REVIEW: Programmed Cell Death via Mitochondria: Different Modes of Dying
M. Bras, B. Queenan, and S. A. Susin*
Apoptose et Systeme Immunitaire, Institut Pasteur, CNRS-URA 1961, 25 rue du Dr. Roux, 75015 Paris, France; fax: +33-1-4061-3186; E-mail: susin@pasteur.fr* To whom correspondence should be addressed.
Received September 13, 2004
Programmed cell death (PCD) is a major component of normal development, preservation of tissue homeostasis, and elimination of damaged cells. Many studies have subdivided PCD into the three categories of apoptosis, autophagy, and necrosis based on criteria such as morphological alterations, initiating death signal, or the implication of caspases. However, these classifications fail to address the interplay between the three types of PCD. In this review, we will discuss the central role of the mitochondrion in the integration of the cell death pathways. Mitochondrial alterations such as the release of sequestered apoptogenic proteins, loss of transmembrane potential, production of reactive oxygen species (ROS), disruption of the electron transport chain, and decreases in ATP synthesis have been shown to be involved in, and possibly responsible for, the different manifestations of cell death. Thus, the mitochondria can be viewed as a central regulator of the decision between cellular survival and demise.
KEY WORDS: apoptosis, ATP, autophagy, Bcl-2, mitochondria, necrosis-like PCD, ROS
Abbreviations: AIF) apoptosis-inducing factor; BH) Bcl-2 homology domain; HtrA2) high temperature requirement protein A2; OPA1) optic atrophy type 1 protein; Smac/DIABLO) second mitochondria-derived activator of caspase/direct IAP binding protein with low pI; TNF) tumor necrosis factor; TRAIL) TNF-related apoptosis-inducing ligand; XIAP) X-linked inhibitor of apoptosis.
Cell death as an essential aspect of the normal functioning of
multicellular organisms has been recognized to some degree since
antiquity. The fact that some structures were transitory and ultimately
committed to disappear was known since Galen observed the transitional
state of the fetal arterial duct, which allows the direct circulation
of blood from the pulmonary artery to the aorta while bypassing the
fetal lung. The disappearance of this structure at birth is a necessary
physiological development. Obviously, Galen did not directly address
cell death, the concept of the cell not being introduced into the
scientific consciousness until 1839 by Schleiden and Schwann. However,
almost immediately after the identification of the cell came a
description of cell death in Vogt's 1842 observations on amphibian
metamorphosis [1]. Officially recognized in 1871 as
both a pathological and physiological occurrence [2], the phenomenon of cell death was explored for an
entire century before a satisfactory explanation of its function was
forwarded. This may be due in part to focus of early investigations on
the conditions or stimuli resulting in cellular demise. The observation
of cell death during development led to its characterization by certain
investigators as a programmed event [3-6], whereas those investigators addressing this
phenomenon in the context of cellular insult maintained cell death to
be merely a passive response to the damage incurred. Moreover, the
myriad stimuli resulting in cell death--hormone withdrawal,
glucocorticoid treatment, chemical treatment, cellular injury,
irradiation, hypoxia, DNA damage, death receptor binding,
etc.--provided conflicting reports of the extent and means of cell
death.
In 1972, Kerr, Wyllie, and Currie forwarded a theory of cell death that reconciled the two schools of thought. Defining necrosis as a “violent” form of cell death initiated by environmental stimuli and resulting in the rapid disruption of cellular homeostasis, Kerr et al. presented the term “apoptosis” as an alternative, programmed form of cell death [7]. From the Greek term for the “dropping off” of petals or leaves from a healthy plant, apoptosis provided a highly regulated form of cell death complimenting mitosis and cell growth, while allowing for the rapid and less tightly controlled necrotic cell death observed in response to numerous stimuli.
Subsequent work went into the further classification of these types of cell death. Based on morphological alterations observed across tissue types and environmental conditions, these classifications proved more useful than those based on the numerous external stimuli resulting in cellular demise. Although specific works concerning the usage of nuclear phenotypes [8], the fate of the cytoskeleton [9], caspase activation, or alterations to intracellular organelles [10] have provided insights into the mechanisms of cell death, for the purposes of this review we will follow Clarke's classification of programmed cell death (PCD) according to lysosomal involvement [11].
An expansion of Schweichel and Mercer's 1973 lysosomal characterization of cell death, Clarke's model names apoptosis as type I PCD, marked by cell shrinkage, oligonucleosomal DNA fragmentation, chromatin condensation leading to the appearance of pyknotic nuclei, and controlled disintegration of the cell into so-called apoptotic bodies. As in Schweichel and Mercer's system, this form of cell death involves heterophagocytosis, with no apparent involvement of the cell's own lysosomes. Biochemical evidence has indicated the caspase family of cysteine protease as well as certain proteins of the mitochondria to be mediators of type I PCD.
By contrast, type II PCD under both classification systems is marked by the autophagocytosis of cellular organelles. The Greek word autophagy meaning “self-eating”, this form of cell death is characterized primarily by the formation of autophagic vacuoles, as well as by the dilation of the mitochondria and the endoplasmic reticulum (ER) and the slight enlargement of the Golgi. There is some controversy as to the origin of the autophagic vacuoles, with Golgi [12, 13], ER [13], and the inward blebbing of the plasma membrane suggested [11, 13]. Regardless of their origin, these double-membraned autophagic vesicles ultimately fuse with lysosomes where the sequestered cytoplasmic components may be degraded prior to heterophagocytosis of cellular remains.
Type III, or necrosis-like, PCD is characterized by the absence of lysosomal implication and can be subdivided into two categories (III A and III B types). Both are marked by swelling of the intracellular organelles, breakdown of the plasma membrane, and disintegration of the cytoplasm although type III B PCD involves a more moderate destruction of the cytoplasm, in conjunction with dilation of the cisternae of the rough ER and the ability of the dying cell to be heterophagocytosed [11].
Although these classification systems provide a useful tool in the study of cell death, they do not reflect the sophisticated interplay between the forms of PCD. Observed morphologies are quickly identified as one of the three characterized forms of cell death in the hopes of providing immediate therapeutic applications. However, the commendable desire to develop new strategies in the treatment of cancer and neurodegenerative diseases may, in fact, hinder a deeper understanding of cell death as an integration of multiple pathways.
Indeed, biochemical investigations into the mechanisms of cellular demise have provided evidence for the centrality of the mitochondrion in integrating the plethora of cell death signals. Initially considered as the source of cellular energy, the mitochondrion has revealed itself to be a key regulator of the decision between life and death. Jacobsen and Duchen claim the discoveries of the past ten years dictate a new mitochondrial biology [14], encompassed in the principle that “what nourishes me, destroys me.” It hardly seems unreasonable to think that the organelle that provides the energy necessary for cell survival should also play a central role in dictating death. In fact, the mitochondrion has been shown not merely to receive and coordinate cell death signals but to generate them.
MITOCHONDRIAL RELEASE OF APOPTOGENIC PROTEINS
One of the best characterized mechanisms used by mitochondria to induce cell death is the release of pro-apoptotic proteins into the cytosol (Fig. 1, see color insert). Mitochondria are very specialized organelles containing an outer membrane (OM) separated from an inner membrane (IM) by an intermembrane space (IMS) containing many proteins implicated in cell death induction following their release from mitochondria. These IMS proteins include caspase-independent death effectors such as nucleases and/or proteases, as well as caspase activators. In addition, certain pro-apoptotic proteins seem to be sequestered in the mitochondrial cristae [15], the pleiomorphic involutions of the IM that increase the surface area for the electron transport chain.
Cytochrome c, the first molecule shown to be released from mitochondria in the induction of apoptosis, normally functions in energy production but, upon release from mitochondria, complexes with apoptosis protease-activating factor 1 (Apaf-1), dATP, and procaspase-9 to form the apoptosome [16-20]. This high molecular weight complex with seven-fold symmetry can then activate downstream effector caspases, which dictate the apoptotic response [21]. Cytochrome c binds Apaf-1 through the latter molecule's WD-40 repeats, increasing the affinity of Apaf-1 for dATP and inducing a subsequent conformational change that exposes the caspase-recruitment domains (CARD) of Apaf-1. These domains interact with procaspase-9 to generate a holoenzyme capable of activating caspases-3 and -7. A proteolytic cascade is then initiated with the cleavage of procaspases -2, -6, -8, and -10, leading to the dismantling of the cell [22, 23].Fig. 1. Many cell death stimuli converge at mitochondrial level to induce mitochondrial outer membrane permeabilization (MOMP) and subsequent release of pro-apoptotic proteins including AIF, cytochrome c, Endo G, Omi/HtrA2, and Smac/DIABLO. See text for details.
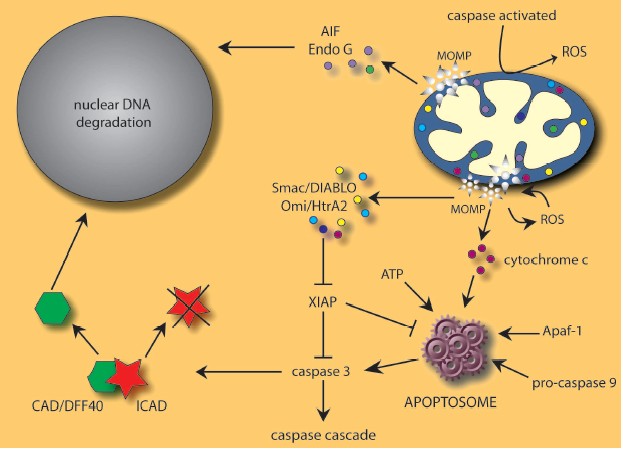
The apoptosome appears to be under the regulation of the inhibitor of apoptosis protein, XIAP, which has been shown to bind the activated forms of caspase-3 and -9, blocking apoptosome-mediated caspase activation [24]. The inhibitory effects of XIAP can be overcome by the antagonizing functions of Smac/DIABLO and Omi/HtrA2, two mitochondrial IMS proteins possessing tetrapeptide IAP binding domains. Released upon apoptotic stimulation, both Smac/DIABLO [25, 26] and Omi/HtrA2 [27-29] antagonize IAP inhibition of caspases, although the latter is also a serine protease that can proteolytically cleave and inactivate IAP proteins. Moreover, recent data has shown that Omi/HtrA2 can induce caspase-independent cell death via its serine protease activity and direct association with cell surface death receptors [30, 31].
In addition to proteins that trigger or enhance caspase activation, proteins such as AIF and endonuclease G are also released from mitochondria and provoke caspase-independent DNA degradation. The flavoprotein AIF translocates from the mitochondria to the nucleus where it is apparently involved in the large-scale DNA fragmentation and peripheral chromatin condensation seen in type I PCD [32, 33]. This activity of AIF, as well as the phosphatidylserine exposure and the disruption of the mitochondrial transmembrane potential, seem to proceed without any intrinsic nuclease activity [32]. The mitochondrial protein endonuclease G (Endo G) appears to work in conjunction with both AIF and the caspase-activated DNase CAD/DFF40 in chromatin condensation and nuclear degradation [34-36]. Although it is unclear to what extent each factor controls large-scale or oligonucleosomal DNA fragmentation, the combined activities of AIF and Endo G as well as CAD provide an efficient means of destroying nuclei under a variety of apoptotic conditions.
HOW DOES IT WORK?
The release of these mitochondrial factors is obviously of critical importance to the progression of cell death. However, the precise molecular mechanism by which apoptogenic proteins are released from mitochondria is still controversial. Different models have been proposed, with many implicating the permeabilization of the OM (Fig. 2, see color insert). The mitochondrial permeability transition pore (PTP), consisting of the ANT protein (Adenine Nucleotide Translocator), the VDAC (Voltage Dependent Anion Channel), the benzodiazepine receptor, and cyclophilin D, is thought to play an important role [37, 38]. Opening of this PTP by various stimuli, including disruption of Ca2+ homeostasis, is thought to result in mitochondrial swelling and rupture of the OM, leading to the nonspecific release of proteins from the IMS [37, 39]. A variation of this model has been proposed upon the demonstration of an interaction of the pro-apoptotic Bcl-2 family member, Bax, with ANT [40] and VDAC [41]. Furthermore, the induction of mitochondrial permeability transition (MPT) has been linked to tBid regulation of the VDAC [42].
