REVIEW: Bioenergetics and Death
B. V. Chernyak*, O. Yu. Pletjushkina, D. S. Izyumov, K. G. Lyamzaev, and A. V. Avetisyan
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (7-095) 939-3181; E-mail: bchernyak@yahoo.com* To whom correspondence should be addressed.
Received September 20, 2004
Specific inhibitors of mitochondrial functions were used in studies on the relation between bioenergetics and programmed cell death. The data of the authors are discussed in the review.
KEY WORDS: mitochondria, respiration, membrane potential, ATP synthesis, apoptosis, necrosis
Abbreviations: ROS) reactive oxygen species; TNF) tumor necrosis factor; DNP) 2,4-dinitrophenol; DOG) 2-deoxyglucose; FCCP) carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone.
In the past decade, two revolutions have dramatically changed our view
on mitochondria. The clear known-from-the-text-books description of
“power plants” producing energy (ATP) was displaced with the
mysterious image of “Pandora's box” determining the fate of
cell. In the previous paradigm, involvement of mitochondria in
pathology was limited to impairment of cellular energetics in genetic
diseases and hypoxic and toxic insults. In the new one, the major role
of byproducts, such as reactive oxygen species (ROS), and side
reactions such as permeability transition were appreciated. The recent
explosion of experimental works demonstrated that various cases of
apoptosis critically depend on the release of specific mitochondrial
proteins into the cytosol. The most important usually is a release of
cytochrome c (one of the components of the respiratory chain)
catalyzing assembly of a large cytosolic complex “apoptosome”
involved in activation of caspases, the major executioners of cell
death. In contrast to the basic bioenergetic principles, the mechanisms
of sensing of apoptotic signals by mitochondria are not well
understood. Mitochondria receive the signals from specific receptors or
from other cellular organelles subjected to physical or toxic insults
(nucleus, reticulum, cytoskeleton) and integrate and transform them to
a unified signal for cell death. The principal mechanisms of these
processes and their connection to bioenergetics of mitochondria are not
well understood.
Almost simultaneously the second revolution overturned the traditional view of the mitochondrion as a small round-shaped organelle. It appeared that the well-known images obtained under the electron microscope were the slices of a variable and dynamic mitochondrial network. These structures were described in detail by L. E. Bakeeva et al. in the laboratory of V. P. Skulachev and received the name of “mitochondrial reticulum” [1]. Skulachev suggested that these cable-like structures were an energy (trans-membrane electric potential) transporting system of the cell [2]. This hypothesis received a strong support in experiments of D. B. Zorov and coworkers where the electrical unity of significant parts of mitochondrial reticulum was demonstrated [3]. Later Skulachev suggested that damage to mitochondria would induce disconnection of fragments of mitochondrial cables to prevent short circuit (uncoupling) in the whole network [4]. This effect could be accompanied with physical fragmentation of the mitochondrial reticulum. In agreement with this idea, fragmentation of mitochondria was observed under various stressful conditions and during apoptosis induced by various stimuli. The interest in this phenomenon strongly grew when it was found that inhibition of fragmentation of mitochondria significantly slowed down the later apoptotic events. The molecular mechanisms of mitochondrial fragmentation were studied for a long time in yeast as a model of organelle division. The major proteins involved in dynamics of mitochondria in yeast were identified but the search for mammalian homologs and determination of their role in apoptosis needs further studies. Relations between bioenergetics of mitochondria and their structural dynamics are of particular interest.
The change in the paradigm of mitochondrial structure and function in the cell coincided with a change in the basic methods in this field. The bioenergetics of mitochondria was investigated using a wonderful arsenal of specific inhibitors. Inhibitors (usually antibiotics) of almost every component of oxidative phosphorylation were found and the mechanisms of inhibition were deciphered. Later studies on chemiosmotic mechanisms appeared possible due to discoveries of different ionophores and membrane-permeable indicators. The final evidences of Mitchellian chemiosmotic principals were presented by V. P. Skulachev in collaboration with E. A. Liberman [5] when they used positive and negative permeable ions for determination of polarity and value of the membrane potential in different models. Later these approaches were employed for development of potential sensitive dyes, measurements of mitochondrial membrane potential in living cells and tissues, and targeting of some reagents (antioxidants, for example) to mitochondria (see the review of Ross et al. in this issue). The new era in mitochondriology coincided with the great successes in genetic engineering and genomics. Hundreds of new proteins involved in mitochondria-related signaling were discovered in a very short time. The studies of their functions were based on powerful approaches using knockout, siRNA, or dominant-negative constructs. For many reasons, these approaches are very difficult to apply to mitochondrial proteins involved in bioenergetic functioning. Even the rare successful attempts, such as knockout of cytochrome c [6], did not help to fill the gap between two areas of research on mitochondria in energy transformation and in cell signaling.
