started 25-30 h after the injection, peaked at 50-75 h and persisted (albeit at a weaker level) until 5-6 days after the injection. This was in good accordance with the trend noted by Tata in his early studies (see Fig. 2a). Injection of T2 at the same dose (25 µg/100 g body weight), on the other hand, induced a temporally different pattern of response. The increase in RMR was of about the same amplitude (about 40%), but in this case the increase started between 6 and 12 h after the injection, peaked at about 24-30 h and had almost disappeared at 48 h. An even more interesting observation to come out of these studies was that whereas administering actinomycin D and T3 together lead to the stimulation of RMR by T3 being almost completely abolished (as indeed was expected from the results of Tata [26]), simultaneously injecting actinomycin D with T2 caused no attenuation at all of the stimulation seen with T2 alone (Fig. 2b).
These studies confirmed the existence of a nuclear-independent mechanism of action for T2 and gave rise to the hypothesis that T2 might mediate some of the short-term effects of TH and might, indeed, be important, in physiological situations requiring additional energy expenditure. One example would be exposure to cold, a situation in which, to counteract increased heat dispersion, extra energy expenditure is required. In fact, TH play important roles in cold-acclimation processes, as evidenced by hypothyroid rats surviving cold for only 3-4 days. Interestingly, in cold-exposed rats both T2 and T3 improve cold tolerance but via different mechanisms [30]. That study showed that when injected into cold-exposed hypothyroid rats, both T2 and T3 increased the energy expenditure of the animals and stimulated the oxidative capacity (expressed as cytochrome c oxidase (COX) activity) of metabolically very active tissues such as heart, skeletal muscle, liver, and brown adipose tissue. However, whereas the stimulation by T3 was principally due to an action (nuclear-mediated) on the trophism of these tissues, the effect of T2 was probably due to its direct interaction with mitochondria, thus enhancing the oxidative capacity of the tissues.Fig. 2. Changes in the resting metabolic rate (RMR) of hypothyroid rats following administration of iodothyronines (a) and following the simultaneous administration of iodothyronines and actinomycin D (b) (adapted from Moreno et al., 1997). The broken line in figure (a) represents data adapted from Tata (1963).
An important study showing an in vivo metabolic effect of T2 was carried out by Cimmino et al. [31]. In this study, they investigated the effect of chronic treatment with either T2 or T3 on the daily energy expenditure of unanaesthetized, unrestrained hypothyroid rats, as well as on amino acid and lipid metabolism. The hypothyroidism was induced by a simultaneous administration of PTU and IOP [29], which, as indicated above, is of advantage in allowing the observed effects to be attributed to the iodothyronine actually administered (T2 or T3). Daily energy expenditure was measured by continuous monitoring of O2 consumption and CO2 production. Compared to that of normal euthyroid rats, daily energy expenditure was lower in hypothyroid animals, while administration of either T2 or T3 to hypothyroid animals restored the normal euthyroid values. Apart from confirming that T2 is a metabolically active iodothyronine, the most intriguing observation to come out of this study was that T2 is able to induce a significant stimulation of lipid beta-oxidation, which was evaluated by measuring the 13CO2 recovered in the breath after the injection of octanoic acid-1-13C. All the above-cited in vivo results were obtained by administering T2 to hypothyroid rats (with deiodinase enzymes inhibited). Indeed, administration of T2 to normal euthyroid animals results in a slight or nonexistent change in RMR [29]. The explanation for this apparent discrepancy is obscure at the moment and several factors may underlie it. Thus, it may be that in normal euthyroid rats: 1) T2 is rapidly metabolized; 2) T2 does not adequately enter the cells; 3) to reach the cellular targets, T2 needs to be formed from a precursor such as T3 (even if a biochemical pathway leading to the formation of T2 from T3 has not yet been demonstrated either in vivo or in vitro); 4) the metabolic state of the animal (diet composition) may also play a role in permitting the metabolic effects of T2 to be revealed. In an attempt to clarify this situation, and in particular point 3, a recent study set out to evaluate the time course of the calorigenic effect (change in RMR) seen after administration of either T3 or T2 to normal euthyroid rats (N rats), with the data being compared with those obtained after acute administration of the same iodothyronines to hypothyroid rats treated with PTU and IOP (P + I rats) [32]. In the same study, the authors compared the time course of the change in RMR with the time course of the changes in the serum and hepatic levels of T2. Acute injection of T3 had an effect on RMR that was evident 25 h earlier in N rats than in P + I animals (see Fig. 3).
In N rats injected with T3, the first phase of the change in RMR almost overlapped that observed when T2 was injected into P + I animals (hypothyroid animals with deiodinase enzymes inhibited) (Fig. 3a). The simultaneous injection of actinomycin D inhibited mostly the late part of the effect seen in N + T3 animals, the early phase being only slightly affected (Fig. 3a). In N rats, an acute inhibition of deiodinase enzymes (4 h before iodothyronine administration), on the other hand, resulted in a marked reduction of the early part of the effect of T3, indicating that deiodination was necessary for it to produce the early part of its effect in full (Fig. 3b).Fig. 3. Changes in resting metabolic rate (RMR) in euthyroid (N) and hypothyroid (P + I) rats after administration of iodothyronines (a), and effect of acute inhibition of the activity of the deiodinases on the changes in RMR seen after acute administration of T3 to N rats (b) (adapted from Moreno et al., 2002).
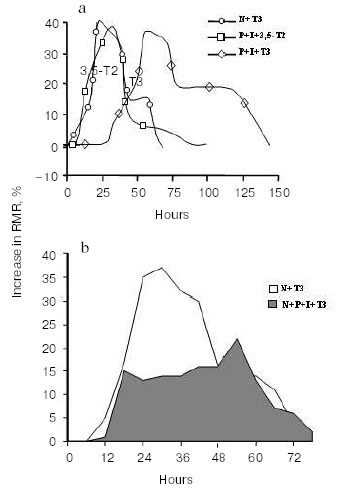
An important observation to come out of that study [32] was that after T3 injection, the maximal increase in RMR and the maximal increase in T2 concentration in the liver occurred at the same time (at about 25 h after the injection). Taken as a whole, these results strongly indicate that: 1) part of the early change in RMR after T3 injection in N animals is due to its conversion to T2, and 2) in vivo, 3,5-diiodo-L-thyronine is formed from T3. The second observation is important since such a conversion T3 --> 3,5-T2 has never been observed in vitro.
STUDIES in vitro
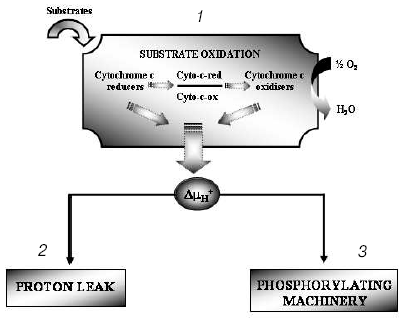
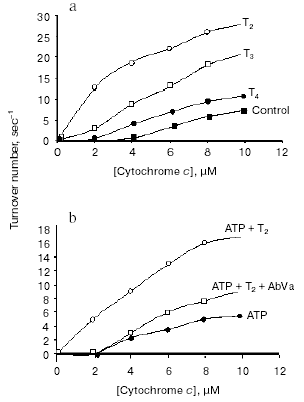
The hypothesis that mitochondria might be the target for the action of T2 is supported by many observations, although in vitro direct addition of T2 to isolated mitochondria does not always result in a stimulation of mitochondrial activities. In fact, an effect of T2 after its addition in vitro has been shown only when a whole homogenate is used. In liver homogenate from hypothyroid rats, a significant increase in COX activity has been observed following the addition of T2, with a half-maximal increase at nanomolar concentrations [33]. In that study, the maximal stimulation of COX was by 66% and this was increased to 93% when PTU was added to the medium. Similar experiments performed on isolated mitochondria gave negative results but, when mitochondria were isolated from liver homogenate preincubated with T2, the organelles showed a significantly higher COX activity. These results led the authors to postulate the presence of a cytoplasmic factor that might mediate the effect of T2. This idea has been supported by further data showing, by photoaffinity labeling, the presence of specific binding sites for T2 in rat liver cytosol [34]. Three proteins with apparent molecular masses of 86, 66, and 38 kD were able to attach covalently to [125I]3,5-T2 after UV irradiation of rat liver cytosol. The 38 kD protein, which showed the greatest affinity for T2 and bound it in the absence of NADPH, was also able to bind T3 (but only in a NADPH-dependent manner). This protein may serve either as a T2/T3 reservoir or as a T2/T3 translocator that works in a cellular redox state-dependent manner (NADP/NADPH ratio). Specific binding sites for T2 have also been detected in rat liver mitochondria [35] but, as emphasized by the authors [36], the data concerning mitochondrial sites need to be interpreted with some caution because of the limitations inherent in such studies. Due to the inner-ring labeling procedure, in fact, the 3,5-125I-T2 used for the measurement of binding parameters had a low specific activity and it was possible to perform studies only over a narrow range of concentrations. Because of this, there may not be a particularly close correspondence between the real values for binding capacity and affinity