REVIEW: Persister Cells and the Riddle of Biofilm Survival
K. Lewis
Northeastern University, 360 Huntington Avenue, Boston, MA 02115, USA; fax: 617-373-3724; E-mail: k.lewis@neu.edu
Received September 27, 2004
This review addresses a long-standing puzzle in the life and death of bacterial populations--the existence of a small fraction of essentially invulnerable cells. Bacterial populations produce persisters, cells that neither grow nor die in the presence of bactericidal agents, and thus exhibit multidrug tolerance (MDT). The mechanism of MDT and the nature of persisters, which were discovered in 1944, have remained elusive. Our research has shown that persisters are largely responsible for the recalcitrance of infections caused by bacterial biofilms. The majority of infections in the developed world are caused by biofilms, which sparked a renewed interest in persisters. We developed a method to isolate persister cells, and obtained a gene expression profile of Escherichia coli persisters. The profile indicated an elevated expression of toxin-antitoxin modules and other genes that can block important cellular functions such as translation. Bactericidal antibiotics kill cells by corrupting the target function, such as translation. For example, aminoglycosides interrupt translation, producing toxic peptides. Inhibition of translation leads to a shutdown of other cellular functions as well, preventing antibiotics from corrupting their targets, which will give rise to tolerant persister cells. Overproduction of chromosomally-encoded “toxins” such as RelE, an inhibitor of translation, or HipA, causes a sharp increase in persisters. Deletion of the hipBA module produces a sharp decrease in persisters in both stationary and biofilm cells. HipA is thus the first validated persister/MDT gene. We conclude that the function of “toxins” is the exact opposite of the term, namely, to protect the cell from lethal damage. It appears that stochastic fluctuations in the levels of MDT proteins lead to formation of rare persister cells. Persisters are essentially altruistic cells that forfeit propagation in order to ensure survival of kin cells in the presence of lethal factors.
KEY WORDS: persisters, multidrug tolerance, biofilms, death, survival, altruism
The study of biofilms is a rapidly expanding, “hot” field of microbiology. Biofilm research encompasses studies of multispecies communities, cell-cell signaling, virulence, industrial fouling, and bioremediation, to name a few. Yet it is fair to say that the field was propelled to its present prominence due to a singular feature that unites all biofilms, namely, their dramatic tolerance to antimicrobial agents. This tolerance is responsible for recalcitrant human infections, accounting for ~60% of all infectious diseases in the West [1]. A large number of empirical studies documenting biofilm tolerance have been published in the past two decades. At the same time, there has been a paucity of molecular biology research into the mechanism of tolerance. This might seem surprising, given the prominence of the problem, and our generally good understanding of various mechanisms of antibiotic resistance [2].
The reason for this reluctance to tackle the main question of the biofilm field may have stemmed from the suggestion that the problem does not really exist. Biofilms are slow growing, while antibiotics act best against rapidly dividing cells. The action of most beta-lactams, for example, depends stringently upon rapid growth [3]. Slow growth alone, or in combination with possible retardation of antibiotic diffusion into the biofilm, could then explain the observed tolerance [4]. Studies reporting the binding of aminoglycoside antibiotics to the biofilm matrix seemed to support the idea of limited antibiotic access to cells [5-9]. Similarly, it was found that retardation of diffusion combined with active degradation of compounds can effectively protect the biofilm from hydrogen peroxide or a beta-lactam antibiotic [10-13]. At the same time, fluoroquinolones appeared to diffuse freely into a biofilm [10], and these compounds are able to kill non-growing cells. Other substances, such as antiseptics and disinfectants, can also kill non-growing cells, and show considerably lower effectiveness against biofilms [14, 15].
Expression of a possible biofilm-specific resistance mechanism was suggested to contribute to biofilm tolerance [16], and seemed to explain recalcitrance to such compounds as fluoroquinolones or quaternary ammonium antiseptics. This proposition however does not appear realistic. Resistance of biofilms to killing by antimicrobials is dramatic, 100-1000-fold above the minimal lethal dose, and occurs for all antimicrobials tested and in all species examined. This means that all pathogens potentially harbor an essentially perfect multidrug resistance mechanism, but only “choose” to express it when growing as a biofilm. If such a mechanism were to exist, we should have seen mutants that express it in rapidly growing planktonic cultures as well. This is not the case. A susceptible Staphylococcus epidermidis, for example, does not acquire multidrug resistance to “everything” due to a mutation.
But how is it possible that a biofilm can be resistant to killing by all antimicrobials and not harbor a resistance mechanism? Herein lies the paradox, and the riddle, of biofilm resistance.
THE CULPRIT--PERSISTER CELLS
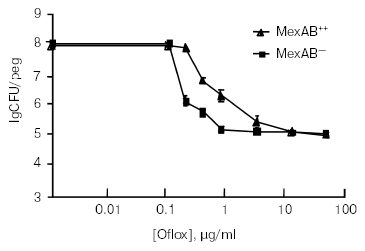
While measuring a dose-response of a Pseudomonas aeruginosa biofilm to ofloxacin [17], we noticed that a small fraction of cells was not eliminated even by very high levels of the antibiotic (Fig. 1). These cells appeared essentially invulnerable. However, the bulk of the biofilm was fairly sensitive. Cells of one of the strains overexpressed a prominent multidrug pump MexAB-OprM, which conferred considerable resistance to the bulk. However, beyond the clinically-achievable concentration, the differences among strains with or without the pump were erased, clearly indicating that something else was responsible for the remarkable survival of these persister cells. But whatever the mechanism, the culprit appeared in clear view. A small fraction of persisters was imparting survival to the biofilm population. Examination of the biofilm literature showed that persisters have been documented in numerous experiments, and ignored [18]. It does indeed seem natural to look at the bulk of cells when searching for a property characteristic of the population. However, in the case of a biofilm, the majority of cells appear unremarkable. It is the persistent minority that deserves attention. The biofilm riddle thus shifts to the study of persisters.
Persisters were actually discovered in a rapidly growing, planktonic population. In 1944, Joseph Bigger noticed that the newly-introduced penicillin was unable to “sterilize” a culture of Staphylococcus [19]. It is interesting to note that the initial focus of that study was to evaluate bactericidal properties of penicillin. Bigger writes: “My results strongly oppose the commonly accepted belief that penicillin is merely bacteriostatic”. It is difficult for us to imagine how the bactericidal property of penicillin might have been missed--the substance rapidly turns a turbid culture into a transparent solution. Bigger plated this transparent-looking medium, and discovered surviving persisters. His experiments indicated that persisters were not mutants, and upon reinoculation produce a population containing a sensitive bulk and new tolerant cells. Lancet did not publish graphs in 1944, and we repeated this important experiment, quantifying survival of E. coli treated with ampicillin [20]. The result clearly showed that surviving persisters regenerate the original population, and are therefore phenotypic variants of the wild type.Fig. 1. Role of the MexAB-OprM pump in resistance of biofilms treated with ofloxacin (Oflox).
If persisters are present in planktonic populations, then how unique is the survival capability of a biofilm? In order to address this question, we compared three populations--logarithmic, stationary, and biofilm cultures of P. aeruginosa [21]. The result was unexpected--the stationary culture produced more persisters, and was more tolerant than the famously tolerant biofilm (Fig. 2, see color insert). In retrospect, this should not be surprising, since a stationary culture has almost no growth, while the biofilm is growing, albeit slowly. We find that persister formation is indeed growth state dependent--an early log culture makes few or no persisters in E. coli, S. aureus, or P. aeruginosa, and the level of persisters increases as the population reaches stationary state. It is important to stress that persisters are not simply non-growing cells--in a stationary culture, fluoroquinolones or mitomycin C kill the bulk, leaving 1-10% intact persisters [21, 22].
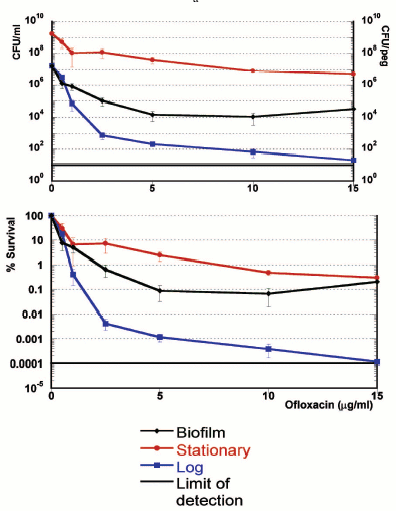
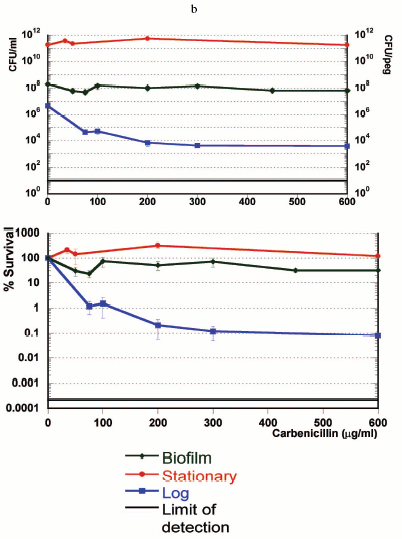
The strong dependence of persister formation on growth state appears as
a typical quorum-sensing phenomenon. We performed a standard test for
quorum sensing, adding spent stationary medium to early log cells and
measuring the rate of persisters. Spent medium did not increase
persisters in E. coli or P. aeruginosa, arguing against
the involvement of quorum sensing (Keren and Lewis, unpublished). We
also noticed that persisters are rapidly lost if a stationary
population is diluted. These observations are consistent with persister
formation being inversely dependent upon the level of metabolic
activity, rather than on a quorum-sensing type signaling molecule.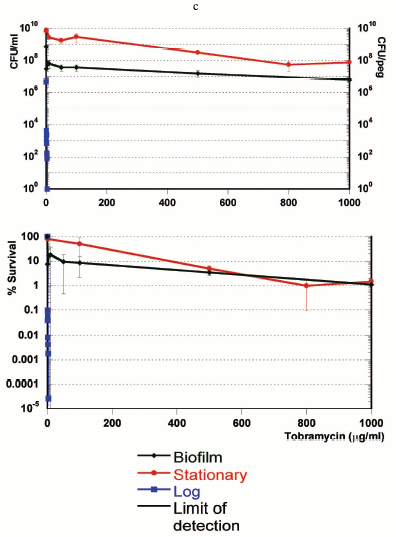
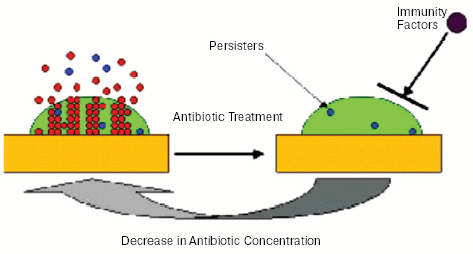
Experiments showing an essential similarity in the tolerance of stationary cells and persisters lead to an obvious question--is drug resistance of biofilms important? The answer would be “no” for a test-tube grown biofilm, and we suggested that tolerance can be much more easily studied with traditional planktonic cultures [21]. But there is every reason to believe that biofilms are considerably more tolerant to antibiotics in vivo, as compared to planktonic cells, irrespective of their growth state. We argued that in vivo, antibiotic treatment will eliminate the bulk of both biofilm and planktonic cells, leaving intact persisters. At this point, the similarity with an in vitro experiment probably ends. The immune system will be able to mop up remaining planktonic persisters, just as it eliminates non-growing cells of a population treated with a bacteriostatic antibiotic (Fig. 3, see color insert). However, the biofilm matrix protects against immune cells [23-25], and its persisters will survive. After antibiotic concentration drops, persisters will repopulate the biofilm, which will shed off new planktonic cells responsible for disease symptoms. This model explains the relapsing nature of biofilm infections, and suggests that understanding persisters will ultimately allow us to eliminate biofilms.