Several years ago with strong support from V. P. Skulachev, we initiated studies on relations between bioenergetics of mitochondria and their role in cell signaling [7-10]. The major tools in these studies were selective inhibitors of mitochondrial functions. Rapidly growing human carcinoma cells HeLa were used as a basic cellular model. These cells have high rate of aerobic glycolysis and high capacity for oxidative phosphorylation, so they were suitable for use of mitochondrial inhibitors.
1. EFFICACY OF THE MITOCHONDRIAL INHIBITORS
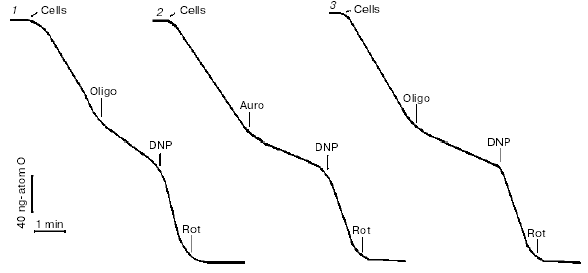
HeLa cells have high respiratory activity, so this assay was used for titration of the effects of the inhibitors. Respiration was strongly suppressed with the inhibitors of mitochondrial FoF1-ATP-synthase indicating high input of coupled respiration (Fig. 1). Maximal respiration rate was induced by uncouplers of oxidative phosphorylation at concentrations of the agents significantly lower than necessary for complete uncoupling. In other words, when respiration was maximally stimulated with uncoupler, ATP synthesis was not completely blocked and steady-state membrane potential was high. At higher concentrations of uncoupler, the membrane potential dropped and ATP synthesis ceased, but simultaneously respiration was inhibited. This effect was related to inhibition of electron transfer in several segments of the respiratory chain and was inevitable for almost all protonophorous uncouplers. In experiments with HeLa cells only one exception was found, a new uncoupler designed by Novo Nordisk (Denmark) (No. 0112-0005, the structure cannot be reported) maximally stimulated respiration at 0.1-0.2 µM (the same concentrations as carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP)) and did not inhibit respiration up to 10-20 µM (at these concentrations FCCP almost completely blocked respiration). One more “special” uncoupler, 2,4-dinitrophenol (DNP), maximally stimulated respiration at 0.1-0.2 mM but did not completely dissipated the membrane potential even at 10-20 time higher concentration due to low solubility in lipids. Inhibitors of the various segments of the respiratory chain (rotenone, antimycin A, myxothiazol, cyanide) completely blocked respiration of HeLa cells. The complete effect of rotenone (an inhibitor of Complex I) (Fig. 1) indicated that cessation of NADH oxidation in the chain prevented formation of succinate in the Krebs cycle and its oxidation by Complexes II-IV. In the absence of rotenone, the contribution of succinate oxidation to total oxidative capacity could be significant.
The inhibitors of ATP synthase (oligomycin, aurovertin), inhibitors of respiration (piericidin, antimycin, myxothiazol), and uncouplers (DNP, FCCP) did not cause any loss in viability of HeLa cells during 24-48 h in the traditional cell culture medium DMEM supplemented with fetal serum (10%) and glucose (25 mM) [7]. High rate of glycolysis completely satisfied energy demands of the cells. This means that inhibitors of oxidative phosphorylation could be safely used in studies of apoptosis if they were completed in 1-2 days. An important exception was found in experiments with rotenone. This classic inhibitor of Complex I induced cell cycle arrest and following apoptosis in HeLa cells at 2 µM concentration, which was necessary for complete inhibition of uncoupled respiration of these cells in the culture medium. The effect of rotenone was not related to inhibition of respiration or specific effect on Complex I, since another inhibitor with the identical specificity namely piericidin did not affect cell cycle or kill the cells. The effect of rotenone is probably targeted on the cytoskeleton, as reported 30 years ago [11].Fig. 1. Test for efficacy of the mitochondrial inhibitors in HeLa (1, 2) and HeLa-Bcl-2 (3) cells. Respiration of the cell suspension was measured polarographically using a Clark-type electrode in the culture medium. Inhibitors were added in the following concentrations: oligomycin (Oligo), 5 µg/ml; aurovertin (Auro), 1 µM; 2,4-dinitrophenol (DNP), 0.2 mM; rotenone (Rot), 4 µM.