constant and those calculated by authors. To acquire further insights into the putative site of action of T2, a study was performed in which top-down elasticity analysis was applied to mitochondria isolated from rats acutely injected with a large dose of T2 [37]. This analysis [38] considers the reactions involved in the energy-transduction apparatus, at the mitochondrial level, to be divided into blocks of reactions: those involved in the formation of proton-motive force (DeltaµH+ producers: substrate oxidation - respiratory chain activity - proton pumps) and those involved in the dissipation of DeltaµH+ (DeltaµH+ consumers: proton leak, redox slip, and ATP synthesis machinery). DeltaµH+ represents the intermediary between these blocks of reactions (Fig. 4). The results of that study [37] showed that T2 at 1 h after its injection affected the overall kinetics of the reactions involved in the oxidation of substrates. It was observed that T2 acted on two distinct blocks of the respiratory chain: namely, complex IV (COX) and the block of reactions involved in the reduction of cytochrome c. The COX complex, indeed, may be one of the targets for the action of T2. Results showing that the addition of T2 to the COX complex isolated from bovine heart stimulated its activity seemed to support this hypothesis [39]. Later, Arnold et al. [40], using photoaffinity labeling procedures, identified subunit Va of the COX complex as the binding site for T2. In the same study, they showed that the action of T2 on the COX complex consisted in abolishing the allosteric ATP inhibition of COX, with the half-maximal effect at 10-7 M (Fig. 5).
Fig. 4. Metabolic-control analysis: the oxidative phosphorylation system is considered to be divided into blocks of reactions: 1) those that are involved in the oxidation of substrates and that thereby generate DeltaµH+; 2) those that dissipate DeltaµH+ without ATP synthesis, which together are called the proton leak (for further details see [38]); 3) those that dissipate DeltaµH+ for the synthesis and export of ATP.
The abolition of the allosteric ATP inhibition was specific for T2 and was prevented by the addition of a monoclonal antibody against subunit Va (“AbVa” in Fig. 5b), thereby confirming that this subunit is a binding site for T2. A more recent and deeper investigation by Kadenbach et al. [41] provided evidence that the addition of T2 to a reconstituted COX complex (in liposomes) led to a decrease in the respiratory control ratio of the complex (the ratio of respiration in presence of uncouplers over that in their absence). The effect was evident in the presence of intraliposomal ATP but not in presence of intraliposomal ADP. The same group concluded that the effect of T2 on cytochrome c oxidase could result in partial uncoupling of oxidative phosphorylation via increased DeltaµH+ (due to a decline in the second mechanism of respiratory control [42]) and decreased H+/e- stoichiometry (intrinsic uncoupling) of COX at higher membrane potentials [43, 44]. Other actions, however, could indirectly involve the mitochondria in the biological effect of T2. Indeed, data from Hummerich at al. [45] suggest an influence of T2 over mitochondrial activity that is mediated by an increase in mitochondrial Ca2+ uptake (which results in an increased activity of these organelles due to an increase in the activity of the mitochondrial dehydrogenases) [46]. This would lead to an increase in the amount of reduced substrates available for the respiratory chain.Fig. 5. Effect of 3,5-diiodothyronine on the coupled respiration of cytochrome c oxidase. a) Cytochrome c oxidase activity in presence and absence of iodothyronines (adapted from Arnold et al., 1998); b) effect of T2 in the abolition of the allosteric inhibition by ATP of the controlled respiration of reconstituted cytochrome c oxidase (adapted from Arnold et al., 1998).