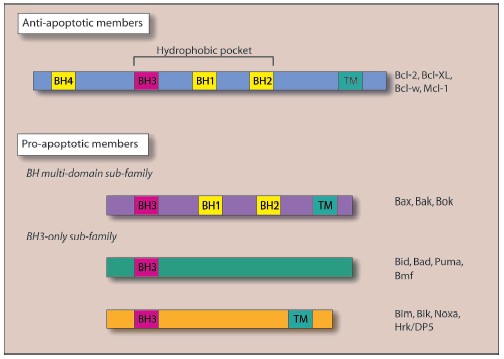
Other Bcl-2 family members have since been shown to have a regulatory function in the release of mitochondrial factors. Figure 3 (see color insert) shows the pro- and anti-apoptotic members of the Bcl-2 family, with the latter group possessing the protective BH4 domain [43-48]. X-Ray analysis of Bcl-XL structure provided a model of the interaction between anti-apoptotic and pro-apoptotic Bcl-2 family proteins: the hydrophobic pocket generated by the alpha-helices of the BH1, BH2, and BH3 Bcl-2 homology domains can interact with the BH3 domain of pro-apoptotic proteins [49, 50]. Sequence homology between Bcl-XL and certain bacterial toxins that function through the formation of membrane channels, (e.g., diphtheria toxin and the colicins [49]) provided the first evidence of the direct regulation of OM permeability by the Bcl-2 protein family [49, 51]. Subsequent investigations into the structure of Bax [52, 53], Bcl-2 [54], and the cleaved form of Bid [55] have revealed similar channel-forming abilities.Fig. 2. The mechanisms responsible for mitochondrial outer membrane permeabilization are still controversial. In fact, many models have been observed: a) The Permeability Transition Pore (PTP). PTP consists of several proteins including Voltage Dependant Anion Channel (VDAC), Adenine Nucleotide Translocator (ANT), cyclophilin D (CypD), and peripheral benzodiazepine receptor (PBR). Release of the apoptotic mitochondrial factors results from PTP opening; b) in a variation of the previous model, pro-apoptotic members of the Bcl-2 family such as Bax interact to VDAC and/or ANT to induce mitochondrial permeabilization; c) release of pro-apoptotic proteins exclusively depends on the balance between pro and anti-apoptotic Bcl-2 family members. In this model, BH3-only proteins (such as Bid, Bim, Puma, and Noxa) play a crucial role in pro-apoptotic Bax and Bak activation.
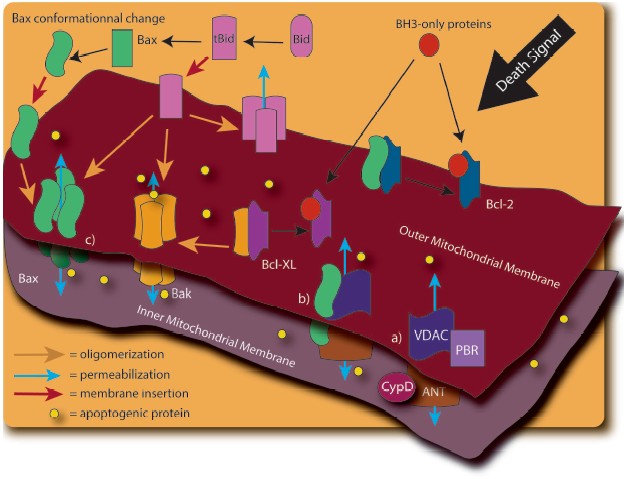
Bax or Bak activation by the cleaved form of the BH3-only protein, Bid (tBid), results in their homo-oligomerization within the OM, with subsequent release of apoptogenic proteins and induction of cell death [56, 57]. These alterations are under the negative regulation of Bcl-2 and Bcl-XL [58-61]. In artificial membranes composed of mitochondrial lipids, the channel-forming ability of Bax under the regulation of tBid induces cytochrome c release from liposomes, an activity dependent on the mitochondrial lipid, cardiolipin, and inhibitable by Bcl-XL [62].Fig. 3. The Bcl-2 family members are potent regulators of cell death and are able to influence the permeability of the mitochondria. According to their Bcl-2 homology domain (BH), they can be subdivided into two categories: anti-apoptotic members and pro-apoptotic members. The BH3-only proteins cannot kill in the absence of Bax and Bak. The BH1, BH2, and BH3 region of the anti-apoptotic and maybe of the BH multi-domain sub-family generate a hydrophobic pocket which can interact with the BH3 domain of the pro-apoptotic proteins. Most of the Bcl-2 family members possess a carboxy-terminal transmembrane domain (TM) implicated in their targeting to intracellular membrane. In Bax and Bcl-w, the C-terminal tail is engaged in the hydrophobic pocket and should fit to allow insertion in the membrane. See Cory et al. for review [115].
An additional level of complexity was recently provided by Douglas Green's laboratory as they demonstrated a new role in cell death induction for the tumor suppressor, p53. p53 is known to activate the transcription of genes such as Noxa, Puma, Bax, Apaf-1, Fas, as well as to repress transcription of Bcl-2 and IAP genes [63-65]. However, independent of its transcriptional control of these genes, p53 has also been shown to engage the apoptotic program by directly activating Bax to permeabilize mitochondria. p53 also facilitated the release of both multi-domain and BH3-only pro-apoptotic proteins previously sequestered by Bcl-XL [66].
The Bcl-2 family appears not merely to regulate mitochondrial permeability but also mitochondrial morphology. In addition to triggering Bax and Bak homo-oligomerization [56, 57], tBid induces a striking remodeling of mitochondrial structure, associated with induction of membrane curvature and mobilization of the cytochrome c stores in the cristae [15]. Although some controversy exists over the mitochondrial localization of cytochrome c, it has been proposed that approximately 85% of mitochondrial cytochrome c is either sequestered in cristae [15] or bound to IM cardiolipin [67, 68].
Mitochondrial remodeling during cell death may therefore represent an interface between the death pathways and the normal regulation of mitochondrial shape and division. Mitochondria are dynamic organelles that exist as a network that often changes shape and subcellular distribution. This interconnected network provides an efficient system for delivering energy between different areas of the cell with its optimal functioning determined by the equilibrium between fission and fusion events [69-71]. Strong evidence suggests that the members of the dynamin family of GTPases are key factors in the regulation of mitochondria morphology. Dynamins are traditionally involved in the scission of a wide range of vesicles and organelles, including clathrin coated vesicles and caveolae [69-71]. Dynamin-related protein 1 (Drp-1), a GTPase that translocates from the cytosol to punctuate foci on the OM [72], seems to control mitochondrial fission, whereas fusion is under the regulation of several proteins including OPA1, which localizes to the IMS tightly bound to the IM [73]. Accumulating evidence implicates the dynamin family proteins in type I PCD induction. Indeed, OPA1 downregulation assays induce mitochondrial fragmentation, cristae remodeling, cytochrome c release, and caspase-dependent apoptosis [74], whereas Drp1 dominant negative mutant overexpression blocks cell death induced by staurosporine [75].
DISRUPTION OF THE ELECTRON TRANSPORT CHAIN AND ROS
PRODUCTION
Although there is some debate concerning its kinetics with respect to cytochrome c release, loss of transmembrane potential is considered a major determinant in the cellular commitment to death. The IM transmembrane potential is often used as an indicator of cellular viability, as the proton gradient across the IM enables the energetically unfavorable production of ATP, the cellular energy source. Thus, disruptions to this transmembrane potential (attributed to MPT of the IM [76]) have severe consequences in mitochondrial respiration, energy production, and, accordingly, cell survival.
The mitochondrial respiratory chain, composed of four complexes coupled to the FoF1-ATPase, functions through the transfer of electrons from the NADH-FADH2 reducing equivalent to molecular oxygen. Electron transport along the respiratory chain generates mitochondrial membrane potential, as protons are pumped out of the matrix across the IM. This electrochemical gradient is essential for ATP synthase activity in the oxidative phosphorylative pathway, as well as for import of mitochondrial proteins and regulation of metabolite transport [77].
A frequent occurrence in the mitochondrial electron transport chain is the escape of an electron, most frequently at complexes I (NADH-ubiquinone oxidoreductase) and III (ubiquinol-cytochrome c oxidoreductase) [78]. The reaction of the renegade electron with molecular oxygen results in the production of an oxygen radical which is normally converted by the cell into hydrogen peroxide or other reactive oxygen species (ROS), including hydroxyl radicals and superoxide anions before elimination [78, 79]. Disproportionate intracellular ROS levels cause significant damage, which may trigger physiological turnover of organelles via autophagy or may result in the complete destruction of the cell via one or more of the PCD pathways. Once again, the mitochondria are particularly involved in the determination of the response to intracellular damage. Incomplete reduction of molecular oxygen during the process of oxidative phosphorylation may induce dramatic cellular damage including lipid peroxidation or DNA damage [80]. ROS production, caused by leakiness of the electron transport chain, has been shown to induce damage to the mitochondrial membrane and a coincidental release of cytochrome c [81]. Interestingly, cytochrome c normally plays a role in the generation of ATP via the electron transport chain. Thus, minor disruptions to mitochondrial respiration may be amplified, resulting in a more rapid induction of cell death. However, type III PCD induced by CD47 ligation is characterized by the loss of the mitochondrial transmembrane potential and ROS generation, but not by the release of cytochrome c [82, 83]. These observations suggest that mitochondrial damage may not be identical across all three forms of PCD and may therefore dictate the cellular response to death stimuli.
Adding an additional level of complexity to the mitochondrial regulation of cell survival is the dual activity of AIF, which both promotes caspase-independent apoptotic DNA fragmentation and maintains normal mitochondrial function in living cells in a manner similar to that of cytochrome c. Structurally, AIF contains an N-terminal FAD-binding domain, a central NADH-binding domain, and a unique C-terminal domain [84, 85]. Furthermore, NADH-oxidase activity of AIF has been demonstrated in vitro, although this activity is seemingly not required for its pro-apoptotic function [86]. Since harlequin mice with an 80% reduction in AIF expression display increased oxidative stress, marked by increased lipid peroxidation and compensatory increases in the activity of catalase and glutathione reductase [87], it has been proposed that AIF may act via its oxyreductase activity as a ROS scavenger under normal conditions [87, 88]. Depending on its localization, AIF could have a dual function: while contained within mitochondria, AIF could act as a ROS scavenger to promote cellular viability whereas, once released into the cytosol, it could enable intracellular ROS accumulation.
Cancer cells undergo increased respiration even under high levels of oxidative stress, an observation partially explained by the frequently hyperpolarized IM of their mitochondria. Both of these findings imply a higher degree of ROS generation and possibly an increased sensitivity to inhibitors of ROS elimination. Huang and coworkers demonstrated an increased sensitivity of leukemic cells to drug-induced apoptosis following treatment with inhibitors of both ROS elimination and complex I of the respiratory chain [89]. Interestingly, such a disruption of the electron transport chain has been linked to direct caspase cleavage of a complex I subunit upon apoptotic stimulation [90], indicating that deliberate impairment of mitochondrial function may be a mechanism of inducing PCD.
MITOCHONDRIAL ATP PRODUCTION
It follows that disruptions to the mitochondrial electron transport chain would result in diminished ATP production and consequently in a striking perturbation of the bioenergetic state of the cell. This disturbance has repercussions on a number of levels, both indicating the necessity of cell death as well as dictating the means to that end [91]. The inhibition of ATP production has been observed in both type I and type III PCD. However, this phenomenon occurs relatively late in type I PCD, as the complete apoptotic program involves the energy-dependent formation of the apoptosome and hydrolysis of macromolecules. By contrast, type III PCD is characterized by an early loss of ATP synthesis and seems to proceed in conditions of low cytosolic ATP levels [37]. Thus ATP seems to play an active role in determining the cell death pathway engaged under various metabolic conditions. Leist et al. found that depletion of cellular ATP levels by an ATP synthase inhibitor, oligomycin, mediated the switch from apoptosis to necrosis in leukemic cells [92]. Cells treated with known apoptosis inducers, staurosporin (STP) and anti-CD95, died exclusively by necrosis when pretreated with oligomycin. Both the characteristic laddering of DNA and exposure of phosphatidylserine on the outer leaflet of the plasma membrane were not observed when cells were depleted of ATP below a critical level, but both apoptotic activities were restored upon glucose addition. Similar results were obtained with inhibitors of complex III of the respiratory chain [93].
As mentioned above, direct caspase cleavage of a complex I subunit of the mitochondrial respiratory chain has been found in the apoptotic pathway [90]. ROS production as well as loss of transmembrane potential and respiratory ability was prevented by expression of a noncleavable subunit of this complex. Thus, damage to the mitochondrial respiratory chain may not be merely a consequence of PCD, but can be specifically induced as a critical component of certain forms of cell death. Of particular interest is the discovery that the pro-apoptotic BH3-only protein, BAD, is an important regulator of glucokinase activity and, accordingly, mitochondrial respiration [94]. The Bcl-2 family of proteins may therefore regulate cell death through control of respiratory function, as well as mitochondrial permeability and morphology.
ATP dependency has been observed for the autophagic type II pathway, seemingly at the lysosomal level [95], perhaps providing a possible determinant in the degree of autophagy observed. Autophagy is the primary means of physiological organelle turnover [96], helping to maintain the balance between protein synthesis and degradation. Furthermore, autophagic PCD has been recently revealed to be a possible defense mechanism against non-lethal cellular insult [13, 97]. However, the signal to proceed from the normal degradation of damaged or aged organelles to the complete autophagic destruction of the cell remains to be determined. Once again, evidence exists for the importance of the mitochondria in this decision, as Beclin-1 and Hspin1--both implicated in the autophagic pathway--interact with Bcl-2 [98, 99]. Furthermore, yeast expression of Bax can induce a form of cell death sharing characteristics of both apoptosis and autophagy [100].
Given the preferential sequestration of mitochondria in autophagic vacuoles and the ATP-dependent nature of type II PCD, it seems plausible that the mitochondria may determine the degree of autophagic degradation. Guimaraes and Linden [101] propose the mitochondrial permeability transition (MPT) to be the critical determinant in the execution of cell death, as the permeabilization of mitochondria has been observed in apoptosis, autophagy, and necrosis. Limited MPT was suggested to result in autophagy, with more extensive permeabilization inducing apoptosis. This milder mitochondrial damage would provide sufficient energy for ATP-dependent autophagic and apoptotic PCD. However, MPT in the vast majority of mitochondria would stimulate a necrotic response [101], produced when the extensive mitochondrial damage resulted in complete uncoupling of oxidative phosphorylation. Such uncoupling was suggested by Lemasters et al. to result in ROS production, as well as uncontrolled hydrolysis of ATP by the inner membrane ATPase [102].
CONCLUSION AND PERSPECTIVES
It becomes increasingly obvious that attempts to sequester observed cell death phenomena into one of three neatly-defined categories may, in fact, hinder a deeper understanding of the cell death machinery. Emerging evidence seems to indicate the interdependence, rather than the autonomy, of the programmed cell death pathways. Although certain authors maintain that apoptosis, autophagy, and necrosis define a continuous spectrum of cell death events [92, 93, 101, 103, 104], it can be argued that the pathways may be simultaneous means to the same end.
Apoptosis and necrosis have been shown to be more tightly linked than once believed, when the two determined the dichotomy of cell death. FAS receptor ligation, which normally induces the classical caspase-mediate apoptotic pathway, induces necrosis in caspase-8 deficient Jurkat cells [105]. The paradigm anti-apoptotic proteins, Bcl-2 and Bcl-XL, have been shown to confer protection against both apoptosis and necrosis [106], while mitochondrial insertion of the BH3-only protein, BNIP3, induces necrosis-like PCD [107]. Necrosis has even been shown to serve as a substitute for caspase-mediate apoptosis during development. [108].
Similarly, the relationship between autophagy and apoptosis has become more complicated. The classic autophagic inhibitor 3-methyladenine (3-MA) sensitizes colon cancer cells to apoptosis [109], implying that the two pathways may complement each other. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) mediates lumen formation in the MCF-10A cell line by simultaneously inducing apoptosis and autophagy [110]. Furthermore, early 3-MA treatment of leukemic cells prevents autophagosome formation, as well as DNA fragmentation and cytolysis in TNF-alpha-dependent apoptosis, whereas late 3-MA treatment does not, suggesting that early stages of autophagy may sometimes be required for apoptosis [111].
One of the most interesting examples of the interplay between the three major cell death pathways is the tamoxifen treatment of MCF-7 cells. Bursch et al. [112] found that high doses of tamoxifen induced a necrotic cell death that was not inhibited by estradiol treatment. However, at lower doses an estradiol-inhibitable autophagic cell death was found to precede the appearance of apoptotic nuclear condensation. Moreover, both autophagic vacuole formation and apoptotic nuclear condensation were prevented by 3-MA treatment [112].
These examples indicate the necessity of further research into the kinetics of cell death activation as well as execution. Besides, the metabolic considerations mentioned above, considerations of rate-dependency of the PCD pathways must be addressed. Is the simultaneous activation of two or more cell death pathways a more efficient route to cellular dismantling? Expression of either (anti-apoptotic) Bcl-XL or a (anti-autophagic) dominant-negative TRAIL receptors delayed lumen formation in MCF-10A cells, although only simultaneous expression prevented cell death [110]. Although this would seem to indicate a synergistic, or at least cooperative, relationship between the two pathways, autophagy has been proposed to delay apoptosis by sequestering the mitochondrial stores of cytochrome c, thus providing a window of opportunity for non-lethally damaged cells to recover [109]. Similar evidence that autophagy can promote cell survival in the face of caspase-mediated apoptosis was presented by Inbal et al. [113]. In cells undergoing caspase-dependent nuclear fragmentation, TNF-receptor 1 induced a caspase-independent autophagy which, when inhibited, increased sensitivity to apoptosis. Are back-up cell death pathways automatically activated but not detected due to a faster execution of the so-called dominant pathway? Marsden et al. suggest the apoptosome to be an amplifier, not an essential component, of the caspase-dependent apoptotic pathway [114]. This would place the formation of the apoptosome, dependent on cytosolic levels of dATP, cytochrome c, Apaf-1, and caspase-9, as the rate-limiting step with slower, alternate pathways only detected in the absence of these apoptosome components.
Thus, investigations into the preferential, as opposed to exclusive, activation of cell death pathways may aid in the understanding of the mechanisms of PCD. Although the interconnected nature of the cell death mechanisms initially seems to frustrate efforts to find therapeutic applications in the treatment of cancer and neurodegenerative diseases, it is exactly this sophisticated interplay that serves as a defense against these debilitating conditions. Moreover, while the centrality of the mitochondria may help illustrate the importance of an integrative approach to cell death research, this approach should be applied to other endeavors. Just as it should not be assumed that cell death proceeds exclusively via one pathway, it should not be assumed that it proceeds exclusively via one organelle.
We thank Cecile Delettre, Rana Moubarak, Nadine Robert, and Victor J. Yuste for stimulating discussions and suggestions. This work was supported by grants from the Association pour la Recherche sur le Cancer (contract No. 4812), Fondation de France, Fondation pour la Recherche Medicale, Ligue contre le Cancer, and the Pasteur-Weizmann Scientific Council (to S. A. S.). M. Bras was supported by a PhD fellowship from MENRT.
REFERENCES
1.Clarke, P. G., and Clarke, S. (1995) Nature,
378, 230.
2.Virchow, R. (1871) Die Cellularpathologie in
ihrer Begrundung auf physiologische Gewebelehre, 4, Auflage.
Hirschwald, Berlin.
3.Glucksmann, A. (1951) Biol. Rev. Cambridge Phil.
Soc., 26, 59-86.
4.Saunders, J. W. (1966) Science, 154,
604-612.
5.Lockshin, R. A., and Williams, C. M. (1964) J.
Insect Physiol., 10, 643-649.
6.Lockshin, R. A., and Williams, C. M. (1965) J.
Insect Physiol., 11, 123-133.
7.Kerr, J. F. R., Wyllie, A. H., and Currie, A. R.
(1972) Br. J. Cancer, 26, 239-257.
8.Jaattela, M. (2004) Oncogene, 23,
2746-2756.
9.Bursch, W., Ellinger, A., Gerner, C. H., Frohwein,
U., and Schulte-Hermann, R. (2000) Ann. NY Acad. Sci.,
926, 1-12.
10.Ferri, K. F., and Kroemer, G. (2001) Nat. Cell
Biol., 3, E255-263.
11.Clarke, P. G. H. (1990) Anat. Embryol.,
181, 195-213.
12.Schweichel, J. U., and Merker, H. J. (1973)
Teratology, 7, 253-266.
13.Ogier-Denis, E., and Codogno, P. (2003)
Biochim. Biophys. Acta, 1603, 113-128.
14.Jacobsen, J., and Duchen, M. R. (2001) Cell
Death Diff., 8, 963-966.
15.Scorrano, L., Ashiya, M., Buttle, K., Weiler, S.,
Oakes, S. A., Mannella, C. A., and Korsmeyer, S. J. (2002) Dev.
Cell., 2, 55-67.
16.Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and
Wang, X. (1996) Cell, 86, 147-157.
17.Li, P., Nijhawan, D., Budihardjo, I.,
Srinivasula, S. M., Ahmad, M., Alnemri, E. S., and Wang, X. (1997)
Cell, 91, 479-489.
18.Zou, H., Henzel, W. J., Liu, X., Lutschg, A., and
Wang, X. (1997) Cell, 90, 405-413.
19.Cain, K., Brown, D. G., Langlais, C., and Cohen,
G. M. (1999) J. Biol. Chem., 274, 22686-22692.
20.Cain, K., Bratton, S. B., Langlais, C., Walker,
G., Brown, D. G., Sun, X. M., and Cohen, G. M. (2000) J. Biol.
Chem., 275, 6067-6070.
21.Acehan, D., Jiang, X., Morgan, D. G., Heuser, J.
E., Wang, X., and Akey, C. W. (2002) Mol. Cell., 9,
423-432.
22.Slee, E. A., Harte, M. T., Kluck, R. M., Wolf, B.
B., Casiano, C. A., Newmeyer, D. D., Wang, H. G., Reed, J. C.,
Nicholson, D. W., Alnemri, E. S., Green, D. R., and Martin, S. J.
(1999) J. Cell Biol., 144, 281-292.
23.Van de Craen, M., Declercq, W., van den brande,
I., Fiers, W., and Vandenabeele, P. (1999) Cell Death Diff.,
6, 1117-1124.
24.Bratton, S. B., Walker, G., Srinivasula, S. M.,
Sun, X. M., Butterworth, M., Alnemri, E. S., and Cohen, G. M. (2001)
EMBO J., 20, 998-1009.
25.Verhagen, A. M., Ekert, P. G., Pakusch, M.,
Silke, J., Connolly, L. M., Reid, G. E., Moritz, R. L., Simpson, R. J.,
and Vaux, D. L. (2000) Cell, 102, 43-53.
26.Du, C., Fang, M., Li, Y., Li, L., and Wang, X.
(2000) Cell, 102, 33-42.
27.Suzuki, Y., Imai, Y., Nakayama, H., Takahashi,
K., Takio, K., and Takahashi, R. (2001) Mol. Cell, 8,
613-621.
28.Verhagen, A. M., Silke, J., Ekert, P. G.,
Pakusch, M., Kaufmann, H., Connolly, L. M., Day, C. L., Tikoo, A.,
Burke, R., Wrobel, C., Moritz, R. L., Simpson, R. J., and Vaux, D. L.
(2002) J. Biol. Chem., 277, 445-454.
29.Van Loo, G., van Gurp, M., Depuydt, B.,
Srinivasula, S. M., Rodriguez, I., Alnemri, E. S., Gevaert, K.,
Vandekerckhove, J., Declercq, W., and Vandenabeele, P. (2002) Cell
Death Differ., 9, 20-26.
30.Blink, E., Maianski, N. A., Alnemri, E. S.,
Zervos, A. S., Roos, D., and Kuijpers, T. W. (2004) Cell Death
Differ., 8, 937-939.
31.Li, W., Srinivasula, S. M., Chai, J., Li, P., Wu,
J. W., Zhang, Z., Alnemri, E. S., and Shi, Y. (2002) Nat. Struct.
Biol., 9, 436-441.
32.Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo,
I., Snow, B. E., Brothers, G. M., Mangion, J., Jacotot, E., Costantini,
P., Loeffler, M., Larochette, N., Goodlett, D. R., Aebersold, R.,
Siderovski, D. P., Penninger, J. M., and Kroemer, G. (1999)
Nature, 397, 441-446.
33.Susin, S. A., Daugas, E., Ravagnan, L., Samejima,
K., Zamzami, N., Loeffler, M., Costantini, P., Ferri, K. F.,
Irinopoulou, T., Prevost, M. C., Brothers, G., Mak, T. W., Penninger,
J., Earnshaw, W. C., and Kroemer, G. (2000) J. Exp. Med.,
192, 571-580.
34.Li, L. Y., Luo, X., and Wang, X. (2001)
Nature, 412, 95-99.
35.Widlak, P., Li, L. Y., Wang, X., and Garrard, W.
T. (2001) J. Biol. Chem., 276, 48404-48409.
36.Enari, M., Sakahira, H., Yokoyama, H., Okawa, K.,
Iwamatsu, A., and Nagata, S. (1998) Nature, 391,
43-50.
37.Kim, J. S., He, L., and Lemasters, J. J. (2003)
Biochem. Biophys. Res. Commun., 304, 463-470.
38.Belzacq, A. S., Vieira, H. L., Kroemer, G., and
Brenner, C. (2002) Biochimie, 84, 167-176.
39.Zamzami, N., Susin, S. A., Marchetti, P., Hirsch,
T., Gomez-Monterrey, I., Castedo, M., and Kroemer, G. (1996)
J. Exp. Med., 183, 1533-1544.
40.Marzo, I., Brenner, C., Zamzami, N.,
Jurgensmeier, J. M., Susin, S. A., Vieira, H. L., Prevost, M. C., Xie,
Z., Matsuyama, S., Reed, J. C., and Kroemer, G. (1998) Science,
281, 2027-2031.
41.Shimizu, S., Narita, M., and Tsujimoto, Y. (1999)
Nature, 399, 483-487.
42.Rostovtseva, T. K., Antonsson, B., Suzuki, M.,
Youle, R. J., Colombini, M., and Bezrukov, S. M. (2004) J. Biol.
Chem., 279, 13575-13583.
43.Borner, C., Martinou, I., Mattmann, C., Irmler,
M., Schaerer, E., Martinou, J. C., and Tschopp, J. (1994) J. Cell
Biol., 126, 1059-1068.
44.Hanada, M., Aime-Sempe, C., Sato, T., and Reed,
J. C. (1995) J. Biol. Chem., 270, 11962-11969.
45.Hunter, J. J., Bond, B. L., and Parslow, T. G.
(1996) Mol. Cell. Biol., 16, 877-883.
46.Huang, D. C., Adams, J. M., and Cory, S. (1998)
EMBO J., 17, 1029-1039.