The possible effects of mitochondrial inhibitors on apoptosis were studied in two models with different signaling pathways leading to cell death. In the first, apoptosis was induced by a cytokine, tumor necrosis factor (TNF), upon binding to specific receptors at the cell surface. In the second, a general inhibitor of protein kinases staurosporine (STS) was used to induce stress-like apoptosis. In the early steps, initiation and transmission of apoptotic signal were different in these models; however, in both cases the release of cytochrome c from mitochondria into cytosol was a critical event and a “point-of-no-return” in the program. The final execution steps were practically identical. During TNF-induced apoptosis, mitochondria were attacked by protein Bid proteolytically activated in cytosol [12]. Staurosporine induced a different signaling pathway where a major component targeted on mitochondria, Bax, is activated by unknown mechanisms [13]. Our experiments demonstrated that in both models the inhibitors of respiration and uncouplers did not affect the release of cytochrome c into cytosol and the following apoptotic events. The important anti-apoptotic mitochondria-located protein Bcl-2, which inhibits the release of cytochrome c [14], also remained fully effective in the presence of the inhibitors of respiration and uncouplers [7, 8]. These data indicated that the release of cytochrome c and some other proteins from intermembrane space during apoptosis is hardly mediated by hyperpolarization of the inner membrane (as suggested [15, 16]) or by any potential-dependent processes, such as Ca2+ accumulation in mitochondria.
These studies revealed an unexpected effect of oligomycin. This inhibitor of the proton channel (Fo) in mitochondrial FoF1-ATPase was found to inhibit TNF-induced release of cytochrome c and apoptosis [8]. STS-induced apoptosis in HeLa cells was not affected by oligomycin. The effect of oligomycin was not related to inhibition of oxidative phosphorylation or to hyperpolarization of the membrane since depolarization with uncouplers did not relieve the inhibition. Moreover, the effect was not directly linked to inhibition of ATPase since another specific inhibitor, aurovertin B, did not affect release of cytochrome c and apoptosis. In contrast to oligomycin, this inhibitor was targeted to the catalytic (F1) component of the enzyme. The well-known non-mitochondrial target of oligomycin, Na+/K+-ATPase of the plasma membrane, was not responsible for the effects described. It was shown that a selective inhibitor of this enzyme, ouabain, did not inhibit apoptosis.
Interestingly, the selective effect of oligomycin on TNF-induced apoptosis correlated with the effect of cyclosporin A, the inhibitor of the permeability transition pore (PTP). This agent (in combination with trifluoperazine, which enhance the effect) inhibited TNF-induced release of cytochrome c and apoptosis while it did not affect STS-induced apoptosis in HeLa cells. A possible role of the PTP in apoptosis has been discussed in a great number of contradictory works. It seems clear that the PTP opening can results in release of cytochrome c due to osmotic swelling of matrix and disruption of the outer mitochondrial membrane. However, in some models of apoptosis this mechanism is not enough for release of cytochrome c, while in some models its role seems improbable. It can be suggested that oligomycin inhibited the concerned action of Fo and PTP in release of cytochrome c from intermembrane space of mitochondria during TNF-induced apoptosis.
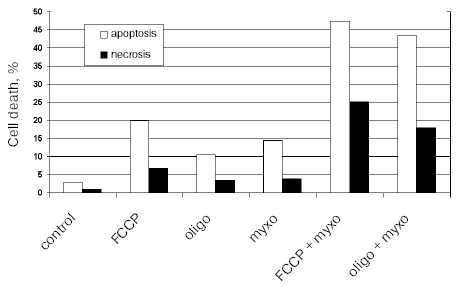
The effective inhibition of oxidative phosphorylation with the inhibitors in use did not induce apoptosis in HeLa cells probably due to general high resistance of these cells to apoptotic stimuli. The anti-apoptotic defense strongly depends on growth factors in the serum. To reveal possible specific induction of apoptosis with the mitochondrial inhibitors we have excluded serum from the culture medium. Apoptosis was increased only slightly during 24 h and more prolonged incubation resulted in significant necrosis. Significant apoptosis was observed when a respiratory inhibitor (myxothiazol) was combined with uncoupler (FCCP) or with oligomycin (Fig. 2). This effect was probably caused by excessive production of reactive oxygen species (ROS) in the initial segments of the respiratory chain. In fact, myxothiazol caused overreduction of Complex I components and catalyzed one electron reduction of oxygen. Uncoupler or oligomycin under these conditions caused dissipation of the membrane potential further stimulating ROS production and apoptosis. ROS generation by Complex I was suggested earlier to be important for cell death in various pathologies including ischemia/reperfusion [17], Parkinson disease, etc. [18] (see also the review by Starkov et al. in this issue).
