
|
Study of Tissue-Specific CpG Methylation of DNA in Extended Genomic LociT. L. Azhikina* and E. D. SverdlovShemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (7-095) 330-6538; E-mail: tanya@humgen.siobc.ras.ru* To whom correspondence should be addressed.
|
Submitted December 29, 2004
Modern approaches for studies on genome functioning include investigation of its epigenetic regulation. Methylation of cytosines in CpG dinucleotides is an inherited epigenetic modification that is responsible for both functional activity of certain genomic loci and total chromosomal stability. This review describes the main approaches for studies on DNA methylation. Under consideration are site-specific approaches based on bisulfite sequencing and methyl-sensitive PCR, whole-genome approaches aimed at searching for new methylation hot spots, and also mapping of unmethylated CpG sites in extended genomic loci.
KEY WORDS: DNA methylation analysis, bisulfite sequencing, methyl-sensitive PCR (MS-PCR), whole-genome approaches, Non-methylated Genomic Sites Coincidence Cloning (NGSCC)
Methylation of cytosines in the 5´-position of the pyrimidine ring is an extremely important epigenetic modification of the eukaryotic genome that affects various cell processes. Methylation of DNA is associated with compaction of chromatin [1-3], repression of transcription of the inactivated X chromosome of mammalian females [4], genomic imprinting of some specific genes [5], and also with chromosomal stabilization [6]. Methylation is shown to play an important role in regulation of gene expression during embryogenesis of mammals and further cell differentiation [3, 7, 8].
Carcinogenesis is also accompanied by changes in methylation of genomic DNA [9]. These changes include an aberrant local hypermethylation of promotors of various tumor suppressor genes, which results in their silencing, and whole-genome hypomethylation accompanied by activation of oncogenes, retrotransposons, and genomic instability. The role of methylation is shown in development of neurodegenerative diseases, such as Rett [10] and Prader-Willi [11] syndromes.
Modern studies of genome functioning have to include its epigenetic component. This review describes basic approaches used for studies on DNA methylation in the Laboratory of Structure and Functions of Human Genes, Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences.
Now it is well known that DNA of vertebrates is mainly methylated at CpG dinucleotide sequences. But contents of 5-methylcytosine in DNA were under study long before this had been established. 5-Methylcytosine (m5C) was first detected in DNA of higher eukaryotes in 1948 by Hotchkiss [12]. He fractionated by paper chromatography nitrogen bases and nucleosides resulting by hydrolysis of calf thymus DNA with HCl and detected an “anomalous” base. Because of similarity of the absorption spectrum of this base to that of cytosine, it was named epicytosine and later identified as m5C. Similar data were obtained virtually concurrently by Visher and Chargaff [13].
Later, various modifications for chromatographic separation of nitrogen bases and their nucleos(t)ide derivatives were proposed, which provided for significantly increased resolution and more reliable quantitative assessments [14-19]. Progress in analysis of biopolymers by mass-spectrometry ensured its use for the sensitive detection (from 0.01% in 1-10 µg) of minor bases [20, 21]. Methylation of DNA is also studied by photometric [22], fluorimetric [23], enzymatic [24, 25], and immunochemical [26] approaches.
The variety of approaches for identifying the total amount of m5C in the genome allows us to choose the most adequate one with respect to labor-saving and the required sensitivity. All these approaches can be used for studies of whole-genome demethylation and de novo methylation of DNA during gametogenesis, embryogenesis, tissue differentiation, or neoplasia. But obviously, data obtained by these approaches are insufficient for analyzing the specific distribution of methylated sequences in the genome and changes in its pattern. Data on the methylation status of specific genomic sequences would be much more informative for studies on the role of this modification of DNA during different processes in the cell. To obtain such data, special approaches have been developed. Some of them are discussed in the present review.
These approaches are mainly based on two strategies: 1) chemical modification of bases with agents discriminating cytosine and 5-methylcytosine, and 2) use of methyl-sensitive restriction enzymes. Other approaches are also described, e.g., affinity columns based on DNA-binding domains of methyl-binding proteins (methyl-CpG-binding domains (MBD)) during fractionation of nucleic acids by the level of their methylation (this possibility was first shown in [27]) or microarray technology for whole-genome detection of tissue-specific methylation of CpG islands [28].
DETERMINATION OF SPECIFIC METHYLATION BY CHEMICAL
MODIFICATION
Sequencing of DNA based on specific chemical modification of nitrogen bases (hydrazinolysis of pyrimidines and interaction of purines with dimethylsulfate) and the subsequent degradation with piperidine of the polynucleotide chain in the modified sites was elaborated more than 25 years ago [29]. Unlike cytosine, m5C does not react with hydrazine [30]; therefore, the polynucleotide chain is not degraded in the m5C position. On electrophoretic separation of chemical degradation products in a high-resolution gel, methylated cytosine residues can be discriminated from cytosine residues by absence of the corresponding fragment. However, just such negative identification associated with artifacts is a fundamental disadvantage of this approach. Recently KMnO4 was proposed to be used as an additional agent because it interacts with m5C and thus provides for degradation of the polynucleotide chain [31]. Afterwards, such approaches were replaced by more efficient modifications of bisulfite sequencing.
Bisulfite sequencing was proposed in 1992 by Australian researchers [32] and is based on the reaction of bisulfite anion (HSO3-) with cytosine resulting in production of a sulfonated derivative, which is rapidly deaminated. During the subsequent replication, uracil generated instead of cytosine will produce hydrogen bonds with adenine, and this will cause transition in this site in one of the daughter chains. 5-Methylcytosine is sulfonated incomparably more slowly.
The specimen under study (Fig. 1) is treated with bisulfite, then desulfonated under alkaline conditions, and then each chain is PCR-amplified separately (these chains lost their complementation after deamination of all unmethylated cytosines). Then either the joined amplificate [33] or individual preliminary cloned sequences are sequenced. In this case, m5C manifests itself in the radioautograph as a positive signal corresponding to cytosine. This approach has some advantages, in particular, it is very sensitive (from 10-12 g, usually <=10-9 g DNA [34]), provides for a separate analysis of two chains, as well as the possibility to study methylation status of individual DNA molecules that allows us to study more finely changes in methylation of specific sequences.
However, bisulfite treatment of DNA has some limitations and shortcomings, e.g., a decrease in the sensitivity because of partial degradation of DNA caused by its apurinization at acidic pH and also incomplete deamination of DNA. A number of approaches have been developed to ameliorate these drawbacks [31, 35-38].Fig. 1. Bisulfite sequencing. DNA is denatured and treated with sodium bisulfite, and then PCR is performed with primers specific to one of two modified chains. The resulting amplificate is sequenced immediately or after preliminary cloning of individual sequences.
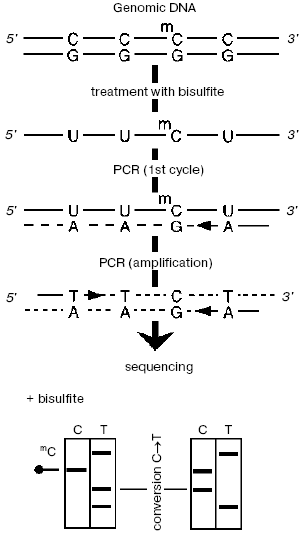
Sequencing of genomic DNA pretreated with sodium bisulfite can be used as a technological principle for large-scale epigenetic studies. Therefore, the Human Epigenome Consortium (http://www.epigenome.org) responsible for the project of identification and cataloging of methylation patterns of genomic DNA of all human genes in the main tissues uses bisulfite sequencing as the major approach for obtaining information about DNA methylation state.
METHYL-SENSITIVE RESTRICTION ENZYMES
The ability of methyl-sensitive restriction enzymes possessing the CpG dinucleotide-containing recognition site to cut only unmethylated sites is used for various approaches [39-41]. Parallel hydrolysis of DNA with such a restriction enzyme and its methyl-insensitive isoschizomer can present information about the number and distribution of the corresponding sites in the specimens containing 5-methylcytosine. Most often, the pair MspI/HpaII is used which recognizes the CCGG sequence, with HpaII sensitive to methylation of the second C in the site.
Methylation in plants is studied using isoschizomers EcoRII/BstN1, which cut the CCNGG site. BstN1 is methyl-sensitive. At present, more than 300 methyl-sensitive restriction endonucleases are known, and ~30 of them have methyl-insensitive isoschizomers [42].
Methyl-sensitive restriction endonucleases were first used by Bird and Southern for analyzing methylation of genomic DNA during studies on methylation of amphibian rRNA genes [43]. The endonucleases MspI/HpaII used by these authors differently hydrolyzed somatic and oocytic rDNA, and this was shown to be associated with the different degree of their methylation. In general, genomic DNA cut with methyl-sensitive restriction enzymes can be analyzed using Southern-hybridization with specific probes, PCR, or both these approaches. Southern-hybridization allows us to rapidly analyze many specimens at a rather sufficient sensitivity (from 10% of methylated sequences in 10 µg of the initial DNA).
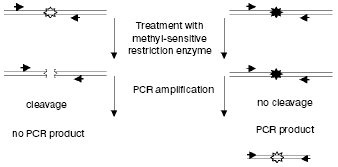
PCR-amplification of the genomic region under study becomes more sensitive after pretreatment of the sample with endonuclease (from 0.1% of methylated sequences can be detected in 10 ng of the initial DNA) [44]. Only the initially methylated and thus non-hydrolyzed fragment of DNA will be amplified exponentially (Fig. 2). The presence of methylated and unmethylated sequences in the initial specimen can be quantitatively assessed by the so-called competitive PCR when the same pair of primers is used for amplification of both the genomic and the exogenously added identical DNA discriminated by size [45]. Adaptors preliminary ligated to hydrolysis products can also be used, and PCR can be performed with one specific primer and with the other identical to the adaptor's sequence (the ligation mediated PCR (LM-PCR)). In this case, both hydrolyzed and non-hydrolyzed (methylated) fragments will be amplified in the site under study, and the resulting ratio of the amplification products will correspond to their presence in the initial mixture [46].
Nevertheless, it is obvious that these approaches require that the genomic region under study be presequenced. It is also essential that by these approaches methylation is studied not of the whole sequence but only of its regions that contain the recognition site of the endonuclease used. These approaches are also associated with possible artifacts caused by incomplete hydrolysis of DNA.Fig. 2. Use of methyl-sensitive restriction endonucleases for analyzing methylation of specific CpG sites. Genomic DNA containing unmethylated (white asterisks) and methylated (black asterisks) CpG sites is restricted and analyzed by PCR with primers (shown by arrows) flanking the site under study.
APPROACHES FOR STUDY AND COMPARISON OF METHYLATION OF SPECIFIC
GENOMIC SEQUENCES
Studies on DNA methylation are designed either to determine the methylation status of a certain nucleotide sequence (e.g., studies on changes in methylation of CpG islands of tumor suppressor genes), or to search for new specifically methylated genomic sites responsible for crucial functional changes in the organism.
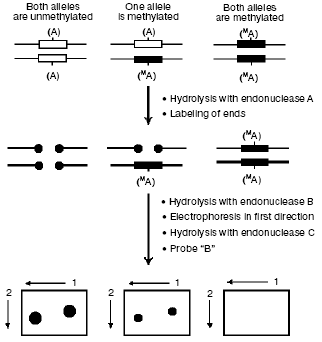
The first group includes the already described bisulfite sequencing, methyl-sensitive PCR (methylation-specific PCR (MSP)) [47], and their combinations. Among other methods [48-53], attention should be paid to the wide-scale whole-genome analysis of DNA methylation by restriction landmark genomic scanning with involvement of methyl-sensitive restriction endonucleases (RLGS-M) [54] (Fig. 3). In this case a specimen is initially hydrolyzed with a methyl-sensitive restriction endonuclease (A, most often NotI which recognizes the palindrome octanucleotide 5´-GC.GGCCGC-3´), and the fragments produced are radiolabeled and hydrolyzed with another restriction enzyme (B). Then the hydrolyzate is fractionated by electrophoresis, and the agarose gel is treated with the third endonuclease (C) and separated in the other direction. The resulting radioautograph presents a characteristic two-dimensional picture (pattern) of the restricted fragments. Increase or decrease in the intensity of a definite spot is associated with hypo- or hypermethylation, respectively, of the endonuclease A recognition site. Fragments of interest can be eluted from the gel and used for their mapping in the genome under study. NotI used as enzyme A allows us to purposefully study gene-containing regions of the genome, because ~90% of all sequences recognized by NotI are located in CpG islands [55], the majority of which, in turn, is associated with genes and their promotor regions in mammalian genomes.
However, along with obvious virtues, this approach also has shortcomings, in particular, it requires a large amount of DNA (more than 10 µg) to obtain a radioautograph of sufficient quality and is rather laborious because of preparation and analysis of radioautographs (software has been recently developed which automates the treatment of results obtained by the RLGS-M approach [56, 57]).Fig. 3. The RLGS-M approach. Specimens of DNA are first hydrolyzed with a methyl-sensitive endonuclease A and then the sticky ends are completed in the presence of radiolabeled nucleoside triphosphates. Then the preparation is hydrolyzed with endonuclease B and fractionated by electrophoresis in the first direction; then the gel is treated with the endonuclease C and separated in the other direction. Comparing the spot intensities in radioautographs from different specimens, one can identify differentially labeled sequences.
However, even improved [58], the approach remains tedious and is not widely used.
Methylation status of two specimens can also be compared by subtractive hybridization (the methylation-sensitive representational difference analysis (MS-RDA)). This approach includes comparison by subtractive hybridization of two simplified genomic fractions, which are HpaII amplicons of the restricted genomic DNA. And during the amplification the mixture is enriched with fragments of 300-600 bp, which are mainly hypomethylated (and as a result hydrolyzed by endonuclease) regions of the genome. The subsequent 2-3 rounds of subtractive hybridization result in a set of sequences with different methylation status in the two DNA specimens under comparison. This approach was first used to search for differentially methylated sequences in normal liver parenchyma and hepatocellular carcinoma [59, 60]. This approach can also be used in searching for genes capable of imprinting [61], identification of tissue-specific expressed sequences [62], whole-genome analysis of changes in the methylation status under conditions of foreign DNA insertions (transgenes, retroviruses, DNA-transposons [63]).
But “simplification” of specimens during their amplification is obviously associated with a loss of some of differentially methylated sequences, and the fraction analyzed is produced rather occasionally.
In general, whole-genome approaches cannot give comprehensive data because of the complexity of the eukaryotic genome. Moreover, they present for the researcher excess information that is difficult to analyze.
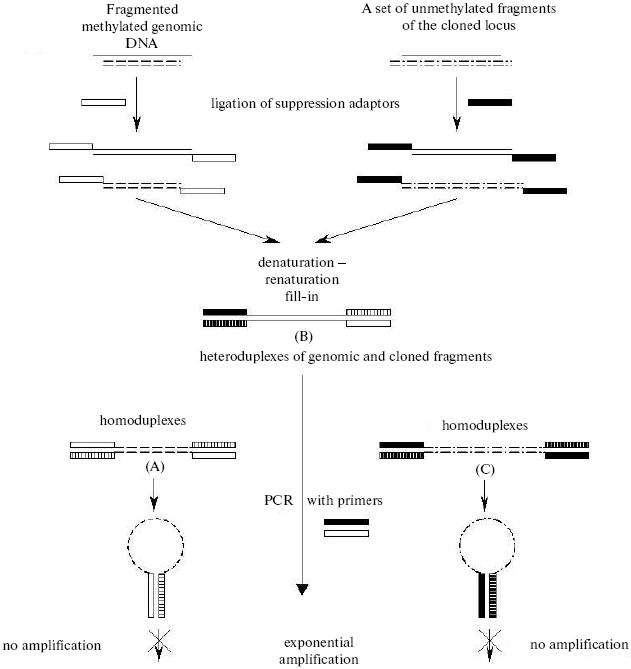
Recently a tendency appeared for limiting the whole-genome analysis by extended polygenic loci possessing common regulatory systems. We have developed an approach for mapping unmethylated CpG sites on megabase regions of the genome (Non-methylated Genomic Sites Coincidence Cloning (NGSCC) [64]), which allows us to map sites independently of their position with respect to the CpG island. The approach is based on cloning identical nucleotide sequences belonging to different fragmented DNA pools (Fig. 4). As the DNA pools for comparison, we used the genomic DNA from a certain tissue fragmented with the methyl-sensitive restriction enzyme HpaII and the complete set of the HpaII restricted fragments of DNA from the cloned locus. Two types of suppression oligonucleotide adaptors were ligated to the resulting pools. The joint denaturation and renaturation of both pools of the fragments resulted in generation of heteroduplexes of identical nucleotide sequences from different pools (duplexes B in Fig. 4). These heteroduplexes can be isolated from the mixture using the effect of a selective PCR suppression caused by the presence of different suppression adaptors [65]. The isolated fragments belong to the genomic locus under study and contain the unmethylated CpG site specific for the tissue under study. The cloning, determination of nucleotide sequence of these fragments, and mapping of unmethylated CpG sites in the genome result in the distribution profile of unmethylated sites in the human genome locus under study. Such a tissue-specific distribution is characteristic for a definite tissue or stage in the organism's development; therefore, it has to represent its functional features.
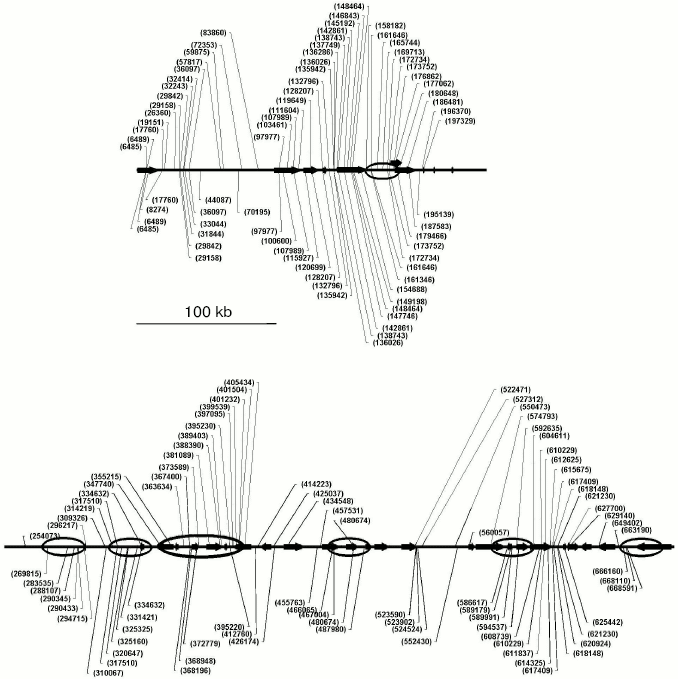
By the NGSCC approach the distribution profiles of unmethylated CpG dinucleotides (in the HpaII sites) were obtained on the 1 Mb length D19S208-COX7A1 locus of chromosome 19. Figure 5 presents the site distribution along the locus, which was obtained for two tissues: normal testis parenchyma and seminoma (germinogenic tumor). For each tissue, a tendency was revealed for clusterization of unmethylated sites. Moreover, the normal and tumor tissues were different in the methylation profiles. Regions of the locus with the tissue-specific difference in methylation are presented in Fig. 5 as ovals. Such regions are located in both gene-rich and noncoding areas of the locus.Fig. 4. Scheme for cloning identical sequences based on PCR selective suppression. The fragments unique for each of the initial sets (A and C) are shown as dashed and dash-dotted lines, the common fragments (B) are shown as solid lines. Two types of suppression adaptors are shown as black or white rectangles, and the complementary sequences are presented as hatched rectangles of the corresponding color.
Thus, the NGSCC approach results in the set of unmethylated restriction sites (URS), which are distributed lengthwise the genomic locus under study, independently of location of CpG islands. Such URS can be considered as markers with the distribution reliably describing the tissue-specific methylation profile inside the extended locus. This profile is unique and characterizes just the tissue under study and its functional state.Fig. 5. Distribution of unmethylated restriction sites (URS) along the locus under study for the normal parenchyma (above the axis) and seminoma (under the axis). Positions of unmethylated sites are shown by lines with numbers corresponding to coordinates on chromosome 19. The annotated genes are indicated with horizontal arrows. Regions of tissue-specific methylation are shown with ovals, indicating the different distribution of unmethylated sites in genomes of the normal and tumor tissues.
We have reviewed only some of a great variety of approaches that were and are used now for studies on DNA methylation in higher eukaryotes. These approaches are different in informativity and give qualitatively different data: from the total amount of 5-methylcytosine in DNA to the specific pattern of its distribution in the genome under investigation. It should be noted that only a few of these approaches are used widely. This is obviously associated with differences in sensitivity, requirements for labor intensity, and reliability. And finally, the choice of approach is most essentially determined by its suitability for the specific purpose of the study.
The work was supported by the Scientific School program of the Russian Federation President (project NSh 2006.2003.4), program of the Ministry of Industry, Science, and Technology (project RF 43.073.1.1.1509), and program of the Russian Academy of Sciences on Molecular and Cellular Biology.
REFERENCES
1.Bird, A. P., and Wolffe, A. P. (1999) Cell,
99, 451-454.
2.Ng, H. H., Zhang, Y., Hendrich, B., Johnson, C. A.,
Turner, B. M., Erdjument-Bromage, H., Tempst, P., Reinberg, D., and
Bird, A. (1999) Nat. Genet., 23, 58-61.
3.Bird, A. (2002) Genes Dev., 16,
6-21.
4.Avner, P., and Heard, E. (2001) Nat. Rev.
Genet., 2, 59-67.
5.Reik, W., and Walter, J. (2001) Nat. Rev.
Genet., 2, 21-32.
6.Hsieh, J., and Fire, A. (2000) Annu. Rev.
Genet., 34, 187-204.
7.Thomassin, H., Flavin, M., Espinas, M. L., and
Grange, T. (2001) EMBO J., 20, 1974-1983.
8.Li, E. (2002) Nat. Rev. Genet., 3,
662-673.
9.Jones, P. A., and Baylin, S. B. (2002) Nat. Rev.
Genet., 3, 415-428.
10.Amir, R. E., van den Veyver, I. B., Wan, M.,
Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999) Nat. Genet.,
23, 185-188.
11.Xin, Z., Tachibana, M., Guggiari, M., Heard, E.,
Shinkai, Y., and Wagstaff, J. (2003) J. Biol. Chem., 278,
14996-15000.
12.Hotchkiss, R. D. (1948) J. Biol. Chem.,
175, 315-332.
13.Visher, E., and Chargaff, E. (1948) J. Biol.
Chem., 176, 703-714.
14.Vanyushin, B. F., Belozersky, A. N., Kokurina, N.
A., and Kadirova, D. X. (1968) Nature, 218,
1066-1067.
15.Rubery, E. D., and Newton, A. A. (1971)
Analyt. Biochem., 42, 149-154.
16.Razin, A., and Sedat, J. (1977) Analyt.
Biochem., 77, 370-377.
17.Burtis, C. A. (1970) J. Chromatogr.,
51, 183-194.
18.Kuo, K. C., McCune, R. A., Gehrke, C. W.,
Midgett, R., and Ehrlich, M. (1980) Nucleic Acids Res.,
8, 4763-4776.
19.Patel, C. V., and Gopinathan, K. P. (1987)
Biochem. Biophys. Res. Commun., 142, 334-340.
20.Razin, A., and Cedar, H. (1977) Proc. Natl.
Acad. Sci. USA, 74, 2725-2728.
21.Singer, J., Schnute, W. C., Jr., Shively, J. E.,
Todd, C. W., and Riggs, A. D. (1979) Analyt. Biochem.,
94, 297-301.
22.Vanyushin, B. F., Tkacheva, A. N., and
Belozersky, A. N. (1970) Nature, 225, 948-949.
23.Oakeley, E. J., Schmitt, F., and Jost, J. P.
(1999) Biotechniques, 27, 744-746, 748-750, 752.
24.Sheid, B., Srinivasan, P. R., and Borek, E.
(1968) Biochemistry, 7, 280-285.
25.Wu, J., Issa, J. P., Herman, J., Bassett, D. E.,
Jr., Nelkin, B. D., and Baylin, S. B. (1993) Proc. Natl. Acad. Sci.
USA, 90, 8891-8895.
26.Miller, O. J., Schnedl, W., Allen, J., and
Erlanger, B. F. (1974) Nature, 251, 636-637.
27.Cross, S. H., Charlton, J. A., Nan, X., and Bird,
A. P. (1994) Nat. Genet., 6, 236-244.
28.Huang, T. H., Perry, M. R., and Laux, D. E.
(1999) Hum. Mol. Genet., 8, 459-470.
29.Maxam, A. M., and Gilbert, W. (1977) Proc.
Natl. Acad. Sci. USA, 74, 560-564.
30.Church, G. M., and Gilbert, W. (1984) Proc.
Natl. Acad. Sci. USA, 81, 1991-1995.
31.Rein, T., Natale, D. A., Gartner, U., Niggemann,
M., DePamphilis, M. L., and Zorbas, H. (1997) J. Biol. Chem.,
272, 10021-10029.
32.Frommer, M., McDonald, L. E., Millar, D. S.,
Collis, C. M., Watt, F., Grigg, G. W., Molloy, P. L., and Paul, C. L.
(1992) Proc. Natl. Acad. Sci. USA, 89, 1827-1831.
33.Paul, C. L., and Clark, S. J. (1996)
Biotechniques, 21, 126-133.
34.Clark, S. J., Harrison, J., Paul, C. L., and
Frommer, M. (1994) Nucleic Acids Res., 22, 2990-2997.
35.Raizis, A. M., Schmitt, F., and Jost, J. P.
(1995) Analyt. Biochem., 226, 161-166.
36.Rein, T., DePamphilis, M. L., and Zorbas, H.
(1998) Nucleic Acids Res., 26, 2255-2264.
37.Olek, A., Oswald, J., and Walter, J. (1996)
Nucleic Acids Res., 24, 5064-5066.
38.Rein, T., Zorbas, H., and DePamphilis, M. L.
(1997) Mol. Cell. Biol., 17, 416-426.
39.Cedar, H., Solage, A., Glaser, G., and Razin, A.
(1979) Nucleic Acids Res., 6, 2125-2132.
40.Singer, J., Roberts-Ems, J., and Riggs, A. D.
(1979) Science, 203, 1019-1021.
41.Antequera, F., Tamame, M., Villanueva, J. R., and
Santos, T. (1984) J. Biol. Chem., 259, 8033-8036.
42.McClelland, M., Nelson, M., and Raschke, E.
(1994) Nucleic Acids Res., 22, 3640-3659.
43.Bird, A. P., and Southern, E. M. (1978) J.
Mol. Biol., 118, 27-47.
44.Singer-Sam, J., Grant, M., LeBon, J. M., Okuyama,
K., Chapman, V., Monk, M., and Riggs, A. D. (1990) Mol. Cell.
Biol., 10, 4987-4989.
45.Singer-Sam, J., LeBon, J. M., Tanguay, R. L., and
Riggs, A. D. (1990) Nucleic Acids Res., 18, 687.
46.Steigerwald, S. D., Pfeifer, G. P., and Riggs, A.
D. (1990) Nucleic Acids Res., 18, 1435-1439.
47.Herman, J. G., Graff, J. R., Myohanen, S.,
Nelkin, B. D., and Baylin, S. B. (1996) Proc. Natl. Acad. Sci.
USA, 93, 9821-9826.
48.Gonzalgo, M. L., and Jones, P. A. (1997)
Nucleic Acids Res., 25, 2529-2531.
49.Sadri, R., and Hornsby, P. J. (1996) Nucleic
Acids Res., 24, 5058-5059.
50.Xiong, Z., and Laird, P. W. (1997) Nucleic
Acids Res., 25, 2532-2534.
51.Bianco, T., Hussey, D., and Dobrovic, A. (1999)
Hum. Mutat., 14, 289-293.
52.Reyna-Lopez, G. E., Simpson, J., and
Ruiz-Herrera, J. (1997) Mol. Gen. Genet., 253,
703-710.
53.Huang, T. H., Laux, D. E., Hamlin, B. C., Tran,
P., Tran, H., and Lubahn, D. B. (1997) Cancer Res., 57,
1030-1034.
54.Costello, J. F., Fruhwald, M. C., Smiraglia, D.
J., Rush, L. J., Robertson, G. P., Gao, X., Wright, F. A., Feramisco,
J. D., Peltomaki, P., Lang, J. C., Schuller, D. E., Yu, L., Bloomfield,
C. D., Caligiuri, M. A., Yates, A., Nishikawa, R., Su Huang, H.,
Petrelli, N. J., Zhang, X., O'Dorisio, M. S., Held, W. A., Cavenee, W.
K., and Plass, C. (2000) Nat. Genet., 24, 132-138.
55.Lindsay, S., and Bird, A. P. (1987)
Nature, 327, 336-338.
56.Rouillard, J. M., Erson, A. E., Kuick, R.,
Asakawa, J., Wimmer, K., Muleris, M., Petty, E. M., and Hanash, S.
(2001) Genome Res., 11, 1453-1459.
57.Takahashi, K., Nakazawa, M., and Watanabe, Y.
(2001) Genome Inform. Ser. Workshop Genome Inform., 12,
212-221.
58.Plass, C., Weichenhan, D., Catanese, J.,
Costello, J. F., Yu, F., Yu, L., Smiraglia, D., Cavenee, W. K.,
Caligiuri, M. A., deJong, P., and Held, W. A. (1997) DNA Res.,
4, 253-255.
59.Ushijima, T., Morimura, K., Hosoya, Y., Okonogi,
H., Tatematsu, M., Sugimura, T., and Nagao, M. (1997) Proc. Natl.
Acad. Sci. USA, 94, 2284-2289.
60.Kaneda, A., Takai, D., Kaminishi, M., Okochi, E.,
and Ushijima, T. (2003) Ann. N. Y. Acad. Sci., 983,
131-141.
61.Peters, J., Wroe, S. F., Wells, C. A., Miller, H.
J., Bodle, D., Beechey, C. V., Williamson, C. M., and Kelsey, G. (1999)
Proc. Natl. Acad. Sci. USA, 96, 3830-3835.
62.Yuan, L., Shan, J., De Risi, D., Broome, J.,
Lovecchio, J., Gal, D., Vinciguerra, V., and Xu, H. P. (1999) Cancer
Res., 59, 3215-3221.
63.Muller, K., Heller, H., and Doerfler, W. (2001)
J. Biol. Chem., 276, 14271-14278.
64.Azhikina, T., Gainetdinov, I., Skvortsova, Y.,
Batrak, A., