REVIEW: Proton Transfer Dynamics at Membrane/Water Interface and Mechanism of Biological Energy Conversion
A. Y. Mulkidjanian1,2*, D. A. Cherepanov3, J. Heberle4, and W. Junge1
1Division of Biophysics, Department of Biology/Chemistry, University of Osnabrueck, D-49069 Osnabrueck, Germany; fax: +49-541-9692871; E-mail: mulkidjanian@biologie.uni-osnabrueck.de2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119899 Moscow, Russia
3Institute of Electrochemistry, Russian Academy of Sciences, Leninsky pr. 31, 117071 Moscow, Russia
4Research Center Juelich, IBI-2: Structural Biology, D-52425 Juelich, Germany
* To whom correspondence should be addressed.
Received September 30, 2004
Proton transfer between water and the interior of membrane proteins plays a key role in bioenergetics. Here we survey the mechanism of this transfer as inferred from experiments with flash-triggered enzymes capturing or ejecting protons at the membrane surface. These experiments have revealed that proton exchange between the membrane surface and the bulk water phase proceeds at >=1 msec because of a kinetic barrier for electrically charged species. From the data analysis, the barrier height for protons could be estimated as about 0.12 eV, i.e., high enough to account for the observed retardation in proton exchange. Due to this retardation, the proton activity at the membrane surface might deviate, under steady turnover of proton pumps, from that measured in the adjoining water phase, so that the driving force for ATP synthesis might be higher than inferred from the bulk-to-bulk measurements. This is particularly relevant for alkaliphilic bacteria. The proton diffusion along the membrane surface, on the other hand, is unconstrained and fast, occurring between the neighboring enzymes at less than 1 µsec. The anisotropy of proton dynamics at the membrane surface helps prokaryotes diminish the “futile” escape of pumped protons into the external volume. In some bacteria, the inner membrane is invaginated, so that the “ejected” protons get trapped in the closed space of such intracellular membrane “sacks” which can be round or flat. The chloroplast thylakoids and the mitochondrial cristae have their origin in these intracellular structures.
KEY WORDS: ATP synthesis, membrane potential, chemiosmotic coupling, alkaliphilic bacteria, chloroplasts, mitochondria, bacterial membranes
A retrospective account of studies on proton transfer dynamics at the membrane surface might be an appropriate contribution to this special issue in honor of Vladimir Skulachev, who has published seminal works in this field. Below we focus on the mechanisms of proton transfer both across and along the membrane/water interface as inferred from pulse experiments with light-triggered enzymes ejecting or capturing protons at the membrane surface. We consider these data in their relation to the mechanism of energy conversion in the living cell.
It is widely accepted that the transmembrane difference in the electrochemical potential of hydrogen ions (DeltaµH+) is a major intermediate in the cellular energy transduction [1-3]. DeltaµH+ is generated by redox- or light-driven proton pumps. It is utilized by the energy-consuming enzymes, the ATP synthase in the first line and by secondary transporters in the second. In some bacteria, DeltaµH+ is functionally replaced/complemented by the sodium potential (DeltaµNa+) (see [4] for a review). Still the majority of bacteria, and, importantly, the plant chloroplasts and the animal mitochondria use only DeltaµH+. Mitchell coined the term protonmotive force (pmf) [2]:
pmf = DeltaµH+/F = Deltapsi - (2.3RT/F)·DeltapH, (1)
where Deltapsi is the transmembrane electrical potential difference, and DeltapH is the pH difference between the two sides of the membrane, namely the positively charged side p and the negatively charged side n. DeltapH was initially conceived by Mitchell as the difference existing between the two bulk phases separated by the membrane [2]. Williams, however, challenged this notion by arguing that in bacteria the p-phase corresponds to the infinitely extended external space. If protons are extruded into this “Pacific Ocean”, they would be diluted and the entropic component of the pmf would be lost [5]. This argument is particularly important when considering alkaliphilic bacteria, such as Bacillus firmus, which keep their internal pH about 3 pH units more acidic than the ambient one (see [6] for a review). As |Deltapsi| in these bacteria hardly increases above 200 mV [7], the straightforward application of Eq. (1) yields a pmf around zero. In this relation, several authors have speculated about a localized, surface-to-surface coupling (see [8-10] for reviews). Kell, in particular, has considered the possibility that the ejected protons readily spread over the membrane surface but are somehow prevented from prompt equilibration with the bulk, so that the local pH at the membrane surface (pHS) might differ from pH in the adjacent bulk (pHB) at steady state [11]. Michel and Oesterhelt came to the same conclusion after revealing a poor correlation between (i) the ATP yield as measured in whole cells of halobacteria and (ii) the sum of measured Deltapsi and DeltapHB [12]. The suggestion that the steady-state pHS at the outer p-surface of cells could stay lower than the pH of the external medium would lead to reasonable pmf values even in alkaliphilic bacteria (for reviews see [8, 13, 14]).
STUDIES OF THE PULSED PROTON TRANSFER ACROSS THE MEMBRANE/WATER
INTERFACE
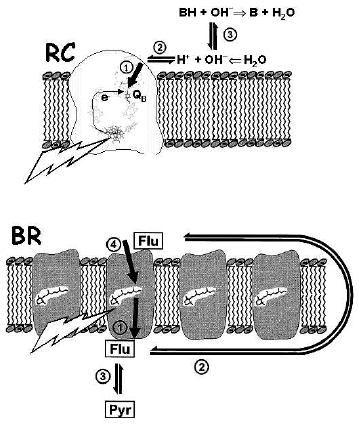
Proton diffusion in water is very fast [15], so that a difference between proton activity at the membrane surface and in the bulk water phase can exist only if the free exchange of protons is impeded by a kinetic barrier at the interface. The first experimental indication of such a barrier was obtained upon studying the flash-induced reduction and protonation of the secondary quinone (QB) in diverse photosynthetic reaction centers (RC) (for review see [16, 17], and the figure, top, for the scheme). It was found that the proton disappearance from the bulk water phase, as reported by hydrophilic pH indicators, was distinctly retarded compared to the estimated time of QB reduction, both in the RCs of purple phototrophic bacteria [18-20] and in photosystem II (PSII) of green plants [21]. These experiments, however, could not discriminate whether protons were impeded (i) on their way from the bulk water to the membrane surface or (ii) during their penetration through the protein towards the buried QB molecule. This ambiguity was clarified by Lel' Drachev, Andrey Kaulen, and Vladimir Skulachev who studied the flash-induced proton transfer by bacteriorhodopsin (BR) sheets ([22], see also the figure, bottom). They followed not only the spectral changes of BR proper and of the pH indicator p-nitrophenol in the solution, but also used capacitive voltammetry to trace, via voltage changes, the transmembrane proton movement. It was found that the proton delivery from the buried retinal cofactor to the p-surface followed the formation of the M intermediate of the BR photocycle, whereas the protonation of the water-dissolved pH indicator was distinctly retarded. These experiments localized the kinetic barrier between the membrane surface and the bulk water phase. The acceleration of the p-nitrophenol protonation by added hydrophilic pH buffers also indicated that the kinetic barrier passed through the water phase [22].
The proton transfer in the opposite direction, from the bulk water phase into the protein, was tracked with native membrane vesicles of phototrophic bacteria Rhodobacter sphaeroides and Rhodobacter capsulatus. It was found that proton transfer from the surface to QB, as traced by electrochromic absorption changes, followed the reduction of QB at ~0.1 msec, whereas the response of diverse pH indicators in the solution was retarded up to 0.5-1 msec ([29], see the figure, top). Hence, the kinetic barrier between the surface and the bulk water phase is present also from the n-side of the coupling membrane.Heberle and Dencher studied the same flash-induced proton release from BR by using two pH indicators, namely fluorescein, which was covalently bound to the surface, and pyranine dissolved in the solution ([23, 24], see the figure, bottom, for the experiment scheme). Fluorescein was protonated at ~0.1 msec, concomitant with the formation of the M-state, whereas pyranine was protonated much slower, at ~0.8 msec [23-25]. The delayed proton transfer from the BR surface into the bulk water phase was thereafter confirmed in several other labs [26-28].
INTERFACIAL POTENTIAL BARRIER: PROPERTIES
The slow rate of proton equilibration between the surface of biological membranes and the bulk water phase was initially attributed to the damping effect of immobile pH buffers at the surface, i.e., the ionizable lipid and protein groups [30, 31]. On the “macroscopic” level, the ability of surface buffers to retard the propagation of a proton pulse was addressed both experimentally [32, 33] and theoretically [30, 31, 34, 35]. On the “microscopic” level, it has been shown that the replacement of particular amino acid residues at the n-surface of BR membranes affected the enzyme kinetics [36]. These data indicated the contribution of the surface exposed amino acids in the efficient collection/trapping of protons (further evidence of such involvement can be found in review [37]). However, if the surface pH buffers were alone responsible for the proton retardation, mobile hydrophilic pH buffers or pH indicators were expected to accelerate proton equilibration when added at concentrations of >1-5 µM, i.e., when they could kinetically compete with free protons [34, 29, 35, 38]. As a rule, this was not the case. Only the monoanionic p-nitrophenol accelerated the proton relaxation already when added at 25 µM [22]. The di-anions such as phosphate, bromcresol purple, or MES were efficient only when added at >100 µM (see [16, 20, 28, 29, 38, 39] and references cited therein). Pyranine, which carries four negative charges, did not accelerate the proton exchange [28, 39]. This apparent dependence on the electric charge of the mobile pH buffer points to a kinetic barrier of electrostatic nature. As elaborated in more detail elsewhere [38, 40, 41], the exact physical picture of the barrier might be rather complex. It may result both from the dielectric saturation of water in the vicinity of a charged surface [40] and from dielectric overscreening [41].
Although the rigorous physical description of the interfacial barrier is not yet feasible, its properties can be inferred from experimental data. In particular, the proton transfer across the interfacial barrier was characterized by weak pH dependence and high activation energy of 30-50 kJ/mol [20, 29, 39, 42]. As argued elsewhere [29], these features indicate the participation of neutral water as an intermediate proton carrier. Apparently, the protons/hydroxyls and/or pH buffers in the bulk fail, because of the potential barrier, to reach the “newborn” surface protons/proton vacancies before the latter interact with molecules of neutral water (as depicted for the RC case in the figure, top). Water is abundant at the surface, but the activation barrier for its protonation/deprotonation is about 50 kJ/mol at neutral pH [29].
In a further attempt to reveal the properties of the interfacial potential barrier, the factors, which determine the rate of pulsed protonic relaxation at the interface of spherical membrane vesicles, were analyzed by solving a system of diffusion equations and by comparing the solution with the experimental data. The modeling showed that the rate of proton exchange between the membrane surface and the bulk water is determined by the pH buffering capacity of the surface, the height of the potential barrier, and the vesicle size [38]. The calculated dependence on the vesicle size corroborates the experimental data as obtained with whole bacterial cells. Several authors [30, 43, 44] have shown that protons appeared at the p-surface of cells and spheroplasts of purple phototrophic bacteria Rb. sphaeroides and Rb. capsulatus at tau < 5 msec, as followed by electrochromic shift of carotenoid pigments (which correlate with the absorbance changes of an amphiphilic, membrane-bound pH indicator neutral red [45]). These protons, however, were sensed by hydrophilic pH indicators in the bulk water phase only at 30-70 msec [30, 43, 44]. The disruption of spheroplasts into smaller vesicles accelerated the response of pH dyes by an order of magnitude [44]. Hence, the proton retardation was more pronounced in whole cells than in the case of smaller vesicles and BR sheets (see above).
The same modeling revealed that the proton relaxation rate is accelerated by added pH buffer once its concentration exceeds a certain “threshold”. The “threshold” value depends on the barrier height but is independent both of the vesicle size and the surface buffering capacity. This feature helped to “extract”, from the experimental data [38], the values of the barrier height, as “felt” by different penetrating ions. The barrier height was found to depend almost linearly on the electric charge and to vary between 0.09 eV for p-nitrophenol and MES (with charge of -1) and more than 0.36 eV for pyranine (with charge of -4). The barrier height for protons proper was found to be about 0.12 eV [38].
Considering the situation at steady state, it was possible to show, by solving the Smoluchowski equation for protons spreading away from proton “pumps” at the surface, that at typical values of proton pump density and of their turnover rate a potential barrier of 0.12 eV can yield a steady-state surface pHS of ~6.0. Importantly, this value of pHS was independent of pH in the bulk water phase [40]. The latter feature might help to understand the bioenergetics of alkaliphilic bacteria: the pH value at the surface of living cells could be much lower than in the surrounding medium. It is noteworthy that the surface buffering capacity does not matter at steady state [32], so that the proton activity (concentration) at the surface is determined only by the height of the potential barrier and by the size of the object. It is possible to say that due to the interfacial potential barrier and the relatively large size of bacterial cells, the proton concentration at the outer surface of respiring bacteria is higher than in the surrounding medium.
As discussed in more detail elsewhere [40], protons that are released by the pumps to the p-surface can either move along the surface to the nearby “pmf consumer”, e.g., an ATP synthase, or escape over the barrier into the bulk phase. The rate of the former, productive reaction is determined by the protonic conductance of the “consumers”. The rate of the futile proton escape is just proportional to the proton concentration at the surface. In the simplest case, a gradual acidification of the surface would lead to the relative increase in the futile proton escape. It seems more lucrative to block the pumps before the futile proton efflux across the barrier reached remarkable values. In this relation, it is noteworthy that the cytochrome bc1 (bf) complexes, which serve as “hubs” in the vast majority of electron transfer chains, remarkably slow down already at pH < 6.5 due to the back-pressure control from the generated pmf (see [46] and references therein). Because of this dynamic feedback, pHS at the p-surface is unlikely to drop below ~6.5, which prevents the futile proton flux into the bulk.
LATERAL PROTON TRANSFER
In late 60s, Skulachev suggested that DeltaµH+ can be used by the cell as a transportable form of power that is transmitted along extended membrane profiles (see [47] and references therein). While there is little doubt that Deltapsi can promptly propagate along the membrane, the rate of lateral proton transfer, which, in turn, determines the rate of lateral DeltapH propagation, remained controversial. Here again, the pulsed experiments with BR-containing membranes were useful in providing quantitative information [25, 48]. The experimental scenario is illustrated in the figure, bottom. The pH indicator fluorescein (Flu) was covalently bound either to Lys129 at the extracellular (EC) surface or to Cys36 at the cytoplasmic (CP) surface. After pulsed light excitation of BR a proton was released to the EC membrane surface at ~100 µsec. The fluorescein at the CP surface got this proton, after its lateral transfer around the edge of the PM, at ~200 µsec, although the size of the BR-sheets was pretty large, on the order of 1 µm [25]. In similar experiments of Alexiev and coworkers, the rate of lateral proton transfer was even faster [49]. Serowy and coworkers [50] “launched” light-triggered “caged” protons from lipid-soluble carriers used to imitate membrane enzymes. In this case as well, protons were promptly transferred along the surface with a diffusion coefficient of 5.8*10-5 cm2*sec-1, only two times smaller than in the bulk water [50]. The fast proton transfer along the surface is crucial not only for such elongated systems as large filamentous mitochondria [47, 51] and cyanobacterial trichoms [52]. The study of the proton-transporting H+-ATP-synthase of Rb. capsulatus revealed that the proton delivery to this enzyme did not limit the turnover even at pH 10.0 and even if the enzyme was decoupled and turned over with a time constant as small as ~5 msec [53]. From the data analysis, the time of proton delivery to Fo, required to yield its turnover at milliseconds, could be estimated as <=1 µsec [53, 54]. As argued above, proton transfer across the interfacial barrier proceeds by three orders of magnitude slower. Hence, the route along the membrane surface is likely to dominate upon the proton transfer between the neighboring proton “sources” and “sinks”.
IMPLICATIONS FOR ENERGY CONVERSION
The above surveyed experimental data, which are discussed in more detail elsewhere (see [25, 29, 38, 40, 48]), specify the mechanism of proton coupling in biological membranes as follows.
The core mechanism, as initially invented by nature and still operative in the majority of bacteria, is based on the ability of the redox- and light-driven proton pumps to electrically charge the inner cellular membrane, alkalize the internal n-surface and acidify the external p-surface of the cell. Because of the interfacial potential barrier, proton equilibration between the surface and the bulk water occurs more slowly than the proton diffusion along the surface. As a result, (i) the majority of the ejected protons are “consumed” by the nearby ATP-synthases before equilibrating with the bulk water and (ii) the proton activity at the membrane surface might deviate from the respective activity in the adjoining bulk phase. Thus in vivo the driving force beyond the ATP synthesis can be better defined as:
pmf = Deltapsi - 2.3RT*DeltapHS.
In this simple case, the reaction space that is relevant for the energy coupling is the bacterial cell proper plus the adjoining water layer with a thickness of ~1 nm. The bathing solution surrounding the cell seems to be involved as a counterproductive sink for the escaped protons, at best.
Apparently, nature has continuously tried to diminish this futile proton escape by “drawing” the segments of the inner bacterial membrane inside the cell, so that ejected protons were trapped in the closed space of such membrane invaginations. Being driven by selection pressure, the inventions of such intracellular structures happened in different lineages and led, in particular, to the formation of thylakoids in cyanobacteria and to the development of the intracellular vesicular structures in purple photosynthetic bacteria. The cyanobacterial thylakoids were retained in plant chloroplasts, whereas the intracellular vesicles of purple bacteria, because of the evolutionary relatedness of the latter to mitochondria [55], might have led the way to the mitochondrial cristae. It is noteworthy that Yaguzhinskii and coworkers have recently shown that pHS differs from pH in the bulk phase in respiring mitochondria as well [56, 57].
The described mechanism provides a coherent picture of electrochemical energy transduction and reconciles Mitchell's idea of DeltaµH+ as a driving force for ATP synthesis both with the existence of localized membrane acidic domains as suggested by Williams [5] and with the experimentally established anisotropy of proton dynamics at the surface [11, 12, 24-26]. Although apparently deviating from Mitchell's initial concept of delocalized bulk-to-bulk coupling, the here outlined mechanism is in full correspondence with the latest, less known notion of Peter Mitchell who wrote, in his last review, that the surfaces of the coupling membranes serve as “two proton conducting zones, P and N, in which the major part of the proton current that flows between the proticity producing and consuming modules is localized” [58].
We are thankful to L. Drachev, M. Gutman, L. I. Krishtalik, N. Dencher, D. Oesterhelt, V. Skulachev, R. J. P. Williams, and L. S. Yaguzhinsky for useful discussions. D. C. and A. M. would especially like to acknowledge numerous encouraging conversations with the late Andrey Kaulen.
This work was supported by the Alexander von Humboldt Foundation, the Volkswagen Foundation, INTAS (2001-736), and by grants from the Deutsche Forschungsgemeinschaft (Mu-1285/1, Ju-97/13, SFB 431-P15, 436-RUS-113/210).
REFERENCES
1.Mitchell, P. (1961) Nature, 191,
144-148.
2.Mitchell, P. (1966) Physiol. Rev.,
41, 445-502.
3.Skulachev, V. P. (1977) FEBS Lett.,
74, 1-9.
4.Skulachev, V. P. (1992) Eur. J. Biochem.,
208, 203-209.