Fig. 2. Cell death induced by mitochondrial inhibitors in HeLa cells. Cells were incubated for 24 h in culture medium without serum. Inhibitors were added in the following concentrations: FCCP, 10 µM; oligomycin (oligo), 5 µg/ml; myxothiazol (myxo), 2 µM. Apoptosis was determined by chromatin condensation after staining with Hoechst 33342, and necrosis by staining of nuclei with propidium iodide.
3. PROGRAMMED CELL DEATH INDUCED BY ENERGY DEPRIVATION
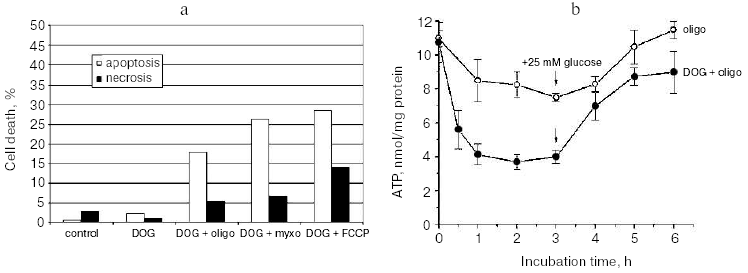
The high concentration of glucose used in the cell culture medium was significantly higher than the physiological level. Presumably, the limited supply of the glycolytic substrates is important for various pathological states and especially for development of rapidly growing solid tumors. To model these conditions we have decreased concentration of glucose to 5 mM and supplied the medium with 5 mM 2-deoxyglucose (DOG), a non-metabolized analog. These conditions did not cause any decrease in viability of HeLa cells indicating the high capacity of oxidative phosphorylation. As expected, mitochondrial inhibitors caused almost complete necrotic cell death during 24 h in this model. However, if the combined treatment with DOG and mitochondrial inhibitors for 3 h was followed by 24 h recovery in the high-glucose medium (without DOG) significant apoptotic cell death was observed (Fig. 3a). A 48-h cultivation resulted in almost complete cell death with strong prevalence of apoptosis over necrosis. In the described model cell death did not significantly depend on the nature of the mitochondrial inhibitor, indicating that apoptosis was induced by the temporary energy deprivation. The content of ATP dropped to 30-40% of the initial level in 0.5 h and remained constant for the next 3-5 h. Removal of DOG caused rapid restoration of ATP level even when the mitochondrial inhibitors were present (Fig. 3b) [9].
After 3-fold decrease in cellular ATP level, it still remained in a range of 1-3 mM which is much higher (10-100 times) than the values of Michaelis constants for the majority of ATP-consuming enzymes. Even in combination with accompanying increase in ADP and AMP level, this decrease in ATP hardly caused significant damage to the cellular structures and irreversible block of the critical systems of homeostasis. It seems more probable that specific ATP-meter(s) detected temporary limited ATP depletion and transformed this signal into signal for apoptosis. Tuning of these sensors should be very precise. When the ATP depletion procedure was prolonged to 5 h the following recovery still restored the ATP level but resulted mostly in necrotic cell death after 24-48 h. A similar switch to necrosis was observed when ATP depletion was increased by more complete inhibition of glycolysis. The mitochondrial inhibitors in combination with DOG added to the medium depleted of glucose caused rapid fall in cellular ATP to less then 10% of the initial level. After 3 h, the medium was changed to complete glucose-rich DMEM and cellular ATP was restored almost completely in 0.5 h, but the following 48 h cultivation resulted in massive necrotic death. Thus temporary ATP depletion can be a trigger of programmed cell death with both necrotic and apoptotic features.Fig. 3. a) HeLa cell death induced by temporary ATP depletion. Cells were incubated for 3 h in low glucose (5 mM) culture medium with 2-deoxyglucose (DOG, 5 mM) and with mitochondrial inhibitors (concentrations as in Figs. 1 and 2). Than medium was changed to the full (25 mM glucose) culture medium with the same inhibitors but without DOG, and cells were incubated for 21 h more. Apoptosis and necrosis were determined as in Fig. 2. b) ATP was determined in the cells treated as described in Fig. 3a. ATP was extracted and measured using luciferase reagent (Pharmacia, Sweden) according to the manufacturer's instructions.