The effects of T2 at the cellular level are not limited to mitochondria. Huang et al. [47], who compared the effects of several thyroid hormone analogs on sodium currents (I-Na) in neonatal rat myocytes, observed that T4, T3, and T2 all significantly increased current density (to a similar extent) relative to the control. In contrast, thyroid hormone analogs with an altered side group on the inner iodophenyl ring (including Tetrac, Triac, and D-T3) had no effect on I-Na, nor did reverse T3 (rT3), monoiodothyrosine (MIT), or thyrosine. The effects of T3 and T2 were not blocked by propranolol, indicating that their effects are not mediated through beta-adrenergic signaling pathways. The authors suggested that the acute effects of TH and their analogs on cardiac I-Na might be mediated by a non-genomic thyroid hormone receptor with a unique structure-activity relationship. A study has also been made of the rapid non-genomic effects of TH and T2 on membrane transport systems in chick embryo hepatocytes such as the Na+/H+ exchanger and the system A of amino acid transport [48]. In that study, the authors presented evidence for rapid non-genomic effects of TH and suggested that the short-term effects of TH may have a role to play during fetal development and cell differentiation. T2 was able to mimic some of the TH effects but with a lower efficiency.
STUDIES ON ENZYME EXPRESSIONS AND ACTIVITIES IN NON-MAMMALIAN
SPECIES
T2 has also been shown to be able to affect some important TH target enzymes such as lipogenic enzymes (malic enzyme and glucose-6-phosphate dehydrogenase (G6PD)) and deiodinase enzymes [49-51]. T2 and T3 were almost equipotent at downregulating Trbeta2 mRNA in GH3 cells but doses of T2 and T3 that were equivalent in their induction of malic enzyme mRNA did not produce equivalent suppressions of circulating thyroid-stimulating hormone (TSH), with T2 being only 27% as effective as T3 [49]. A weak effect of T2 on the TSH level was also observed after its chronic administration in rats; indeed, only at very high doses of T2 were the TSH levels significantly lower (even then they were still within the normal range) [50]. An interesting observation is that T2 is more potent than T3 at directly stimulating G6PD, the former being effective at a dose as low as 1 µg/100 g body weight (3-5 times more potent) [51]. In that study, T2 did not affect G6PD mRNA and its effect on G6PD activity was not attenuated by inhibitors of protein synthesis, thus indicating, once again, a non-nuclear-mediated effect. Moreover, D1 activity in the rat anterior pituitary is transiently increased after a single injection of T2, while in a reaggregate culture of anterior pituitary, T2 stimulates D1 at 24 h after its application, dose-dependently [52].
The biological effects of T2 are not restricted to mammalian species. An effect of T2 on mitochondrial activities has been demonstrated in liver and muscle from various non-terrestrial vertebrates [53-57]. In the goldfish Carassius auratus, a stimulation of pyruvate-fuelled mitochondrial respiration (both in liver and in muscle) has been shown following a 5-min in vitro incubation with 0.3 nM T2 [54]. Such a rapid increase in substrate oxidation by T2 (and also by T3) may be important in mediating diurnal changes in mitochondrial metabolism. An interesting observation has been made by Garcia-G et al. [53] who examined the effect of short-term exposure to T4, T3, and T2 (0.1 µM; 12 or 24 h) on D1 and D2 activities and expressions (at the mRNA level) in killifish (a teleost) liver. They observed that although none of the three iodothyronines had any effect on D1 activity, they all decreased D2 activity. In contrast to those of the other iodothyronines, the effect of T2 was already evident after a 12-h treatment, those of T4 and T3 being evident only after a 24-h treatment. T2 was also able to affect D2 transcription and the authors suggested that in view of its different time course of action, the mechanism actuated by T2 is different from that actuated by the other iodothyronines.
