
|
Methylation of DNA -- One of the Major Epigenetic MarkersS. V. Salozhin*, E. B. Prokhorchuk, and G. P. GeorgievInstitute of Gene Biology, Russian Academy of Sciences, ul. Vavilova 34/5, 119334 Moscow, Russia; fax: (7-095) 135-4105; E-mail: salozhin@co.ru* To whom correspondence should be addressed.
|
Received November 4, 2004
Regulation of gene expression is a complex process. It includes a great number of steps from control of mRNA synthesis to posttranslational modification of proteins. Epigenetic events play essential roles in regulation of transcription. In this review, we concentrate on methylation of DNA as one of the important epigenetic marks. It is well known that DNA methylation is associated with closed chromatin state and, therefore, repressed, inactive genes. Here we describe major processes that depend on DNA methylation: imprinting, X-inactivation, and oncogenesis. Also we describe a number of known methyl-DNA-binding proteins and links between methylation of DNA and higher-order chromatin structure.
KEY WORDS: DNA methylation, epigenetics, imprinting, methyl-DNA-binding proteins
Symmetric methylation of cytosine in CpG dinucleotides is one of the widespread modifications in animal genomes. It is associated with “closed” (inactive) chromatin state and, therefore, negative regulation of transcription. To date, this modification has been found both in invertebrates (Drosophila melanogaster [1]) and in chordates (from Ciona intestinalis [2] to mammals).
It is known that DNA methylation plays an important role in so-called “epigenetic” regulation of gene expression--regulation that is not directly dependent on primary structure of DNA, but is maintained by many protein or non-protein factors (like histone modification, chromosome territory, etc., i.e., “epi”-genomic factors).
DNA methylation affects gene expression directly or indirectly. Some transcriptional factors (i.e., Sp1) can interact only with non-methylated DNA sequences, whereas methylation of cytosine abolishes interaction [3]. This in turn leads to less effective transcription of certain genes. On the other hand, there is a different mechanism of action of CpG methylation. So-called MBD (methyl-DNA-binding domain) proteins [4] specifically recognize modified sequences and attract large multiprotein complexes that can change chromatin conformation from “opened” to “closed”.
In the genome of vertebrates, approximately 80% of all CpG-dinucleotides are subject to methylation. Exceptions to this rule are so-called “CpG-islands”--short (1000-1500 bp) regions of DNA with high density of CpGs. Usually these regions are associated with regulatory sequences and do not undergo methylation during either early development or establishment of tissue-specific expression [5].
It is generally accepted that DNA methylation is a unidirectional process. If any sequence acquires CpG methylation then this modification becomes stable and will be inherited after cell division. So, both daughter DNA molecules will have same pattern of methylation. However, CpG methylation is much more dynamic during early embryonic development.
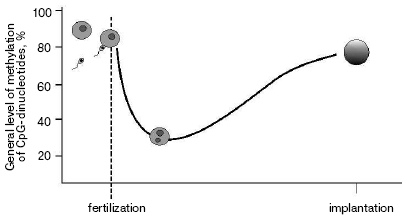
As a result of gametogenesis, DNA in sperm or oocytes become almost fully methylated. Fertilization starts the process of active demethylation of the male genome [6] followed by de novo methylation (Fig. 1). At the same time passive demethylation of the female genome occurs. The normal level of methylation is reached by the time of implantation. Such “failure” in gross methylation of the genome is quite strong in embryogenesis of the house mouse Musculus musculus, much less during the development of Xenopus laevis [7], and was not reported for zebra fish Danio rerio [8]. Although some modifications associated with establishment of tissue-specific expression still happen during later stages of embryogenesis, major changes in DNA methylation level are finished by the time of implantation.
The most popular explanation of heritability of methylation during cell divisions is the suggestion of a key role of maintenance methyltransferase DNMT1 in this process [9]. During replication DNMT1 simply “copies” pattern of methylation modifying cytosines on newly synthesized chain in accordance with modifications on the initial strand. So-called de novo methyltransferases DNMT3a and DNMT3b are responsible for re-methylation of the genome during early embryonic development and changes in overall methylation in gametogenesis [10].Fig. 1. Level of methylation of male genome after fertilization.
But this theory has some weak points. It has been shown, for example, that CpG-methylation can be maintained even without DNMT1 [11]. Moreover, there is no common opinion concerning what factors direct the de novo methylation process.
Epigenetic regulation of gene expression will be discussed later with examples of genomic imprinting and X-inactivation. Another important function of DNA methylation is transcriptional repression of retrotransposones and other mobile elements. It was shown, for example, that expression of murine retrotransposone element IAP can be increased up to 100-fold in the absence of DNA methylation [12]. This repression is necessary because expression of mobile element can lead either to genetic rearrangements or increment of “transcriptional noise” and as a consequence to the genesis of different disorders.