Apoptosis induced by temporary ATP depletion had all the signs of stress-induced apoptosis. At the early steps translocation of Bax from cytosol to mitochondria, formation of large Bax-containing aggregates, release of cytochrome c, and activation of caspases was observed. Overexpression of Bcl-2 inhibited release of cytochrome c and apoptosis but did not affect necrosis, indicating low input of “secondary necrosis” in these models. Apoptosis was prevented by inhibition of caspases with zVADfmk and simultaneously necrosis was stimulated, suggesting that caspases not only catalyzed apoptosis but also inhibited necrosis induced by energy deprivation. In contrast to apoptosis, the signaling resulting in necrosis remained poorly characterized. An example of necrosis induced by rotenone and glucose deprivation in myogenic cells was recently found to be dependent on activation of the stress-activated protein kinase (JNK) [19]. In HeLa cells, neither apoptosis nor necrosis was sensitive to inhibitors of JNK or p38 (another stress-activated protein kinase). The nature of putative ATP-meter(s) also remained mysterious. The candidate sensors includes mTOR, a protein kinase with exceptionally high Km(ATP) and AMP-activated protein kinase (AMPK) which is allosterically activated by AMP. The both kinases are involved in regulation of gene expression in response to nutrient starvation [20, 21] but their role in induction of apoptosis is purely speculative. Interestingly, activation of AMPK upon inhibition of mitochondrial ATP synthesis appeared to be significantly stronger than in mitochondria-independent models even at the same increase in AMP [22]. Probably this overstimulation of AMPK caused induction of apoptosis in our model of energy deprivation.
4. INHIBITION OF BIOENERGETIC FUNCTIONS CAUSES MORPHOLOGICAL
CHANGES AND DEGRADATION OF MITOCHONDRIA
Mitochondrial inhibitors in the presence of glucose induced dramatic changes in mitochondrial structure independently from ATP depletion or apoptotic events [10, 23]. Similar changes were observed in a great number of works and one of the first description was made by D. B. Zorov in V. P. Skulachev's laboratory (see the review by Zorov et al. in this issue). In HeLa cells, the inhibitors of respiration and uncouplers induced fragmentation of mitochondrial reticulum after a significant lag-phase (3-5 h). Later the fragments were transformed to small round bodies due to further fragmentation and changes in the form of the organelles. The following swelling of the mitochondria was clearly visible in the presence of uncouplers. Washout of uncoupler caused slow (24 h) restoration of mitochondrial reticulum, and the process was blocked by the inhibitor of protein synthesis emetine.
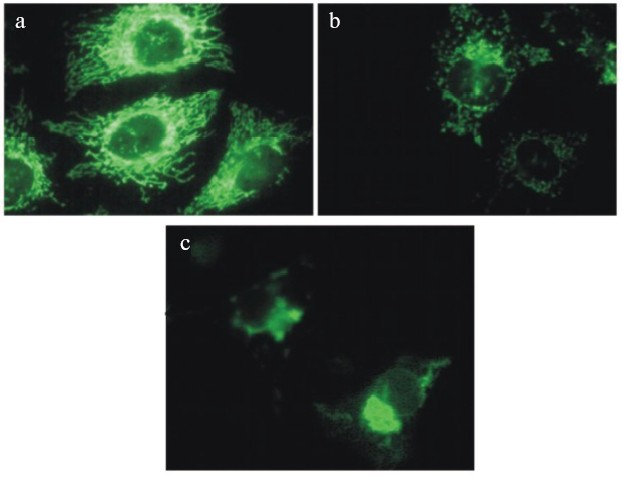
The most rapid fission and the following transitions of mitochondrial network were induced by combined treatment with respiratory inhibitors and uncouplers (see color insert, Fig. 4) indicating the possible role of ROS produced by the respiratory chain. This suggestion is in good agreement with the observations of mitochondrial transitions induced by hydrogen peroxide (0.1-0.4 mM) in the same cell line [23]. In the both models no release of cytochrome c from mitochondria or any visible signs of apoptosis were observed until the final steps of mitochondrial transitions including swelling. Similar changes in morphology of mitochondria were observed during apoptosis induced by TNF or STS and in both models release of cytochrome c happened with significant delay after complete fission of mitochondria. It was clear that both processes were completed rapidly (5-15 min) in comparison with the prolonged lag-period after initiation of apoptosis. Recently, it was found [24] that fission of mitochondria during apoptosis depended on translocation of dynamin-related protein (Drp1) from cytosol to some local sites at the surface of mitochondria. Inhibition of this process not only prevented fission of mitochondria but also strongly inhibited apoptosis. There is no evidence that mitochondrial transitions induced by the inhibitors or by hydrogen peroxide included the same molecular mechanisms, but it appears clear that fission of mitochondria can be necessary but is not sufficient for apoptosis. It is known that mitochondria are subjected to continuous fission and fusion in the living cell. It seems possible that inhibition of mitochondrial functions interrupted fusion more than stimulated fission [25].