CONCLUSIONS AND PERSPECTIVES
The studies performed so far on the biological effects of T2, which are summarized in the table, raise a number of important questions and open new perspectives on some aspects of the metabolic effects of TH. The first conclusion that can be drawn is that the previous common assumption that T3 is the only active iodothyronine has to be reconsidered. This is important for several reasons; for example, when administering TH to animals, we need to pay attention to which thyroid hormone to use and in which thyroid state of the animal to administer it. This is particularly important in animals that have been made hypothyroid, because the effects on deiodination mechanisms differ among surgical thyroidectomy and the various chemicals (PTU, methymazole, or IOP) used to induce hypothyroidism (see [13]). Indeed, if deiodinase enzymes are fully active we cannot be sure that all the effects observed after the administration/addition of a certain iodothyronine are caused by that iodothyronine itself. In addition, the studies reported in this article may provide an explanation for the conflicting results concerning a direct effect of TH on isolated mitochondria. In fact, in isolated mitochondria--depending on the microsomal contamination (which in turn depends on the methods used in mitochondrial preparation; see [13])--TH may be converted to another iodothyronine and a stimulatory effect may be more or less evident. As far as T2 is concerned, the most important problem that remains to be elucidated is the physiological relevance of its actions. The first point to be considered before we can conclude that it has a physiological relevance is the concentration used in studies on the biological effects of T2. The in vivo studies have been carried out using both acute and chronic administration of T2, and its effects have been compared with those of T3. In both cases (acute and chronic), the doses of T2 and T3 have been comparable and have ranged from 2.5 µg/100 g body weight to 10 µg/100 g body weight in chronic studies, while in acute studies the dose used has been 25 µg/100 g body weight for both T3 and T2. From an analysis of the literature, it seems to me that the doses of T2 used in chronic studies were within the range of those used in most studies concerning the metabolic effects of TH. Only in one study did the doses range more widely, from 2.5 µg/100 g body weight to 200 µg/100 g body weight [48]. The same conclusion can be drawn for acute studies, in which a single dose of 25 mg/100 g body weight has been used. Indeed, this dose is the minimal one needed to show a clear-cut effect of TH on basal metabolic rate [4, 5, 58]. Another related aspect is the concentrations of T2 in the serum and tissues; however, little is known about these parameters, especially in rats. From a unique study on T2 levels in euthyroid rats, it seems that the serum level is of the order of 4.6 pM, while the hepatic level is of the order of 10 fmol/g (0.4 pmol/g in the liver of 16-day-old rat fetuses). In rats, the serum level of T2 increases after T3 injection (from 4.6 to 24.1 pM after 12 h). This last result strongly indicates that in vivo, T2 is indeed formed from T3 (even though this pathway has never been demonstrated in vitro).
Short summary of the effects of T2 and corresponding
references

The last consideration about the biological effects of T2 I want to discuss is: does T2 simply mimic the effects of T3 (by nonspecific and T3-related pathways) or are its effects specific ones involving pathways distinct from those actuated by T3? From an analysis of the available data, it seems to me that the second possibility is the more realistic. Indeed: a) most of the effects of T3 are attenuated by inhibitors of protein synthesis, whereas those of T2 are not; b) the effects of the two iodothyronines differ in their time course, with those due to T2 being more rapid that those due to T3; c) the metabolic effects of T3 are inhibited (both in vitro and, the early effect, in vivo) by the simultaneous use of inhibitors of deiodinases, whereas the effects of T2 are not; and d) the doses of T2 used in most studies are too low to induce nonspecific effects due to its interaction with the nuclear receptors for T3. These nuclear sites show a very low affinity for T2 (about 1,000 times less than that for T3 [49]) and thus, at least in the in vivo studies, the dose of T2 needed to reproduce or mimic the effects of T3, via the nucleus, would be around 1,000 times higher for T2 (in 100 g body weight). Finally, some important points to be investigated in future studies concern the biochemical pathways involved in the formation of T2 and in its clearance from serum and tissues.