At the same time, another hypothesis explaining such repression exists. mRNA that arises during transcription of so-called “junk DNA” can degenerate with production of small double-strand RNA molecules. This leads to siRNA-dependent methylation of retrotransposones and other mobile elements [13]. There is a possibility that this effect is the first step in the process of de novo methylation of DNA.
It is worth mentioning that a small part of CpG-islands undergo methylation in a normal organism. These CpG-islands belong to imprinted genes or genes on inactivated X-chromosome. Promotors associated with such CpG-islands remain silent during the whole life of the organism. But if methylation of CpG-islands of X-liked genes occurs during early embryonic development [14], methylation status of imprinted genes remains invariable from gametogenesis and is not subject to passive or active demethylation.
Genomic imprinting. There are a number of genes whose expression depends on what allele they are located, paternal or maternal. Two alleles of imprinted genes differ in such features as DNA methylation, chromatin structure (histone modifications, nuclease hypersensitivity), and time of replication [15]. The important characteristics of imprinting are heritability in cell divisions and reversibility in gametogenesis. Usually imprinted genes organized in big clusters on the chromosome. Expression of several genes in one cluster can be ruled by a common regulation element.
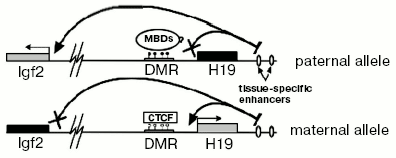
One of best known models used for investigation of imprinting is the system of H19/IGF2 genes. H19 and IGF2 are two genes located approximately 90 kb apart. H19-gene is transcribed only when it is located on the maternal chromosome. Vice versa, IGF2-gene is expressed only from the paternal chromosome. A key role in regulation of these genes is played by a 2 kb DNA fragment that is located immediately before the promotor of H19 [16]. This fragment is called a Differentially Methylated Region (DMR) because it is heavily methylated on the paternal but not methylated on the maternal chromosome.
There is a common model explaining maintenance of imprinting in the H19/IGF2 locus. Although the mechanism providing imprinting of the H19 gene is not exactly clear and is rather autonomous (i.e., does not need any external factor except maintenance DNA-methyltransferase DNMT1 and, possibly, methyl-DNA-binding proteins), expression of IGF2 depends on a methylation-sensitive insulator between its promotor and tissue-specific enhancers (Fig. 2). A crucial role in insulation is played by a protein called CTCF (CCCTC-binding factor). It is an evolutionarily conserved protein belonging to DNA-binding proteins that have a so-called “zinc fingers” domain. CTCF can act either as a transcriptional factor or as a protein interacting with many different insulators [17]. It was shown that CTCF has four binding sites in murine H19 DMR and interacts only with the maternal allele [18]. Absence of CTCF leads to bi-allelic expression of IGF2 and, moreover, acquiring of DNA methylation on the maternal allele [19].
Although H19 DMR is a major regulator element in the H19/IGF2-locus, there are a number of additional domains regulating imprinting in this locus. For example, one additional DMR is located just upstream from IGF2 (DMR1) and another one inside of one of the IGF2 introns (DMR2). Both DMR1 and DMR2 have DNA methylation only on the paternal allele. But unlike H19 DMR deletion, DMR2 deletion does not lead to reactivation of the silent maternal allele. Oppositely, it leads to less efficient transcription of IGF2 [20]. So, DMR2 serves as a positive regulator of expression of IGF2.Fig. 2. Scheme of maintenance of imprinting in the H19/IGF2 locus. White and black circles designate, respectively, non-methylated and methylated cytosine residues in CpG-dinucleotides.
As a rule, genes expressed from the paternal allele enforce cell growth and differentiation and genes expressed from the maternal allele have the opposite effect. As an example, deletion of the expressed from paternal chromosome genes PEG1/MEST or PEG3 leads to restriction of growth. Deletion of H19 results in overgrowth of the fetus [21].
Normal expression of imprinted genes is very important for ontogenesis. Any faulty expression can lead to tumorigenesis or such genetic disorders like Prader-Willy, Angelman [22], and Bechwith-Wiedemann syndromes [23].
X-Inactivation. Since in mammals, females have two and males have only one X-chromosome, it is necessary to equalize expression of X-linked genes [24]. There are two types of X-inactivation: imprinted which occurs in extra-embryonic tissues and random that can be seen in all embryonic tissues. In contrast to other known cases of imprinting, X-inactivation covers the entire chromosome. This process is controlled by the X-inactivation center (Xic) that is responsible for both random and nonrandom X-inactivation.