During prolonged treatment with the uncouplers alone or in combination with the respiratory inhibitors, fragmented mitochondria gathered near the nucleus and formed several large clusters (Fig. 4). Similar processes were observed in cells treated with hydrogen peroxide and in apoptotic cells. Electron microscopy revealed swollen and partially degraded mitochondria in these clusters. This observation was in agreement with the significant decrease in the total mass of mitochondrial material and content of the specific mitochondrial proteins. The selective elimination of mitochondria from apoptotic cells was recently described under complete inhibition of final steps of apoptosis with inhibitor of caspases [26]. The major role of autophagy in this process was suggested. We did not observe accumulation of autophagosomes in our model, so probably the mechanism of depletion from mitochondria was different. The preliminary data indicated the possible expulsion of mitochondrial aggregates from the cell. This process resembled the final steps of differentiation of erythroblasts and some other cells whose mature forms are depleted of mitochondria.Fig. 4. Changes in morphology of mitochondria in HeLa cells treated with mitochondrial inhibitors: a) control cells; b, c) cells treated with antimycin (2 µM) and DNP (0.4 mM) for 8 (b) and 24 h (c). Mitochondria were stained with Mitotracker Green (Molecular Probes, USA).
When HeLa cells were treated for 48-72 h with uncouplers in combination with antimycin or myxothiazol, a significant (50-70%) fraction of the cells died but the rest of the population was viable, without any signs of apoptosis and with very low mitochondrial content. The cell death was not related to energy deprivation and probably depended on hyperproduction of ROS in mitochondria. The viable cells depleted of mitochondria probably had selective advantage under these conditions. Elimination of mitochondria and the selective pressure could be responsible for low content of mitochondria in some rapidly growing tumors [27] and development of tumors resistant to chemotherapy. Depletion of mitochondria (“mitochondrial death”) as a possible mechanism for protection against hyperproduction of ROS was postulated by Skulachev in 1999 [28] and was named “mitoptosis”. Future studies would show whether the observations described above are in agreement with these predictions and what the role of these phenomena in cell physiology is.
We would like to cordially thank V. P. Skulachev for his constant attention and support of our work. Our birthday congratulations and wishes of many years of fruitful work in a glory of science.
We are grateful to all the members of our laboratory for invaluable help in the past and in the present times.
The work was supported by grants from the Russian Foundation for Basic Research (RFBR) (Nos. 04-04-49484, 02-04-48843), RFBR-NOW (The Netherlands), RFBR-GFEN (China), Ludwig Cancer Research Institute (USA), and Fund “Paritet” (Russia).
REFERENCES
1.Bakeeva, L. E., Chentsov, Yu. S., and Skulachev, V.
P. (1978) Biochim. Biophys. Acta, 501, 349-369.
2.Skulachev, V. P. (2001) Trends Biochem.
Sci., 26, 23-29.
3.Amchenkova, A. A., Bakeeva, L. E., Chentsov, Y. S.,
Skulachev, V. P., and Zorov, D. B. (1988) J. Cell Biol.,
107, 481-495.
4.Severina, I. I., Skulachev, V. P., and Zorov, D. B.
(1988) J. Cell Biol., 107, 497-501.
5.Skulachev, V. P., Sharaf, A. A., and Liberman, E.
A. (1967) Nature, 216, 718-719.
6.Li, K., Li, Y., Shelton, J. M., Richardson, J. A.,
Spencer, E., Chen, Z. J., Wang, X., and Williams, R. S. (2000)
Cell, 101, 389-399.
7.Shchepina, L. A., Popova, E. N., Pletjushkina, O.
Y., and Chernyak, B. V. (2002) Biochemistry (Moscow), 67,
222-226.
8.Shchepina, L. A., Pletjushkina, O. Y., Avetisyan,
A. V., Bakeeva, L. E., Fetisova, E. K., Izyumov, D. S., Saprunova, V.
B., Vyssokikh, M. Y., Chernyak, B. V., and Skulachev, V. P. (2002)
Oncogene, 21, 8149-8157.
9.Izyumov, D. S., Avetisyan, A. V., Pletjushkina, O.
Y., Sakharov, D. V., Wirtz, K. W., Chernyak, B. V., and Skulachev, V.
P. (2004) Biochim. Biophys. Acta, 1658, 141-147.
10.Lyamzaev, K. G., Izyumov, D. S., Avetisyan, A.
V., Yang, F., Pletjushkina, O. Y., and Chernyak, B. V. (2004) Acta
Biochim. Pol., 51, 553-562.
11.Brinkley, B. R., Barham, S. S., Barranco, S. C.,
and Fuller, G. M. (1974) Exp. Cell Res., 85, 41-46.
12.Luo, X., Budihardjo, I., Zou, H., Slaughter, C.,
and Wang, X. (1998) Cell, 94, 481-490.
13.Cory, S., Huang, D. C., and Adams, J. M. (2003)
Oncogene, 22, 8590-8607.
14.Yang, J., Liu, X., Bhalla, K., Kim, C. N.,
Ibrado, A. M., Cai, J., Peng, T. I., Jones, D. P., and Wang, X. (1997)
Science, 275, 1129-1132.
15.Vander Heiden, M. G., Chandel, N. S., Schumacker,
P. T., and Thompson, C. B. (1999) Mol. Cell, 3,
159-167.
16.Vander Heiden, M. G., Li, X. X., Gottleib, E.,
Hill, R. B., Thompson, C. B., and Colombini, M. (2001) J. Biol.
Chem., 276, 19414-19419.
17.Paradies, G., Petrosillo, G., Pistolese, M., Di
Venosa, N., Federici, A., and Ruggiero, F. M. (2004) Circ. Res.,
94, 53-59.
18.Barnham, K. J., Masters, C. L., and Bush, A. I.
(2004) Nat. Rev. Drug Discov., 3, 205-214.
19.Gabai, V. L., Meriin, A. B., Yaglom, J. A., Wei,
J. Y., Mosser, D. D., and Sherman, M. Y. (2000) J. Biol. Chem.,
275, 38088-38094.
20.Dennis, P. B., Jaeschke, A., Saitoh, M., Fowler,
B., Kozma, S. C., and Thomas, G. (2001) Science, 294,
1102-1105.
21.Hardie, D. G. (2003) Endocrinology,
144, 5179-5183.
22.Fryer, L. G., Parbu-Patel, A., and Carling, D.
(2002) J. Biol. Chem., 277, 25226-25232.
23.Skulachev, V. P., Bakeeva, L. E., Chernyak, B.
V., Domnina, L. V., Minin, A. A., Pletjushkina, O. Y., Saprunova, V.
B., Skulachev, I. V., Tsyplenkova, V. G., Vasiliev, J. M., Yaguzhinsky,
L. S., and Zorov, D. B. (2004) Mol. Cell Biochem.,
256/257, 341-358.
24.Frank, S., Gaume, B., Bergmann-Leitner, E. S.,
Leitner, W. W., Robert, E. G., Catez, F., Smith, C. L., and Youle, R.
J. (2001) Dev. Cell, 1, 515-525.
25.Legros, F., Lombes, A., Frachon, P., and Rojo, M.
(2002) Mol. Biol. Cell., 13, 4343-4354.
26.Xue, L., Fletcher, G. C., and Tolkovsky, A. M.
(2001) Curr. Biol., 11, 361-365.
27.Cuezva, J. M., Krajewska, M., de Heredia, M. L.,
Krajewski, S., Santamaria, G., Kim, H., Zapata, J. M., Marusawa, H.,
Chamorro, M., and Reed, J. C. (2002) Cancer Res., 62,
6674-6681.
28.Skulachev, V. P. (1999) Mol. Aspects Med.,
20, 139-184.