Fig. 3. Model of biofilm resistance based on persister survival. An initial treatment with antibiotic kills planktonic cells and the majority of biofilm cells. The immune system kills planktonic persisters, but the biofilm persister cells are protected from host defenses by the exopolysaccharide matrix. After the antibiotic concentration drops, persisters resurrect the biofilm and the infection relapses.
PERSISTERS AS SPECIALIZED ALTRUISTIC SURVIVOR CELLS
It was suggested that persisters represent normal cells at a particular stage in the cell cycle [19, 26, 27]. We found that repeated reinoculation of E. coli maintaining cells in an early log state leads to a complete loss of persisters [20]. Since early log cells undergo a cell cycle but do not produce persisters, we can rule out this particular hypothesis. This simple experiment also suggests that persisters are not formed in response to antibiotic treatment, since early log cells challenged with antibiotics produce no persisters. Neither are persisters cells that temporarily lost their ability to grow due to a reversible defect, such as a stalled replication fork. We would expect defects to occur in early log as well, though no persisters are formed at this stage. What we are left with, then, is an intriguing possibility of persisters representing specialized survivor cells whose production is regulated by the growth stage of the population. It was recently shown that persisters are rare non-growing cells, by directly tracking dividing cells in a log population of E. coli [28]. Persisters are essentially altruistic cells that forfeit rapid propagation, which ensures survival of the population of kin cells [29] in the presence of lethal factors.
THE MECHANISM OF TOLERANCE
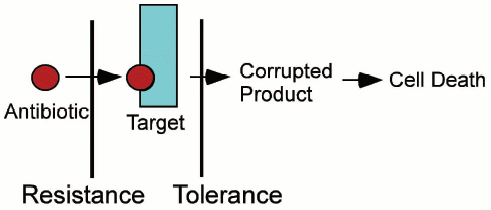
Realizing that persisters are responsible for biofilm tolerance is helpful, but does not solve the problem. Indeed, how can a cell that does not express specific resistance mechanisms be tolerant to all cidal antibiotics? All resistance mechanisms do essentially the same thing--prevent an antibiotic from binding to the target. We hypothesized that tolerance works in a different way, not by preventing antibiotic binding, but by interfering with the lethal action of cidal compounds [22]. Specifically, we proposed that persister/MDT proteins shut down antibiotic targets (Fig. 4, see color insert). It is important to stress that bactericidal antibiotics kill by corrupting the target function, rather than by merely inhibiting it. For example, erythromycin inhibits protein synthesis and is a bacteriostatic antibiotic. Streptomycin, a bactericidal aminoglycoside, causes translational misreading, which apparently produces truncated toxic peptides, leading to cell death. Shutting down the ribosome in a persister cell would produce tolerance to aminoglycosides. Fluoroquinolones act by inhibiting DNA gyrase and topoisomerase IV. The action of fluoroquinolones is selective--the ligase activity is inhibited, while the nicking remains intact. As a result, fluoroquinolones force the enzymes to create DNA lesions. Cell-wall acting antibiotics do not merely inhibit peptidoglycan synthesis, but cause death and cell lysis [30, 31]. By contrast, cells that stop peptidoglycan synthesis due to a lack of nutrients do not usually lyse. Persister proteins could shut down all antibiotic targets, creating a tolerant, dormant persister cell.
Fig. 4. Antibiotic resistance versus tolerance.
PERSISTER GENES
Finding a persister gene appears straightforward--screening a transposon insertion library should produce clones that die completely, or considerably more so than the wild type, when challenged with a cidal antibiotic. Our efforts to obtain such a mutant from Tn insertion libraries of E. coli and P. aeruginosa however were not successful, perhaps due to a considerable background variation in the level of persisters among cultures growing in the wells of a microtiter plate (Spoering and Lewis, unpublished). We therefore turned to a gene expression profile approach.
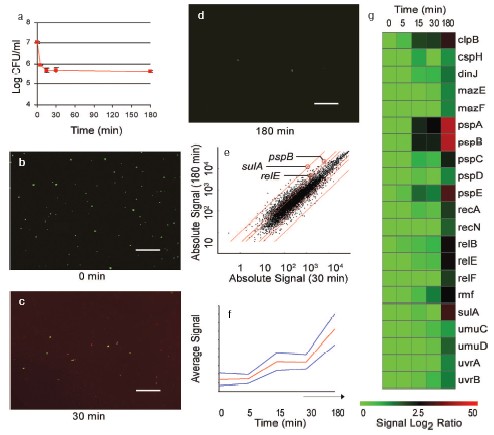
The obvious problem with obtaining an expression profile from persisters is that they first need to be isolated. We reasoned that after treatment with ampicillin, only intact persisters will remain, while the bulk of the cells will lyse. This appeared to be the case, and intact, live persisters were obtained by simple centrifugation. Ampicillin only lyses a rapidly growing, logarithmic culture, where the fraction of persisters is ~10-5, insufficient material for array work. We therefore took advantage of the hipA7 strain [27] that produces ~1% persisters (note that a null hipA mutant was reported to have no phenotype, see below) surviving ampicillin treatment (Fig. 5a, see color insert).
Labeled mRNA from persisters was used to obtain an expression profile with Affymetrix [32] E. coli Genechips. A potential problem with analyzing results of this array was that ampicillin treatment produces massive changes in the gene profile of E. coli [33], which could obscure persister-specific genes. However, we found that most of the changes accompanying ampicillin treatment occurred between 0 and 30 min time points. At 30 min, the population consisted of mostly dead but intact cells (Fig. 5, b and c, see color insert). Further incubation produced complete lysis of dead cells, leaving intact persisters after 3 h with ampicillin (Fig. 5d, see color insert). We therefore used cluster analysis to point to genes that specifically increased following the clearing of dead cells and isolation of persisters. In this way, we would be tracking genes that were elevated in persisters prior to ampicillin treatment. We indeed identified such a cluster (Fig. 5, e-g, see color insert). The cluster contained about 300 genes, including those coding for SOS stress response (recA, sulA, uvrBA, and umuDC), phage-shock (psp); heat and cold-shock (cspH, htrA, ibpAB, htpX, clpB). This expression of stress responses is consistent with a survival function of persisters, but does not in itself explain antibiotic tolerance. In practical terms, this set of ~300 candidate genes was too large to point to specific persister genes.Fig. 5. Gene expression profile of isolated persisters. E. coli HM22 hipA7 cells were grown to mid-exponential and treated with 50 µg/ml ampicillin. a) Samples were taken at indicated times and plated to determine live cells by colony count. b-d) Samples were stained with a LIVE/DEAD kit and visualized with epifluorescent microscopy (bar, 50 µm). Green cells are live, while red cells, stained by normally impermeant propidium iodide, are dead. Note the extensive red background due to cellular debris, as well as dead intact cells in the 30 min sample. e) Scatter plot of absolute gene expression at 180 min vs. 30 min. Red lines indicate 2-, 3-, and 30-fold changes, respectively. f) Cluster analysis of persister gene expression profile obtained with Affymetrix Self-Organizing Map (SOM). The profile was obtained by hybridizing labeled mRNA from samples (0, 5, 15, 30, 180 min) to Affymetrix E. coli gene chips. The cluster shown indicates the expression profile of genes specifically upregulated in persisters (180 min). The red line indicates average signal intensity. g) Heatmap of selected genes from the cluster (f) generated with Spotfire Decisionsite 7.2.
Armed with the tolerance hypothesis, we searched for genes that could shut down cellular functions. A number of genes expressed in persisters appeared to fulfill this criterion. RMF inhibits translation by forming ribosome dimers in stationary state [34]; UmuDC has been reported to inhibit replication [35]; and SulA is an inhibitor of septation [36]. Most striking however was the overexpression of well-characterized chromosomal toxin-antitoxin (TA) modules RelBE, MazEF, and putative TA modules PspAB,CD [37], and DinJ/YafQ, homologous to RelBE. Homologs of these genes are found on plasmids where they constitute a maintenance mechanism [38]. Typically, the toxin is a protein that inhibits an important cellular function such as translation or replication, and forms an inactive complex with the antitoxin. The toxin is stable, while the antitoxin is degradable. If a daughter cell does not receive a plasmid after segregation, the antitoxin level decreases due to proteolysis, leaving a toxin that either kills the cell or inhibits propagation. TA modules are also commonly found on bacterial chromosomes, but their role is largely unknown. MazEF was proposed to serve as a programmed cell death mechanism [39]. However, it was reported recently that MazF and an unrelated toxin RelE do not actually kill cells, but induce stasis by inhibiting translation, a condition that can be reversed by expression of corresponding antitoxins [40, 41]. It was also suggested that MazF and RelE act as attenuators of the stringent response. We reasoned that the ability of “toxin” modules to reversibly block translation makes them excellent candidates for persister genes. By shutting down potential antibiotic targets, toxins will produce tolerant cells.
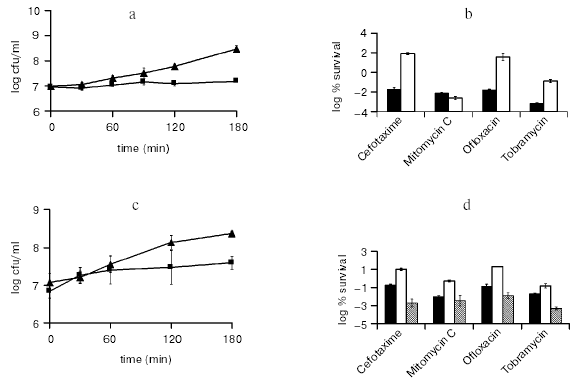
The best-studied TA module in E. coli is RelBE that cleaves mRNA on translating ribosomes, which stalls protein synthesis [42]. We tested the ability of RelE to generate persisters tolerant to antibiotics. Cells expressing RelE from an inducible promoter became highly tolerant to ofloxacin, a fluoroquinolone; to cefotaxime, a cephalosporin cell wall synthesis inhibitor; and to tobramycin, an aminoglycoside protein synthesis inhibitor (Fig. 6, a and b). This result is consistent with the idea of a toxin blocking antibiotic targets. Indeed, RelE directly blocks the ribosome, which would prevent the lethal action of aminoglycosides. Inhibition of translation in turn blocks DNA and cell wall synthesis, which would diminish the ability of antibiotics acting against DNA gyrase and peptidoglycan synthases to corrupt their targets. Interestingly, RelE did not protect cells from mitomycin C, which forms DNA adducts. This would be consistent with the target blocking idea--RelE does not “block” DNA.
In order to observe persisters, RelE cells were plated on a medium containing an inducer of a recombinant RelB antitoxin. Recovery without inducing RelB was observed as well, but slower. This suggests that a TA module has the ability to both produce, and resuscitate persisters.Fig. 6. The effects of toxin overexpression on persister formation. a) RelE was induced (¢) from pKD3035 (pBAD::relE) in MG1 (MC1000 DeltarelBE) at time zero by adding 0.2% arabinose, and MG1 with a blank vector pBAD33 served as the control (p). Samples were removed at indicated time points and plated for colony counts on LB agar plates containing ampicillin (100 µg/ml), chloramphenicol (50 µg/ml), glucose (0.2%, to suppress the pBAD promoter), and 1 mM IPTG to induce RelB expression from pKD3033 (pA1/O4/O3::opSD::relB). b) Cells were cultured and RelE was induced as described above. After 3 h of RelE induction samples were removed and treated with either cefotaxime (100 µg/ml), mitomycin C (10 µg/ml), ofloxacin (5 µg/ml), or tobramycin (25 µg/ml) for 3 h at 37°C with aeration. Before and after the challenge the cells were plated on media as described in (a) for colony counts. The control (MG1/pBAD; ¢) was challenged at a similar cell density to that of the relE induced cells (¢). c) HM22 cells (K12 hipA7) were cultured as described above and at time 0 were moved to 30°C (¢) or kept at 37°C (p). d) HM22 cells were challenged at 30°C (¢) as described in (b). Controls are HM22 (black columns) and HM21 (K12 hipA w.t.; gray columns) cells challenged at 37°C.
Given that RelE expression increased persister production prompted us to revisit what was known about the hipBA locus, a likely TA module. It was discovered in the first screen for genes specifically affecting persistence that was performed by Moyed and coworkers [27]. An ethyl methanesulfonate (EMS)-mutagenized population of E. coli was enriched with cells surviving ampicillin treatment and then screened for colonies producing higher numbers of persisters. Only mutants whose growth was normally inhibited by ampicillin were examined further. This approach led to the identification of a hipA7 (high persistence) mutant producing about 1000-fold more persistent cells as compared to the wild type [26, 43, 44]. This was the first report of a bacterial gene specifically involved in the regulation of persister production and antibiotic tolerance. However, a deletion in either hipA or hipBA appeared to have no phenotype [45]. This suggested that the hipA7 allele creates a complex pleiotropic artifact, leading to an increase in persisters. The pioneering findings of Moyed's group have been largely ignored.
Interestingly, HipBA is a likely TA module. Indeed, HipB and HipA form a complex [27]; overexpression of HipA is “toxic”, leading to arrest of cell division [44]; a hipB mutation could not be obtained due to apparent lethality of free HipA [43]; HipB is a repressor of the operon, which is typical for antitoxins; and a homolog of the chromosomal hipBA operon is found on the Rhizobium symbiotic plasmid pNGR234a where it may play a role in segregation maintenance [46]. It was previously reported that mild induction of recombinant HipA increases production of persisters [27]. We confirmed this observation and found that functional expression of HipA strongly increases persisters tolerant to a range of antibiotics (Fig. 6, c and d) [22].
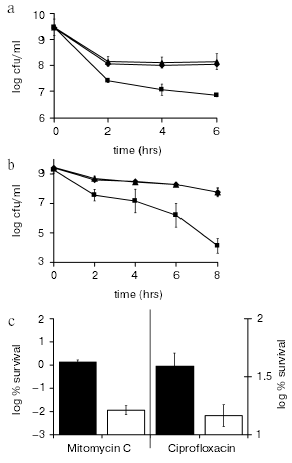
The most important remaining unanswered question regarding HipA was whether it had a functional role in creating persisters in vivo. We reasoned that the level of persisters is particularly high in stationary state, and that might be the growth phase at which HipA exerts its action. A hipBA deletion strain produced 10-100-fold less persisters in stationary state as compared to the wild type (Fig. 7, a and b). The mutant showed considerably lower tolerance when treated with a fluoroquinolone or mitomycin C. This experiment completes the first chapter of the hip saga. hipA is a persister/MDT gene. A biofilm grown from a DeltahipBA strain produced considerably fewer persisters (Fig. 7c), which makes hipA the first validated biofilm tolerance gene as well.
We also tested knockout mutants in other TA modules (mazEF, dinJ-yafQ, psp), and in rmf. None showed a phenotype in either log or stationary cultures. Given that there are >10 TA modules in E. coli [37], which probably are functionally redundant, this is not surprising. What is surprising is that we saw a strong phenotype in the single hipBA knockout strain. On the other hand, the lack of an obvious phenotype of a DeltahipBA in log state (or in stationary minimal medium, Spoering and Lewis, unpublished) points directly to other players obscuring HipBA under these conditions.Fig. 7. The effects of toxin deletion on persister formation. a, b) K12 (wild type, .), KL310 (DeltarelBE; p), and KL312 (DeltahipBA; ¢) were grown to stationary state (16-18 h) and challenged with ofloxacin (5 µg/ml) (a) or mitomycin C (10 µg/ml) (b). At the designated times a sample was removed and plated on LB-agar plates for colony count. c) Biofilms (K12 (w.t.; ¢) and KL312 (DeltahipBA; ¢) were grown at 37°C for 48 h, on LB agar to ~2*109 CFU/biofilm. They were then exposed to LB agar plates containing 20 mM NaNO3 with or without 5 µg/ml mitomycin C (left y-axis); and with or without 5 µg/ml ciprofloxacin (right y-axis). After a 24 h incubation, biofilms were suspended by vortexing and sonicating in LB medium, producing a cell suspension that was plated for colony counts.
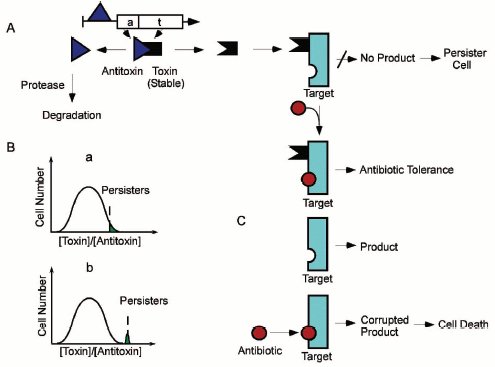
Based on our findings, we proposed the following model of persister production and antibiotic tolerance (Fig. 8, see color insert). The ratio of a toxin/antitoxin (such as HipA/HipB) in a population fluctuates, and rare cells will express relatively high levels of a toxin. Cidal antibiotics bind to a target protein and corrupt its function, generating a lethal product (for example, aminoglycosides interrupt translation, resulting in misfolded peptides that damage the cell). A toxin binds to the target and inhibits the function, leading to tolerance. The antibiotic can bind to the blocked target, but can no longer corrupt its function. Inhibition of translation by a toxin such as RelE will further cause a relative increase in the stable toxin (due to antitoxin degradation) of this and other TA modules, which might have an autocatalytic effect on inhibition of translation, leading to a shutdown of other cellular functions, and to dormant, tolerant persister cells. Our observation of several TA modules upregulated in persisters is consistent with this model. Antitoxins act as repressors of TA modules, so a decrease in antitoxin protein level will cause an induction in transcription, which we observed in the gene profile of persisters.
Our gene profiling data also support the possibility of persisters being dormant. A cluster of approximately 600 genes involved in metabolism and flagellar synthesis was seen to gradually diminish in cells treated with ampicillin, in accordance with our previous findings [33]. Interestingly, the same genes showed a further decrease in isolated persisters, suggesting that these might have been downregulated in persisters prior to the addition of antibiotic. Among these repressed genes were members of the large operons involved in oxidative phosphorylation--NADH dehydrogenase, ATP synthase, and cytochrome O-ubiquinol oxidase. The general theme of this gene cluster appears to be a decrease in non-essential genes and a shutdown of metabolism, both consistent with a dormant phenotype.Fig. 8. A model of persister formation and antibiotic tolerance. A) Chromosomal Toxin/Antitoxin modules are coded by two-gene operons, with the antitoxin usually acting as a repressor. Once synthesized, the toxin and antitoxin form a stable inactive complex. The antitoxin is labile and can be degraded by proteases. B) A random fluctuation in relative toxin/antitoxin concentration (a) will produce free active toxin leading to an inhibition of a target function, such as protein synthesis. Suppression of translation will lead to a decrease in the level of this, as well as additional labile antitoxins, producing a distinct population of persister cells with a set of elevated toxin proteins (b). C) Cidal antibiotics bind to a target protein and corrupt its function. This generates a lethal product (for example, aminoglycosides interrupt translation, resulting in misfolded peptides that damage the cell). A toxin binds to the target and inhibits the function, leading to tolerance. The antibiotic can normally bind to the blocked target, but can no longer corrupt its function. This accounts for antibiotic tolerance of persister cells.
TA modules are widely spread among bacteria [38] and might represent the essential mechanism responsible for persister formation in different species. Production of these phenotypic variants of the wild type appears to be stochastic [47] and resembles individual variations in the chemotactic behavior of E. coli [48, 49]. Persistence is a reversible phenomenon. Overexpression of toxins inhibits protein synthesis strongly but incompletely [40], which may allow for the synthesis of a neutralizing antitoxin. An even more effective mechanism for resuscitation of persisters would be through expression of terminator RNA (tmRNA), which releases ribosomes stalled on mRNA lacking a termination codon. tmRNA has been shown to counter the action of artificially expressed RelE and MazF toxins [40]. Importantly, tmRNA synthesis may proceed in cells with inhibited translation and might therefore trigger the resuscitation process. Interestingly, “toxins” appear not as damaging factors, but as their opposite, proteins whose function is to protect the cell from damage.
Identification of persister genes, including those described in this work, will lead to a better understanding of tolerance in general and of biofilm resistance to killing in particular. Interestingly, “toxins” appear to be their direct opposite, specialized protective proteins. So far, studies aimed at identifying genes responsible for biofilm resistance were limited to the bulk of the population [50], which is fairly susceptible to therapeutic doses of antibiotics or may express resistance genes that act equally well in exponentially growing cells [17, 51]. It is the persister subpopulation however, present in all species studied, which is responsible for the dramatic tolerance of biofilms to unrelated antibiotics [18]. Identification of persister genes is an important first step in understanding recalcitrance of biofilms to antibiotic therapy, and it is likely to shed light on other related but poorly understood phenomena involving a dormant state, such as latent Mycobacterium tuberculosis infection [52, 53], “Viable But Not Culturable Bacteria” [54, 55], and “uncultivable” bacteria [56].
PERSISTERS AND PROGRAMMED CELL DEATH
We originally suggested that persisters might by cells that turn off a programmed cell death (PCD) mechanism [47] (see [57] for a review of PCD in a variety of organisms). There are several cases of well-documented PCD in bacteria, such as mother cell lysis during sporulation in B. subtilis or lysis of the majority of Myxococcus cells, which apparently provides nutrients to for the formation of a fruiting body that produces spores [47]. Whether death from antibiotics is a result of PCD process in bacteria remains an open question. The finding that persisters are dormant cells provides a simple explanation for their survival, which does not depend on the presence of a PCD program.
Our finding of TA module involvement in persister formation seems to be at odds with the reports of the MazEF TA module causing cells death of E. coli under a variety of (but not all) conditions [39, 58]. Similarly to RelE, MazF inhibits translation, and causes reversible inhibition of cell growth [41]. This would suggest that, similarly to RelE and HipA, overexpression of MazF should lead to production of persisters. But why would a mazF mutant have a considerably better survival to heat, nalidixic acid, and mitomycin C, for example [58]?
I suggest that a single underlying mechanism can satisfactorily account for these seemingly irreconcilable differences. We noted above that formation of persisters is reversible. At the same time, it is reasonable to assume that resuscitation of persisters is a probabilistic process that depends on the level of the toxin, on the level of the antitoxin that may be formed, and on other factors that affect the process. In short, there are likely to be “unresuscitatable” persisters, which for all practical purposes are dead. Perhaps antibiotics kill by producing such irreversible persisters? Would death by dormancy be adaptive and amount to a PCD mechanism? These are some of the intriguing questions regarding the biology of persisters that await an answer.
I gratefully acknowledge my mentor and friend, Professor Skulachev, who taught his students the art of defining an intriguing problem.
REFERENCES
1.Licking, E. (1999) Business Week,
98-100.
2.Lewis, K., Salyers A., Taber H., and Wax, R. (2001)
Bacterial Resistance to Antimicrobials: Mechanisms, Genetics,
Medical Practice and Public Health, Marcel Dekker, New York.
3.Tuomanen, E., Cozens, R., Tosch, W., Zak, O., and
Tomasz, A. (1986) J. Gen. Microbiol., 132, 1297-1304.
4.Gilbert, P., Collier, P. J., and Brown, M. R.
(1990) Antimicrob. Agents Chemother., 34, 1865-1868.
5.Gordon, C. A., Hodges, N. A., and Marriott, C.
(1988) J. Antimicrob. Chemother., 22, 667-674.
6.Nichols, W. W., Dorrington, S. M., Slack, M. P.,
and Walmsley, H. L. (1988) Antimicrob. Agents Chemother.,
32, 518-523.
7.Hoyle, B. D., Jass, J., and Costerton, J. W. (1990)
J. Antimicrob. Chemother., 26, 1-5.
8.Shigeta, M., Tanaka, G., Komatsuzawa, H., Sugai,
M., Suginaka, H., and Usui, T. (1997) Chemotherapy, 43,
340-345.
9.Ishida, H., Ishida, Y., Kurosaka, Y., Otani, T.,
Sato, K., and Kobayashi, H. (1998) Antimicrob. Agents
Chemother., 42, 1641-1645.
10.Hassett, D. J., Ma, J. F., Elkins, J. G.,
McDermott, T. R., Ochsner, U. A., West, S. E., Huang, C. T.,
Fredericks, J., Burnett, S., Stewart, P. S., McFeters, G., Passador,
L., and Iglewski, B. H. (1999) Mol. Microbiol., 34,
1082-1093.
11.Elkins, J. G., Hassett, D. J., Stewart, P. S.,
Schweizer, H. P., and McDermott, T. R. (1999) Appl. Environ.
Microbiol., 65, 4594-4600.
12.Anderl, J. N., Franklin, M. J., and Stewart, P.
S. (2000) Antimicrob. Agents Chemother., 44,
1818-1824.
13.Stewart, P. S. (2003) J. Bacteriol.,
185, 1485-1491.
14.Gilbert, P., Alison, D. G., Rickhard, A., Sufya,
N., Whyte, F., and McBain, A. J. (2001) in Biofilm Community
Development: Chance or Necessity? (Gilbert, P., Allison, D. G.,
Brading, M., Verran, J., and Walker, J., eds.) Bioline Press,
Cardiff.
15.Gilbert, P., Das, J., and Foley, I. (1997)
Adv. Dent. Res., 11, 160-167.
16.Costerton, J. W., Stewart, P. S., and Greenberg,
E. P. (1999) Science, 284, 1318-1322.
17.Brooun, A., Liu, S., and Lewis, K. (2000)
Antimicrob. Agents Chemother., 44, 640-646.
18.Lewis, K. (2001) Antimicrob. Agents
Chemother., 45, 999-1007.
19.Bigger, J. W. (1944) Lancet, 11,
497-500.
20.Keren, I., Kaldalu, N., Spoering, A., Wang, Y.,
and Lewis, K. (2004) FEMS Microbiol. Lett., 230,
13-18.
21.Spoering, A. L., and Lewis, K. (2001) J.
Bacteriol., 183, 6746-6751.
22.Keren, I., Shah, D., Spoering, A., Kaldalu, N.,
and Lewis, K. (2005) J. Bacteriol., in press.
23.Leid, J. G., Shirtliff, M. E., Costerton, J. W.,
and Stoodley, A. P. (2002) Infect. Immun., 70,
6339-6345.
24.Jesaitis, A. J., Franklin, M. J., Berglund, D.,
Sasaki, M., Lord, C. I., Bleazard, J. B., Duffy, J. E., Beyenal, M.,
and Lewanowski, Z. (2003) J. Immunol., 171,
4329-4339.
25.Vuong, C., Voyich, J. M., Fischer, E. R.,
Braughton, K. R., Whitney, A. R., DeLeo, F. R., and Otto, M. (2004)
Cell Microbiol., 6, 269-275.
26.Moyed, H. S., and Bertrand, K. P. (1983) J.
Bacteriol., 155, 768-775.
27.Falla, T. J., and Chopra, I. (1998)
Antimicrob. Agents Chemother., 42, 3282-3284.
28.Balaban, N. Q., Merrin, J., Chait, R., Kowalik,
L., and Leibler, S. (2004) Science, 305, 1622-1625.
29.Lewis, K. (1998) J. Theor. Biol.,
193, 359-363.
30.Tomasz, A., Albino, A., and Zanati, E. (1970)
Nature, 227, 138-140.
31.Rice, K. C., and Bayles, K. W. (2003) Mol.
Microbiol., 50, 729-738.
32.Selinger, D. W., Cheung, K. J., Mei, R.,
Johansson, E. M., Richmond, C. S., Blattner, F. R., Lockhart, D. J.,
and Church, G. M. (2000) Nat. Biotechnol., 18,
1262-1268.
33.Kaldalu, N., Mei, R., and Lewis, K. (2004)
Antimicrob. Agents Chemother., 48, 890-896.
34.Wada, A. (1998) Genes Cells, 3,
203-208.
35.Opperman, T., Murli, S., Smith, B. T., and
Walker, G. C. (1999) Proc. Natl. Acad. Sci. USA, 96,
9218-9223.
36.Walker, G. C. (1996) in Escherichia coli and
Samonella. Cellular and Molecular Biology (Neidhardt, F. C., ed.)
ASM Press, Washington, DC, pp. 1400-1416.
37.Brown, J. M., and Shaw, K. J. (2003) J.
Bacteriol., 185, 6600-6608.
38.Hayes, F. (2003) Science, 301,
1496-1499.
39.Sat, B., Hazan, R., Fisher, T., Khaner, H.,
Glaser, G., and Engelberg-Kulka, H. (2001) J. Bacteriol.,
183, 2041-2045.
40.Pedersen, K., Christensen, S. K., and Gerdes, K.
(2002) Mol. Microbiol., 45, 501-510.
41.Christensen, S. K., Pedersen, K., Hansen, F. G.,
and Gerdes, K. (2003) J. Mol. Biol., 332, 809-819.
42.Pedersen, K., Zavialov, A. V., Pavlov, M. Y.,
Elf, J., Gerdes, K., and Ehrenberg, M. (2003) Cell, 112,
131-140.
43.Moyed, H. S., and Broderick, S. H. (1986) J.
Bacteriol., 166, 399-403.
44.Black, D. S., Kelly, A. J., Mardis, M. J., and
Moyed, H. S. (1991) J. Bacteriol., 173, 5732-5739.
45.Black, D. S., Irwin, B., and Moyed, H. S. (1994)
J. Bacteriol., 176, 4081-4091.
46.Falla, T. J., and Chopra, I. (1999)
Microbiology, 145, 515-516.
47.Lewis, K. (2000) Microbiol. Mol. Biol.
Rev., 64, 503-514.
48.Spudich, J. L., and Koshland, D. E., Jr. (1976)
Nature, 262, 467-471.
49.Korobkova, E., Emonet, T., Vilar, J. M., Shimizu,
T. S., and Cluzel, P. (2004) Nature, 428, 574-578.
50.Mah, T. F., Pitts, B., Pellock, B., Walker, G.
C., Stewart, P. S., and O'Toole, G. A. (2003) Nature,
426, 306-310.
51.Drenkard, E., and Ausubel, F. M. (2002)
Nature, 416, 740-743.
52.Mukamolova, G. V., Turapov, O. A., Young, D. I.,
Kaprelyants, A. S., Kell, D. B., and Young, M. (2002) Mol.
Microbiol., 46, 623-635.
53.Tufariello, J. M., Chan, J., and Flynn, J. L.
(2003) Lancet Infect. Dis., 3, 578-590.
54.Colwell, R. R., and Grimes, D. J. (2000)
Nonculturable Microorganisms in the Environment, American
Society for Microbiology, Washington, DC.
55.Bogosian, G., and Bourneuf, E. V. (2001) EMBO
Rep., 2, 770-774.
56.Kaeberlein, T., Lewis, K., and Epstein, S. S.
(2002) Science, 296, 1127-1129.
57.Skulachev, V. P. (2002) Ann. N. Y. Acad.
Sci., 959, 214-237.
58.Hazan, R., Sat, B., and Engelberg-Kulka, H.
(2004) J. Bacteriol., 186, 3663-3669.