47.Shimizu, S., Konishi, A., Kodama, T., and
Tsujimoto, Y. (2000) Proc. Natl. Acad. Sci. USA, 97,
3100-3105.
48.Shimizu, S., Ide, T., Yanagida, T., and
Tsujimoto, Y. (2000) J. Biol. Chem., 275,
12321-12325.
49.Muchmore, S. W., Sattler, M., Liang, H., Meadows,
R. P., Harlan, J. E., Yoon, H. S., Nettesheim, D., Chang, B. S.,
Thompson, C. B., Wong, S. L., Ng, S. L., and Fesik, S. W. (1996)
Nature, 381, 335-341.
50.Sattler, M., Liang, H., Nettesheim, D., Meadows,
R. P., Harlan, J. E., Eberstadt, M., Yoon, H. S., Shuker, S. B., Chang,
B. S., Minn, A. J., Thompson, C. B., and Fesik, S. W. (1997)
Science, 275, 983-986.
51.Minn, A. J., Velez, P., Schendel, S. L., Liang,
H., Muchmore, S. W., Fesik, S. W., Fill, M., and Thompson, C. B. (1997)
Nature, 385, 353-357.
52.Antonsson, B., Conti, F., Ciavatta, A.,
Montessuit, S., Lewis, S., Martinou, I., Bernasconi, L., Bernard, A.,
Mermod, J. J., Mazzei, G., Maundrell, K., Gambale, F., Sadoul, R., and
Martinou, J. C. (1997) Science, 277, 370-372.
53.Schlesinger, P. H., Gross, A., Yin, X. M.,
Yamamoto, K., Saito, M., Waksman, G., and Korsmeyer, S. J. (1997)
Proc. Natl. Acad. Sci. USA, 94, 11357-11362.
54.Schendel, S. L., Xie, Z., Montal, M. O.,
Matsuyama, S., Montal, M., and Reed, J. C. (1997) Proc. Natl. Acad.
Sci. USA, 94, 5113-5118.
55.Schendel, S. L., Azimov, R., Pawlowski, K.,
Godzik, A., Kagan, B. L., and Reed, J. C. (1999) J. Biol. Chem.,
274, 21932-21936.
56.Eskes, R., Desagher, S., Antonsson, B., and
Martinou, J. C. (2000) Mol. Cell Biol., 20, 929-935.
57.Wei, M. C., Lindsten, T., Mootha, V. K., Weiler,
S., Gross, A., Ashiya, M., Thompson, C. B., and Korsmeyer, S. J. (2000)
Genes Dev., 14, 2060-2071.
58.Korsmeyer, S. J. (1999) Cancer Res.,
59, 1693s-1700s.
59.Scorrano, L., and Korsmeyer, S. J. (2003)
Biochem. Biophys. Res. Commun., 304, 437-444.
60.Murphy, K. M., Streips, U. N., and Lock, R. B.
(2000) J. Biol. Chem., 275, 17225-17228.
61.Mikhailov, V., Mikhailova, M., Pulkrabek, D. J.,
Dong, Z., Venkatachalam, M. A., and Saikumar, P. (2001) J. Biol.
Chem., 276, 18361-18374.
62.Kuwana, T., Mackey, M. R., Perkins, G., Ellisman,
M. H., Latterich, M., Schneiter, R., Green, D. R., and Newmeyer, D. D.
(2002) Cell, 111, 331-342.
63.May, P., and May, E. (1999) Oncogene,
18, 7621-7636.
64.Bourdon, J. C., Laurenzi, V. D., Melino, G., and
Lane, D. (2003) Cell Death Differ., 10, 397-399.
65.Schuler, M., and Green, D. R. (2001) Biochem.
Soc. Trans., 29, 684-688.
66.Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L.,
Droin, N. M., Newmeyer, D. D., Schuler, M., and Green, D. R. (2004)
Science, 303, 1010-1014.
67.Petrosillo, G., Ruggiero, F. M., and Paradies, G.
(2003) FASEB J., 17, 2202-2208.
68.Ott, M., Robertson, J. D., Gogvadze, V.,
Zhivotovsky, B., and Orrenius, S. (2002) Proc. Natl. Acad. Sci.
USA, 99, 1259-1263.
69.Karbowski, M., and Youle, R. J. (2003) Cell
Death Differ., 10, 870-880.
70.Bossy-Wetzel, E., Barsoum, M. J., Godzik, A.,
Schwarzenbacher, R., and Lipton, S. A. (2003) Curr. Opin. Cell
Biol., 15, 706-716.
71.Praefcke, G. J., and McMahon, H. T. (2004)
Nat. Rev. Mol. Cell Biol., 5, 133-147.
72.Smirnova, E., Griparic, L., Shurland, D. L., and
van der Bliek, A. M. (2001) Mol. Biol. Cell, 12,
2245-2256.
73.Griparic, L., van der Wel, N. N., Orozco, I. J.,
Peters, P. J., and van der Bliek, A. M. (2004) J. Biol. Chem.,
279, 18792-18798.
74.Olichon, A., Baricault, L., Gas, N., Guillou, E.,
Valette, A., Belenguer, P., and Lenaers, G. (2003) J. Biol.
Chem., 278, 7743-7746.
75.Frank, S., Gaume, B., Bergmann-Leitner, E. S.,
Leitner, W. W., Robert, E. G., Catez, F., Smith, C. L., and Youle, R.
J. (2001) Dev. Cell., 1, 515-525.
76.Hirsch, T., Susin, S. A., Marzo, I., Marchetti,
P., Zamzami, N., and Kroemer, G. (1998) Cell. Biol. Toxicol.,
14, 141-145.
77.Ricci, J. E., Waterhouse, N., and Green, D. R.
(2003) Cell Death Differ., 10, 488-492.
78.Fleury, C., Mignotte, B., and Vayssiere, J. L.
(2002) Biochimie, 842, 131-141.
79.Saybasili, H., Yuksel, M., Haklar, G., and
Yalcin, A. S. (2001) Antioxid. Redox Signal., 3,
1099-1104.
80.Bergamini, C. M., Gambetti, S., Dondi, A., and
Cervellati, C. (2004) Curr. Pharm. Des., 10,
1611-1626.
81.Huang, P., Feng, L., Oldham, E. A., Keating, M.
J., and Plunkett, W. (2000) Nature, 407, 390-395.
82.Roue, G., Bitton, N., Yuste, V. J., Montange, T.,
Rubio, M., Dessauge, F., Delettre, C., Merle-Beral, H., Sarfati, M.,
and Susin, S. A. (2003) Biochimie, 85, 741-746.
83.Manna, P. P., and Frazier, W. A. (2004) Cancer
Res., 64, 1026-1036.
84.Mate, M. J., Ortiz-Lombardia, M., Boitel, B.,
Haouz, A., Tello, D., Susin, S. A., Penninger, J., Kroemer, G., and
Alzari, P. M. (2002) Nat. Struct. Biol., 9, 442-446.
85.Ye, H., Cande, C., Stephanou, N. C., Jiang, S.,
Gurbuxani, S., Larochette, N., Daugas, E., Garrido, C., Kroemer, G.,
and Wu, H. (2002) Nat. Struct. Biol., 9, 680-684.
86.Miramar, M. D., Costantini, P., Ravagnan, L.,
Saraiva, L. M., Haouzi, D., Brothers, G., Penninger, J. M., Peleato, M.
L., Kroemer, G., and Susin, S. A. (2001) J. Biol. Chem.,
276, 16391-16398.
87.Klein, J. A., Longo-Guess, C. M., Rossmann, M.
P., Seburn, K. L., Hurd, R. E., Frankel, W. N., Bronson, R. T., and
Ackerman, S. L. (2002) Nature, 419, 367-374.
88.Lipton, S. A., and Bossy-Wetzel, E. (2002)
Cell, 111, 147-150.
89.Pelicano, H., Feng, L., Zhou, Y., Carew, J. S.,
Hileman, E. O., Plunkett, W., Keating, M. J., and Huang, P. (2003)
J. Biol. Chem., 278, 37832-37839.
90.Ricci, J. E., Munoz-Pinedo, C., Fitzgerald, P.,
Bailly-Maitre, B., Perkins, G. A., Yadava, N., Scheffler, I. E.,
Ellisman, M. H., and Green, D. R. (2004) Cell, 117,
773-786.
91.Izyumov, D. S., Avetisyan, A. V., Pletjushkina,
O. Y., Sakharov, D. V., Wirtz, K. W., Chernyak, B. V., and Skulachev,
V. P. (2004) Biochim. Biophys. Acta, 1658, 141-147.
92.Leist, M., Single, B., Castoldi, A. F., Kuhnle,
S., and Nicotera, P. (1997) J. Exp. Med., 185,
1481-1486.
93.Formigli, L., Papucci, L., Tani, A., Schiavone,
N., Tempestini, A., Orlandini, G. E., Capaccioli, S., and Orlandini, S.
Z. (2000) J. Cell. Phys., 182, 41-49.
94.Danial, N. N., Gramm, C. F., Scorrano, L., Zhang,
C. Y., Krauss, S., Ranger, A. M., Datta, S. R., Greenberg, M. E.,
Licklider, L. J., Lowell, B. B., Gygi, S. P., and Korsmeyer, S. J.
(2003) Nature, 424, 952-956.
95.Plomp, P. J., Gordon, P. B., Meijer, A. J.,
Hoyvik, H., and Seglen, P. O. (1989) J. Biol. Chem., 264,
6699-6704.
96.Seglen, P. O., and Bohley, P. (1992)
Experientia, 48, 158-172.
97.Paglin, S., Hollister, T., Delohery, T., Hackett,
N., McMahill, M., Sphicas, E., Domingo, D., and Yahalom, J. (2001)
Cancer Res., 61, 439-444.
98.Liang, X. H., Jackson, S., Seaman, M., Brown, K.,
Kempkes, B., Hibshoosh, H., and Levine, B. (1999) Nature,
402, 672-676.
99.Yanagisawa, H., Miyashita, T., Nakano, Y., and
Yamamoto, D. (2003) Cell Death Differ., 10, 798-807.
100.Priault, M., Camougrand, N., Kinnally, K. W.,
Vallette, F. M., and Manon, S. (2003) FEMS Yeast Res., 4,
15-27.
101.Guimaraes, C. A., and Linden, R. (2004) Eur.
J. Biochem., 271, 1638-1650.
102.Lemasters, J. J., Nieminen, A. L., Qian, T.,
Trost, L. C., Elmore, S. P., Nishimura, Y., Crowe, R. A., Cascio, W.
E., Bradham, C. A., Brenner, D. A., and Herman, B. (1998) Biochim.
Biophys. Acta, 1366, 177-196.
103.Burlacu, A., Jinga, V., Gafencu, A. V., and
Simionescu, M. (2001) Cell Tissue Res., 306, 409-416.
104.Seoane, A., Dememes, D., and Llorens, J. (2001)
J. Comp. Neurol., 439, 385-399.
105.Kawahara, A., Ohsawa, Y., Matsumura, H.,
Uchiyama, Y., and Nagata, S. (1998) J. Cell Biol.,
143, 1353-1360.
106.Shimizu, S., Eguchi, Y., Kamiike, W., Waguri,
S., Uchiyama, Y., Matsuda, H., and Tsujimoto, Y. (1996)
Oncogene, 12, 2045-2050.
107.Vande Velde, C., Cizeau, J., Dubik, D.,
Alimonti, J., Brown, T., Israels, S., Hakem, R., and Greenberg, A. H.
(2000) Mol. Cell. Biol., 20, 5454-5468.
108.Oppenheim, R. W., Flavell, R. A., Vinsant, S.,
Prevette, D., Kuan, C. Y., and Rakic, P. (2001) J. Neurosci.,
21, 4752-4760.
109.Bauvy, C., Gane, P., Arico, S., Codogno, P.,
and Ogier-Denis, E. (2001) Exp. Cell Res., 268,
139-149.
110.Mills, K. R., Reginato, M., Debnath, J.,
Queenan, B., and Brugge, J. S. (2004) Proc. Natl. Acad. Sci.
USA, 101, 3438-3443.
111.Jia, L., Dourmashkin, R. R., Allen, P. D.,
Gray, A. B., Newland, A. C., and Kelsey, S. M. (1997) Br. J.
Haematol., 98, 673-685.
112.Bursch, W., Ellinger, A., Kienzl, H., Torok,
L., Pandey, S., Sikorska, M., Walker, R., and Hermann, R. S. (1996)
Carcinogenesis, 17, 1595-1607.
113.Inbal, B., Bialik, S., Sabanay, I., Shani, G.,
and Kimchi, A. (2002) J. Cell Biol., 157, 455-468.
114.Marsden, V. S., O'Connor, L., O'Reilly, L. A.,
Silke, J., Metcalf, D., Ekert, P. G., Huang, D. C., Cecconi, F., Kuida,
K., Tomaselli, K. J., Roy, S., Nicholson, D. W., Vaux, D. L., Bouillet,
P., Adams, J. M., and Strasser, A. (2002) Nature, 419,
634-637.
115.Cory, S., Huang, D. C., and Adams, J. M. (2003)
Oncogene, 22, 8590-8607.