REFERENCES
1.Magnus-Levi, A. (1895) Berlin Klein
Wochschr., 32, 650-652.
2.Lardy, H. A., and Feldcott, G. (1951) Ann. NY
Acad. Sci., 54, 636-648.
3.Martius, C., and Hess, B. (1951) Arch. Biochem.
Biophys., 33, 486-487.
4.Tata, J. R., Ernster, L., and Lindberg, O. (1962)
Nature, 193, 1058-1060.
5.Tata, J. R. (1963) Nature, 197,
1167-1168.
6.Oppenheimer, J. H., Koerner, D., Schwartz, H. L.,
and Surks, M. I. (1972) J. Clin. Endocrinol. Metab., 35,
330-333.
7.Oppenheimer, J. H., Schwartz, H. L., Mariash, C.
N., Kinlaw, W. B., Wong, N. C. W., and Freake, H. C. (1987)
Endocrine Rev., 8, 288-308.
8.Yen, P. M. (2001) Physiol. Rev.,
81, 1097-1142.
9.Zhang, J., and Lazar, M. A. (2000) Annu. Rev.
Physiol., 62, 439-466.
10.Lazar, M. A. (2003) J. Clin. Invest.,
112, 497-499.
11.Hulbert, A. J. (2000) Biol. Rev.,
75, 519-631.
12.Goglia, F., Moreno, M., and Lanni, A. (1999)
FEBS Lett., 452, 115-120.
13.Goglia, F., Silvestri, E., and Lanni, A. (2001)
Biosci. Rep., 22, 17-32.
14.Davis, P. J., and Davis, F. B. (1996)
Thyroid, 6, 497-504.
15.Revelli, A., Massobrio, M., and Tesarik, J.
(1998) Endocrine Rev., 19, 3-17.
16.Wehling, M. (1994) Trends Endocrinol.
Metab., 5, 347-353.
17.Buttgereit, F., Wehling, M., and
Burmester, G. R. (1998) Arthritis Rheumatism, 41,
761-767.
18.Davis, P. J., Tillmann, H. C., Davis, F. B., and
Wehling, M. (2002) J. Endocrinol. Invest., 25,
377-388.
19.Horst, C., Rokos, H., and Seitz, H. J. (1989)
Biochem. J., 261, 945-950.
20.Goglia, F., Torresani, J., Bugli, P., Barletta,
A., and Liverini, G. (1981) Pflugers Arch., 390,
120-124.
21.Lanni, A., Moreno, M., Cioffi, M., and Goglia, F.
(1992) Mol. Cell. Endocrinol., 86, 143-148.
22.Lanni, A., Moreno, M., Cioffi, M., and Goglia, F.
(1993) J. Endocrinol., 136, 59-64.
23.O'Reilly, I., and Murphy, M. P. (1992) Acta
Endocrinol. (Copenh.), 127, 542-546.
24.Kvetny, J. (1992) Horm. Metab. Res.,
24, 322-325.
25.Tata, J. R., Ernster, L., and Lindberg, O. (1962)
Nature, 198, 1059-1060.
26.Tata, J. R. (1963) Nature, 197,
1167-1168.
27.Leonard, J., and Koehrle, J. (2000) in Werner
& Ingbar's. The Thyroid: a Fundamental and Clinical Text
(Braverman, L. E., and Utiger, R. D., eds.) Lippincott, Williams
& Wilkins, Philadelphia, pp. 136-173.
28.Lanni, A., Moreno, M., Lombardi, A., and Goglia,
F. (1996) J. Physiol. (London), 494, 831-837.
29.Moreno, M., Lanni, A., Lombardi, A., and Goglia,
F. (1997) J. Physiol. (London), 505, 529-538.
30.Lanni, A., Moreno, M., Lombardi, A., and Goglia,
F. (1998) Pflugers Arch.-Eur. J. Physiol., 436,
407-414.
31.Cimmino, M., Mion, F., Goglia, F., Minaire, Y.,
and Geloen, A. (1996) J. Endocrinol., 149, 319-325.
32.Moreno, M., Lombardi, A., Beneduce, L.,
Silvestri, E., Pinna, G., Goglia, F., and Lanni, A. (2002)
Endocrinology, 143, 504-510.
33.Lanni, A., Moreno, M., Lombardi, A., and Goglia,
F. (1994) Mol. Cell. Endocrinol., 99, 89-94.
34.Moreno, M., Silvestri, E., Lombardi, A., Visser,
T. J., Goglia, F., and Lanni, A. (2003) Endocrinology,
144, 2297-2303.
35.Goglia, F., Lanni, A., Horst, C., Moreno, M., and
Thoma, R. (1994) J. Mol. Endocrinol., 13, 275-282.
36.Lanni, A., Moreno, M., Lombardi, A., de Lange,
P., and Goglia, F. (2001) J. Endocrinol. Invest., 24,
897-913.
37.Lombardi, A., Lanni, A., Moreno, M., Brand, M.,
and Goglia, F. (1998) Biochem. J., 330, 521-526.
38.Brand, M. D. (1997) J. Exp. Biol.,
200, 193-202.
39.Goglia, F., Lanni, A., Barth, J., and Kadenbach,
B. (1994) FEBS Lett., 346, 295-298.
40.Arnold, S., Goglia, F., and Kadenbach, B. (1998)
Eur. J. Biochem., 252, 325-330.
41.Kadenbach, B., Huttemann, M., Arnold, S., Lee,
I., Muhlenbein, N., and Bender, E. (2000) Free Rad. Biol. Med.,
29, 211-221.
42.Kadenbach, B., and Arnold, S. (1999) FEBS
Lett., 447, 131-134.
43.Kadenbach, B. (2003) Biochim. Biophys.
Acta, 1604, 77-94.
44.Kadenbach, B., Arnold, S., Lee, I., and
Huttemann, M. (2004) Biochim. Biophys. Acta, 1655,
400-408.
45.Hummerich, H., and Soboll, S. (1989) Biochem.
J., 258, 363-367.
46.Denton, R. M., and McCormac, J. G. (1985) Am.
J. Physiol., 249, E543-E554.
47.Huang, C. J., Geller, H. M., Green, W. L., and
Craeliu, W. (1999) J. Mol. Cell. Cardiol., 31,
881-893.
48.Incerpi, S., De Vito, P., Luly, P., Spagnolo, S.,
and Leoni, S. (2002) Endocrinology, 143, 1660-1668.
49.Ball, S. G., Sokolov, J., and Chin, W. W. (1997)
J. Mol. Endocrinol., 19, 137-147.
50.Horst, C., Harneit, A., Seitz, H. J., and Rokos,
H. (1995) J. Endocrinol., 145, 291-297.
51.Lombardi, A., Beneduce, L., Moreno, M., Diano,
S., Colantuoni, V., Ursini, M. V., Lanni, A., and Goglia, F. (2000)
Endocrinology, 141, 1729-1734.
52.Baur, A., Bauer, K., Jarry, H., and Koherle, J.
(1997) Endocrinology, 138, 3242-3248.
53.Garcia-G, C., Jeziorski, M. C., Valverde-R, C.,
and Orozco, A. (2004) Gen. Comp. Endocrinol., 135,
201-209.
54.Leary, S. C., Barton, K. N., and Ballantyne, J.
S. (1996) Gen. Comp. Endocrinol., 104, 61-66.
55.Varghese, S., Shameena, B., Sudhakaran, P. R.,
and Oommen, O. V. (2001) Indian J. Exp. Biol., 39,
431-435.
56.Varghese, S., Shameena, B., and Oommen, O. V.
(2001) Comp. Biochem. Physiol. B. Biochem. Mol. Biol.,
128, 165-171.
57.Davey, K. G. (2000) Insect Biochem. Mol.
Biol., 30, 877-884.
58.Tata, J. R., Ernster, L., and Lindberg, O. (1963)
Biochem. J., 86, 408-428.