5.Williams, R. J. P. (1978) Biochim. Biophys.
Acta, 505, 1-44.
6.Krulwich, T. A., Ito, M., Gilmour, R., Sturr, M.
G., Guffanti, A. A., and Hicks, D. B. (1996) Biochim. Biophys.
Acta, 1275, 21-26.
7.Guffanti, A. A., Mann, M., Sherman, T. L., and
Krulwich, T. A. (1984) J. Bacteriol., 159, 448-452.
8.Ferguson, S. J. (1985) Biochim. Biophys.
Acta, 811, 47-95.
9.Cramer, W. A., and Knaff, D. B. (1991) Energy
Transduction in Biological Membranes: a Textbook of Bioenergetics,
Springer, New York.
10.Ferguson, S. J. (1995) Curr. Biol.,
5, 25-27.
11.Kell, D. B. (1979) Biochim. Biophys. Acta,
549, 55-99.
12.Michel, H., and Oesterhelt, D. (1980)
Biochemistry, 19, 4615-4619.
13.Guffanti, A. A., and Krulwich, T. A. (1984)
Biochem. Soc. Trans., 12, 411-412.
14.Kell, D. B. (1986) Meth. Enzymol.,
127, 538-557.
15.Eigen, M. (1963) Angew. Chem., 75,
489-588.
16.Wraight, C. A., Cogdell, R. J., and Chance, B.
(1978) in The Photosynthetic Bacteria (Clayton, R. K., and
Sistrom, W. R., eds.) Academic Press, New York, pp. 471-511.
17.Junge, W., and Jackson, J. B. (1982) in
Photosynthesis (Govindjee, ed.) Vol. 1, Academic Press, New
York, pp. 589-646.
18.Chance, B., Crofts, A. R., Nishimura, M., and
Price, B. (1970) Eur. J. Biochem., 13, 364-374.
19.Codgell, R. J., Jackson, J. B., and Crofts, A. R.
(1972) Bioenerg., 4, 413-429.
20.Petty, K. M., and Dutton, P. L. (1976) Arch.
Biochem. Biophys., 172, 335-345.
21.Auslander, W., and Junge, W. (1974) Biochim.
Biophys. Acta, 357, 285-298.
22.Drachev, A. L., Kaulen, A. D., and Skulachev, V.
P. (1984) FEBS Lett., 178, 331-336.
23.Heberle, J., and Dencher, N. A. (1990) FEBS
Lett., 277, 277-280.
24.Heberle, J., and Dencher, N. A. (1992) Proc.
Natl. Acad. Sci. USA, 89, 5996-6000.
25.Heberle, J., Riesle, J., Thiedemann, G.,
Oesterhelt, D., and Dencher, N. A. (1994) Nature, 370,
379-382.
26.Scherrer, P., Alexiev, U., Marti, T., Khorana, H.
G., and Heyn, M. P. (1994) Biochemistry, 33,
13684-13692.
27.Dioumaev, A. K., Richter, H. T., Brown, L. S.,
Tanio, M., Tuzi, S., Saito, H., Kimura, Y., Needleman, R., and Lanyi,
J. K. (1998) Biochemistry, 37, 2496-2506.
28.Porschke, D. (2002) J. Phys. Chem. B,
106, 10233-10241.
29.Gopta, O. A., Cherepanov, D. A., Junge, W., and
Mulkidjanian, A. Y. (1999) Proc. Natl. Acad. Sci. USA,
96, 13159-13164.
30.Junge, W., and Polle, A. (1986) Biochim.
Biophys. Acta, 848, 265-273.
31.Heberle, J., and Dencher, N. A. (1992) in
Structures and Functions of Retinal Proteins (Rigaud, J. L.,
ed.) John Libbey Eurotext Ltd, pp. 221-224.
32.Junge, W., and McLaughlin, S. (1987) Biochim.
Biophys. Acta, 890, 1-5.