The Xic contains two major genes (XIST and TSIX) coding non-translated mRNAs. TSIX is located in the 3´-region of XIST and its mRNA is antisense transcript for XIST mRNA [14, 25]. Initially, both XIST and TSIX are expressed simultaneously from each of two X-chromosomes. But repression of transcription of TSIX from one X-chromosome leads to increase in the level of XIST expression and then to spreading of XIST mRNA along the X-chromosome, substitution of H2A histone by its specific form, macro-H2A [26], and methylation of lysines 9 and 27 of histone H3 [27]. Inactivation of X-linked genes occurs as inactive chromatin state spreading along the chromosome. It is complex process that involves action of different trans-acting factors, histone modifications, and partially methylation of CpG-islands associated with promotor of these genes [28].
Despite association, XIST and TSIX do not have fully overlapping temporal profiles of expression. So, TSIX cannot be considered as one negative regulator of XIST. There are about 40 potential CTCF-binding sites identified in the 5´-region of TSIX [29]. According to recent data, the following simplified model of maintenance of active status of the X-chromosome can be suggested. Initially TSIX-independent factor causes silencing of XIST. Then CTCF binds to the 5´-region of TSIX and activate its transcription, which leads to complete repression of XIST.
Abnormal methylation in cancer. Major processes associated with changes in DNA methylation come to an end on implantation of the embryo. Future epigenetic events involved in organogenesis, fetus growth, as well as normal vital functions are not so massive. But the picture changes dramatically in the case of tumorigenesis.
There are several epigenetic rearrangements characteristic for cancer: general demethylation [30], hypermethylation of individual genes [31], and hypermethylation of CpG-islands of a number of “housekeeping” genes [32] and tumor suppressor genes. Such hypermethylation often leads to transcriptional repression of these genes and finally to abnormalities in intracellular processes. Moreover, abnormal methylation of DMRs causes loss of imprinting (LOI) that often leads to genesis of embryonic malignancies [33].
Methylation of the promotor of the P16 gene (or INK4A) in many cancers can be an example of abnormal methylation. This protein is inhibitor of cycline-dependent protein kinase. Together with other proteins of the INK-family and retinoblastoma protein Rb it negatively regulates G1- to S-phase transition [34]. So, faulty expression of P16 leads to abnormalities in the cell cycle that positively influence tumor growth. Repression of transcription of another gene (MLH1) coding O6-methylguanine-DNA-methyltransferase causes increase in mutation frequency and consequently faulty expression of other genes [35].
Different types of cancers are associated with methylation of different combination of tumor suppressor genes. For example, in the case of adenocarcinoma methylation of promotor of K-RAS genes can be seen in 80% of patients, p16/INK4A always, P53 in approximately 60%, and DPC4 in ~50%. Promotors of genes DOC2/DAB2 [36], BTAK/AURORA-A [37], and K-RAS become methylated in the case of ovarian carcinoma. These differences can reflect different genesis of various cancers.
It was shown that decrease in expression of tumor suppressor genes correlates with decrease in acetylation and increase in methylation of histones in promotor regions of these genes. Although application of inhibitors of histone deacetylases alone did not lead to reactivation of transcription, the combination of these inhibitors with inhibitors of DNA-methyltransferases restored the normal level of expression [38]. Moreover, application of only inhibitors of DNA-methyltransferases leads to significant decrease in methylation of histones. Also, methyl-DNA-binding proteins MBD2 [39] and MeCP2 [40] specifically interact with abnormally methylated promotors of tumor suppressor genes (i.e., promotors of P14 and P16).
It is interesting that insertion of new SINEs (short interspersed elements) in the non-methylated sequence makes centers of de novo methylation. This can lead to silencing of neighboring tumor suppressor genes and, consequently, to tumorigenesis.
Methyl-DNA-binding proteins. In late 1980s, A. Bird identified the first methyl-DNA-binding activity called MeCP1 (methyl-CpG-binding protein 1) [41]. This activity included two complexes (400 and 800 kD) consisting of different components. It was shown that MeCP1 plays an important role in regulation of expression of reporter genes if these genes had methylated promotor regions. Then a protein called MeCP2 was identified [42]. It had a number of biochemical and functional characteristics different from MeCP1. For example, its molecular weight was about 70 kD and only one symmetrically methylated CpG was sufficient for specific interaction of MeCP2 with DNA sequence (MeCP1 needed 12 symmetrically methylated CpGs). Is was shown that for interaction with methylated DNA an 85-amino-acid domain in the N-terminal part of protein was responsible. This domain was called MBD (methyl-DNA-binding domain). An analogous domain was found for MBD1 protein (formerly PCM1).
In 1998, B. Hendrich and A. Bird published a paper about identification and characterization of a family of methyl-DNA-binding proteins [4]. Three new proteins MBD2, MBD3, and MBD4 were described in addition to previously characterized MBD1 and MeCP2. Expression of all MBD proteins was found in almost all somatic tissues. Moreover, methyl-specific interaction with DNA and co-localization with constitutive heterochromatin were shown for MBD1, MBD2, and MBD4.
MeCP2. This is a protein with molecular weight ~70 kD. It has two functional domains--MBD and TRD (transcription repression domain). TRD is necessary for interaction of MeCP2 with the mSin3A/HDAC nucleosome remodeling complex and plays crucial role in repression of transcription of target genes. MeCP2 can also interact with other transcriptional factors and co-repressors (like SMRT [43]).
MeCP2 is the most intensively studied MBD protein because conditional brain-specific deletion of this factor leads to generation of symptoms analogous to symptoms of Rett syndrome [44]. Some of these symptoms are mental retardation, heavy breathing, stereotyping, memory dysfunctions, etc. Patients suffering from Rett syndrome in most cases have point mutations in the MeCP2 gene. It is now known that MeCP2 regulates expression of BDNF (brain-developed neurotrophic factors) that is important for proper development of the nervous system as well as memory formation, etc.
MBD1. This is the largest member of the MBD family. It consists of ~640 amino acid residues and has molecular weight ~75 kD. MBD1 interacts with DNA in a methylation-dependent manner in EMSA experiments. But it can repress either methylated or non-methylated transitory transfected constructs. Moreover, MBD1, unlike MBD2 and MBD4, can co-localize with regions of constitutive heterochromatin even in cell with aberrant DNA methylation maintenance system (i.e., without DNMT1) [4].
In early works, only two functional domains of MBD1 were described--MBD and a domain consisting of three CxxC-motifs (homologous to motifs found in DNA-methyltransferase DNMT1). But another domain was described quite soon. This domain (TRD, by analogy to transcriptional repression domain of MeCP2) is involved in transcriptional repression of reporter constructs as well as one of the CxxC-motifs [45].
It was shown at the same time that MBD1 can utilize different functional domains for interaction with different proteins. For example, TRD is important for interaction with MCAF (MBD1-containing chromatin-associated factor) [46]. Complexes Suv39h1-HP1alpha [47] and p150-CAF-HP1 [48] interact with the MBD domain. MBD1 can also interact with SETDB1 histone-methyltransferase.
Protein p150-CAF was characterized as a partner of MBD1 in a yeast two-hybrid screen. Complex CAF (chromatin associated factor) takes part in nucleosome assembly after DNA replication and maintenance of active/inactive chromatin state. One of three subunits of this complex, p150, interacts with HP1 protein and is involved in maintenance of inactive heterochromatin. Moreover, p150-CAF can interact with PCNA (proliferating cell nuclear antigen) during DNA replication. PCNA, in turn, interacts with DNMT1. p150-CAF attracts MBD1-SETDB1 complex that leads to methylation of lysine 9 of histone H3 and formation of inactive chromatin.
MBD2 and MBD3. MBD2 and MBD3 are related proteins. There is a hypothesis that genes coding these factors diverged from a common precursor. The genes have common exon-intron structure and amino acid sequences of MBD2 and MBD3 are identical by 70% [49].
But despite these similarities, MBD2 and MBD3 have different functions. MBD3 is a structure subunit of one of the major chromatin-remodeling complexes--NuRD (nucleosome remodeling and histone deacetylase complex) [50], whereas MBD2 is only one of the DNA-binding subunits of MeCP1 [51]. Knockout of MBD3 leads to embryonic lethality in mice immediately after implantation of the embryo [52]. Deletion of MBD2 does not have such serious consequences. Moreover, there are no abnormalities either in imprinting/X-inactivation or repression of mobile elements in MBD2-/- mice. There is only one phenotypic effect of knockout of MBD2--abnormal maternal behavior [52].
MBD4. This is a protein with molecular weight ~60 kD having two functional domains--MBD and a glycosylase domain. MBD4 co-localizes with regions of constitutive heterochromatin and can bind methylated DNA in vitro, but despite other MBD-proteins does not participate in regulation of gene expression.
This protein belongs to the mismatch repair system. Methylated cytosines are so-called “hot spot” of mutagenesis--they can be converted to thymines after spontaneous deamination. MBD4 is involved in processes of recognition and correction of such mutations [53]. Deletion of MBD4 leads to accumulation of mutations and higher frequency of carcinogenesis [54].
Kaiso. This is a unique methyl-DNA-binding protein. It does not have classical methyl-DNA-binding domain and interacts with DNA via a zinc finger domain consisting of three zinc fingers of C2H2 type. Another functional domain of Kaiso is the N-terminal 120 amino acid BTB/POZ-domain [55]. This domain usually located in the N-terminal part of BTB-proteins and serves for homo- or heterodimerization during protein-protein interactions. Most BTB-proteins are transcriptional repressors.
Kaiso was initially identified by a yeast two-hybrid screen as a partner of p120-catenin. p120-catenin is an important predominantly cytoplasmic protein interacting with and stabilizing E-cadherin [56].
It was also shown that Kaiso is a component of double MeCP1 complex. It is a part of a rapidly migrating band called Kaiso-generated band (KGB) [55]. The molecular weight of KGB is ~700 kD. There is a possibility that Kaiso-containing complex serves as an effector in signal-transduction pathway from cell membrane to nucleus and is responsible for repression of target genes in response to extracellular signals.
Target genes of methyl-DNA-binding proteins. Despite the importance of DNA-methylation, not many targets for methyl-DNA-binding proteins genes have been described. This can be partially explained by the fact that even in animals with deletion of any of methyl-DNA-binding proteins genes with altered expression can hardly be found. But there are a number of genes (first of all tumor suppressor genes) whose expression depends on MBDs. Proteins MBD2 and MeCP2 specifically interact with promotor region of P14/P16 genes in the case of abnormal methylation in cancer. But this interaction cannot be considered as a major function of MBDs because the cancer cell is rather artificial than the normal system.
It was shown that T-helpers in MBD2-/- mice produce interleukin-4 (IL4), which is a marker of some limphopoietic precursors. Transcriptional regulation of IL4 is carried out by two antagonistic proteins--MBD2 and Gata3. Gata3 is a positive regulator of IL4 expression and it provides high level of transcription from IL4 promotor in the absence of MBD2 [57].
To date the most important gene whose expression depends on MeCP2 is BDNF. BDNF protein belongs to the family of neurotrophins--factors participating in neuronal survival, memory formation, and a number of other neuron-specific processes. It was shown that expression of BDNF depends on methylation of one CpG-dinucleotide in its promotor region [58]. MeCP2 binds to this CpG and causes repression of BDNF, but after specific extracellular signal, MeCP2 becomes phosphorylated and dissociates from promotor of BDNF [59]. Importantly, this leads to demethylation of the key CpG-dinucleotide. For a long time DNA methylation was thought to be reversible but a very stable DNA modification. The works dedicated to interaction of MeCP2 with BDNF promotor demonstrated that the nature of this modification can be much more labile.
There is another gene whose expression depends on MeCP2. It is a gene coding cyclin A [60]. But this dependence was demonstrated only for cell lines because in a normal organism the correlation between methylation of promotor of CYCLIN A and its expression is absent. Similarly, there is no evidence that MeCP2 takes part in regulation of expression of imprinted genes, while the interaction of MeCP2 with H19 DMR was shown in vivo by chromatin immunoprecipitation.
There is only one characterized target gene for BTB-protein Kaiso. It is a gene coding protein MTA2, which is one of components of NuRD [61]. It was shown that Kaiso represses transcription of this gene. But an interaction of Kaiso with promotor of MTA2 can be detected only in cancer but not in normal cell lines.
Histone code. Recently more and more investigation are being dedicated to histone modifications. Specific modifications of histones associated with changes or maintenance of active/inactive state of gene expression is called the “histone code”.
There are a number of identified histone modifications--methylation, acetylation, phosphorylation, ubiquitination, etc. To date the best-characterized modifications are methylation and acetylation of lysine residues. For example, lysine (K) residues of the N-terminal tail of histone H3 can be methylated in positions K4, K9, K27, and K36. Methylation of K9 and K27 is associated with transcriptional repression, whereas methylation of K4 and K36 correlates with active chromatin state [62]. Methylation of K9 of histone H3 makes possible binding of heterochromatic proteins (such as HP1) with nucleosomes and, consequently, formation of stable heterochromatin.
On the other hand, histone H3 can be acetylated in positions K9, K14, K18, and K27. Acetylation of both H3 and H4 (K5, K8, K12, K16, etc.) is associated with transcriptional activation [63]. But acetylation of histones is not always a mark of active chromatin. For example, acetylation of H2A and H2B in regulatory regions of certain genes correlates with their transcriptional repression.
Histones can also be phosphorylated and ubiquitinated. These modifications are being extensively studied at present. It is not yet possible to say if these modifications are associated with transcriptional activation or repression. Rather, every particular modification of every amino acid residue plays its special role.
So, processes of histone modification reflect a complex multilevel system of regulation of gene expression.
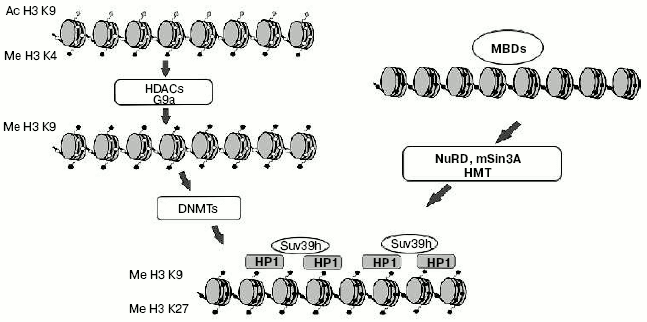
Interaction of DNA methylation and histone modifications. Until recently one of the major questions of epigenetics could be formulated as “What is first: DNA methylation or histone modification?” First suppositions were made soon after identification of MBD-proteins family. It seemed obvious that MBD-proteins interact with methylated DNA and attract complexes containing histone deacetylases. But a number of experiments made on plant and fungi systems refuted this hypothesis. For example, mutation of the gene coding histone methyltransferase in Neurospora crassa leads to general hypomethylation of DNA [64]. On the other hand, deletion of gene CMT3 coding DNA methyltransferase in Arabidopsis does not lead to changes in overall level of histone methylation [65]. These and a number of other works changed common views on the problem of priority of DNA methylation. It is now generally accepted that methylation of histones is the primary event in transcriptional silencing and DNA methylation is less reversible and serves as an “epigenetic lock” (Fig. 3). Nevertheless, in some particular cases DNA methylation can be the first signal for transcriptional repression. For example, the interaction of MeCP2 with methylated regulatory sequences of certain genes leads to increase in methylation of histone H3 (as shown in a paper published by T. Kousaridies) [66]. Moreover, DNA methylation serves as a major epigenetic marker during cell division.
So, DNA methylation is one of the main epigenetic modifications taking part in regulation of gene expression and, consequently, ontogenesis and the normal life of organisms.Fig. 3. Model of heterochromatin formation. White and black circles designate, respectively, non-methylated and methylated DNA; gray rhombi, acetylated lysine residues of histone H3; black pentagons, methylated lysine residues of histone H3.
REFERENCES
1.Hung, M. S., Karthikeyan, N., Huang, B., Koo, H.
C., Kiger, J., and Shen, C. J. (1999) Proc. Natl. Acad. Sci.
USA, 96, 11940-11945.
2.Simmen, M. W., Leitgeb, S., Charlton, J., Jones, S.
J., Harris, B. R., Clark, V. H., and Bird, A. (1999) Science,
283, 1164-1167.
3.Clark, S. J., Harrison, J., and Molloy, P. L.
(1997) Gene, 195, 67-71.
4.Hendrich, B., and Bird, A. (1998) Mol. Cell
Biol., 18, 6538-6547.
5.Antequera, F., and Bird, A. (1993) Proc. Natl.
Acad. Sci. USA, 90, 11995-11999.
6.Kafri, T., Ariel, M., Brandeis, M., Shemer, R.,
Urven, L., McCarrey, J., Cedar, H., and Razin, A. (1992) Genes
Dev., 6, 705-714.
7.Stancheva, I., and Meehan, R. R. (2000) Genes
Dev., 14, 313-327.
8.Macleod, D., Clark, V. H., and Bird, A. (1999)
Nat. Genet., 23, 139-140.
9.Pradhan, S., Bacolla, A., Wells, R. D., and
Roberts, R. J. (1999) J. Biol. Chem., 274,
33002-33010.
10.Okano, M., Xie, S., and Li, E. (1998) Nat.
Genet., 19, 219-220.
11.Pfeifer, G. P., Steigerwald, S. D., Hansen, R.
S., Gartler, S. M., and Riggs, A. D. (1990) Proc. Natl. Acad. Sci.
USA, 87, 8252-8256.
12.Walsh, C. P., Chaillet, J. R., and Bestor, T. H.
(1998) Nat. Genet., 20, 116-117.
13.Kawasaki, H., and Taira, K. (2004) Nature,
431, 211-217.
14.Takagi, N. (2003) Semin. Cell Dev. Biol.,
14, 319-329.
15.Gribnau, J., Hochedlinger, K., Hata, K., Li, E.,
and Jaenisch, R. (2003) Genes Dev., 17, 759-773.
16.Croteau, S., Polychronakos, C., and Naumova, A.
K. (2001) Genesis, 31, 11-16.
17.Filippova, G. N., Fagerlie, S., Klenova, E. M.,
Myers, C., Dehner, Y., Goodwin, G., Neiman, P. E., Collins, S. J., and
Lobanenkov, V. V. (1996) Mol. Cell Biol., 16,
2802-2813.
18.Schoenherr, C. J., Levorse, J. M., and Tilghman,
S. M. (2003) Nat. Genet., 33, 66-69.
19.Kanduri, C., Pant, V., Loukinov, D., Pugacheva,
E., Qi, C. F., Wolffe, A., Ohlsson, R., and Lobanenkov, V. V. (2000)
Curr. Biol., 10, 853-856.
20.Murrell, A., Heeson, S., Bowden, L., Constancia,
M., Dean, W., Kelsey, G., and Reik, W. (2001) EMBO Rep.,
2, 1101-1106.
21.Leighton, P. A., Ingram, R. S., Eggenschwiler,
J., Efstratiadis, A., and Tilghman, S. M. (1995) Nature,
375, 34-39.
22.Nicholls, R. D., and Knepper, J. L. (2001)
Annu. Rev. Genomics Hum. Genet., 2, 153-175.
23.Maher, E. R., and Reik, W. (2000) J. Clin.
Invest., 105, 247-252.
24.Plath, K., Mlynarczyk-Evans, S., Nusinow, D. A.,
and Panning, B. (2002) Annu. Rev. Genet., 36,
233-278.
25.Shibata, S., and Lee, J. T. (2003) Hum. Mol.
Genet., 12, 125-136.
26.Costanzi, C., Stein, P., Worrad, D. M., Schultz,
R. M., and Pehrson, J. R. (2000) Development, 127,
2283-2289.
27.Rougeulle, C., Chaumeil, J., Sarma, K., Allis, C.
D., Reinberg, D., Avner, P., and Heard, E. (2004) Mol. Cell
Biol., 24, 5475-5484.
28.Cohen, D. E., and Lee, J. T. (2002) Curr.
Opin. Genet. Dev., 12, 219-224.
29.Chao, W., Huynh, K. D., Spencer, R. J., Davidow,
L. S., and Lee, J. T. (2002) Science, 295, 345-347.
30.Feinberg, A. P., Gehrke, C. W., Kuo, K. C., and
Ehrlich, M. (1988) Cancer Res., 48, 1159-1161.
31.Feinberg, A. P., and Vogelstein, B. (1983)
Nature, 301, 89-92.
32.Baylin, S. B., Hoppener, J. W., de Bustros, A.,
Steenbergh, P. H., Lips, C. J., and Nelkin, B. D. (1986) Cancer
Res., 46, 2917-2922.
33.Rainier, S., Johnson, L. A., Dobry, C. J., Ping,
A. J., Grundy, P. E., and Feinberg, A. P. (1993) Nature,
362, 747-749.
34.Sherr, C. J. (1996) Science, 274,
1672-1677.
35.Esteller, M., Gaidano, G., Goodman, S. N.,
Zagonel, V., Capello, D., Botto, B., Rossi, D., Gloghini, A., Vitolo,
U., Carbone, A., Baylin, S. B., and Herman, J. G. (2002) J. Natl.
Cancer Inst., 94, 26-32.
36.Yang, D. H., Smith, E. R., Cohen, C., Wu, H.,
Patriotis, C., Godwin, A. K., Hamilton, T. C., and Xu, X. X. (2002)
Cancer, 94, 2380-2392.
37.Gritsko, T. M., Coppola, D., Paciga, J. E., Yang,
L., Sun, M., Shelley, S. A., Fiorica, J. V., Nicosia, S. V., and Cheng,
J. Q. (2003) Clin. Cancer Res., 9, 1420-1426.
38.Cameron, E. E., Bachman, K. E., Myohanen, S.,
Herman, J. G., and Baylin, S. B. (1999) Nat. Genet., 21,
103-107.
39.Magdinier, F., and Wolffe, A. P. (2001) Proc.
Natl. Acad. Sci. USA, 98, 4990-4995.
40.Nguyen, C. T., Gonzales, F. A., and Jones, P. A.
(2001) Nucleic Acids Res., 29, 4598-4606.
41.Meehan, R. R., Lewis, J. D., McKay, S., Kleiner,
E. L., and Bird, A. P. (1989) Cell, 58, 499-507.
42.Lewis, D., Meehan, R., Henzel, J., Maurer-Fogy,
I., Jeppesen, P., Klein, F., and Bird, A. (1992) Cell,
69, 905-914.
43.Klose, R. J., and Bird, A. P. (2004) J. Biol.
Chem., 279, 46490-46496.
44.Johnston, M. V., Mullaney, B., and Blue, M. E.
(2003) J. Child Neurol., 18, 688-692.
45.Ng, H. H., Jeppesen, P., and Bird, A. (2000)
Mol. Cell Biol., 20, 1394-1406.
46.Fujita, N., Watanabe, S., Ichimura, T., Ohkuma,
Y., Chiba, T., Saya, H., and Nakao, M. (2003) Mol. Cell Biol.,
23, 2834-2843.
47.Fujita, N., Watanabe, S., Ichimura, T., Tsuruzoe,
S., Shinkai, Y., Tachibana, M., Chiba, T., and Nakao, M. (2003) J.
Biol. Chem., 278, 24132-24138.
48.Reese, B. E., Bachman, K. E., Baylin, S. B., and
Rountree, M. R. (2003) Mol. Cell Biol., 23,
3226-3236.
49.Hendrich, B., Abbott, C., McQueen, H., Chambers,
D., Cross, S., and Bird, A. (1999) Mamm. Genome, 10,
906-912.
50.Zhang, Y., Ng, H. H., Erdjument-Bromage, H.,
Tempst, P., Bird, A., and Reinberg, D. (1999) Genes Dev.,
13, 1924-1935.
51.Ng, H. H., Zhang, Y., Hendrich, B., Johnson, C.
A., Turner, B. M., Erdjument-Bromage, H., Tempst, P., Reinberg, D., and
Bird, A. (1999) Nat. Genet., 23, 58-61.
52.Hendrich, B., Guy, J., Ramsahoye, B., Wilson, V.
A., and Bird, A. (2001) Genes Dev., 15, 710-723.
53.Hendrich, B., Hardeland, U., Ng, H. H., Jiricny,
J., and Bird, A. (1999) Nature, 401, 301-304.
54.Millar, C. B., Guy, J., Sansom, O. J., Selfridge,
J., MacDougall, E., Hendrich, B., Keightley, P. D., Bishop, S. M.,
Clarke, A. R., and Bird, A. (2002) Science, 297,
403-405.
55.Prokhortchouk, A., Hendrich, B., Jorgensen, H.,
Ruzov, A., Wilm, M., Georgiev, G., Bird, A., and Prokhortchouk, E.
(2001) Genes Dev., 15, 1613-1618.
56.Davis, M. A., Ireton, R. C., and Reynolds, A. B.
(2003) J. Cell Biol., 163, 525-534.
57.Hutchins, A. S., Mullen, A. C., Lee, H. W.,
Sykes, K. J., High, F. A., Hendrich, B. D., Bird, A. P., and Reiner, S.
L. (2002) Mol. Cell, 10, 81-91.
58.Martinowich, K., Hattori, D., Wu, H., Fouse, S.,
He, F., Hu, Y., Fan, G., and Sun, Y. E. (2003) Science,
302, 890-893.
59.Chen, W. G., Chang, Q., Lin, Y., Meissner, A.,
West, A. E., Griffith, E. C., Jaenisch, R., and Greenberg, M. E. (2003)
Science, 302, 885-889.
60.Muller, C., Readhead, C., Diederichs, S., Idos,
G., Yang, R., Tidow, N., Serve, H., Berdel, W. E., and Koeffler, H. P.
(2000) Mol. Cell Biol., 20, 3316-3329.
61.Yoon, H. G., Chan, D. W., Reynolds, A. B., Qin,
J., and Wong, J. (2003) Mol. Cell, 12, 723-734.
62.He, H., and Lehming, N. (2003) Brief. Funct.
Genomic. Proteomic., 2, 234-243.
63.He, H., and Lehming, N. (2003) Brief. Funct.
Genomic. Proteomic., 2, 234-243.
64.Tamaru, H., Zhang, X., McMillen, D., Singh, P.
B., Nakayama, J., Grewal, S. I., Allis, C. D., Cheng, X., and Selker,
E. U. (2003) Nat. Genet., 34, 75-79.
65.Lindroth, A. M., Shultis, D., Jasencakova, Z.,
Fuchs, J., Johnson, L., Schubert, D., Patnaik, D., Pradhan, S.,
Goodrich, J., Schubert, I., Jenuwein, T., Khorasanizadeh, S., and
Jacobsen, S. E. (2004) EMBO J., 23, 4286-4296.
66.Fuks, F., Hurd, P. J., Wolf, D., Nan, X., Bird,
A. P., and Kouzarides, T. (2003) J. Biol. Chem., 278,
4035-4040.