33.Jones, M. R., and Jackson, J. B. (1989)
Biochim. Biophys. Acta, 975, 34-43.
34.Nachliel, E., and Gutman, M. (1996) FEBS
Lett., 393, 221-225.
35.Georgievskii, Y., Medvedev, E. S., and
Stuchebrukhov, A. A. (2002) Biophys. J., 82,
2833-2846.
36.Riesle, J., Oesterhelt, D., Dencher, N. A., and
Heberle, J. (1996) Biochemistry, 35, 6635-6643.
37.Adelroth, P., and Brzezinski, P. (2004)
Biochim. Biophys. Acta, 1655, 102-115.
38.Cherepanov, D. A., Junge, W., and Mulkidjanian,
A. Y. (2004) Biophys. J., 86, 665-680.
39.Heberle, J. (1991) Zeitauflosende Untersuchung
der Protonentranslokationsschritte von bakteriorhodopsin mittels
chemisch-gekoppelter pH-Indikatoren, PhD Thesis, Freien
Universitat, Berlin.
40.Cherepanov, D. A., Feniouk, B. A., Junge, W., and
Mulkidjanian, A. Y. (2003) Biophys. J., 85,
1307-1316.
41.Cherepanov, D. A. (2005) Phys. Rev. Lett.,
in press.
42.Maroti, P., and Wraight, C. A. (1997) Biophys.
J., 73, 367-381.
43.Arata, H., Takenaka, I., and Nishimura, M. (1987)
J. Biochem., 101, 261-265.
44.Jones, M. R., and Jackson, J. B. (1990)
Biochim. Biophys. Acta, 1019, 51-58.
45.Mulkidjanian, A. Y., and Junge, W. (1994) FEBS
Lett., 353, 189-193.
46.Kramer, D. M., Sacksteder, C. A., and Cruz, J. A.
(1999) Photosynth. Res., 60, 151-163.
47.Skulachev, V. P. (2001) Trends Biochem.
Sci., 26, 23-29.
48.Heberle, J. (2000) Biochim. Biophys. Acta,
1458, 135-147.
49.Alexiev, U., Mollaaghababa, R., Scherrer, P.,
Khorana, H. G., and Heyn, M. P. (1995) Proc. Natl. Acad. Sci.
USA, 92, 372-376.
50.Serowy, S., Saparov, S. M., Antonenko, Y. N.,
Kozlovsky, W., Hagen, V., and Pohl, P. (2003) Biophys. J.,
84, 1031-1037.
51.Amchenkova, A. A., Bakeeva, L. E., Chentsov, Y.
S., Skulachev, V. P., and Zorov, D. B. (1988) J. Cell Biol.,
107, 481-495.
52.Severina, I. I., Skulachev, V. P., and Zorov, D.
B. (1988) J. Cell Biol., 107, 497-501.
53.Feniouk, B. A., Kozlova, M. A., Knorre, D. A.,
Cherepanov, D. A., Mulkidjanian, A. Y., and Junge, W. (2004)
Biophys. J., 86, 4094-4109.
54.Cherepanov, D. A., Mulkidjanian, A. Y., and
Junge, W. (1999) FEBS Lett., 449, 1-6.
55.Andersson, S. G. E., Zomorodipour, A., Andersson,
J. O., Sicheritz-Ponten, T., Alsmark, U. C. M., Podowski, R. M.,
Naslund, A. K., Eriksson, A. S., Winkler, H. H., and Kurland, C. G.
(1998) Nature, 396, 133-140.
56.Kozlova, M. V., Gramadskii, K. B., Solodovnikova,
I. M., Krasinskaya, I. P., Vinogradov, A. V., and Yaguzhinskii, L. S.
(2003) Biofizika, 48, 443-452.
57.Solodovnikova, I. M., Yurkov, V. I., Ton'shin, A.
A., and Yaguzhinskii, L. S. (2004) Biofizika, 49,
47-56.
58.Mitchell, P. (1991) Biosci. Rep.,
11, 297-344.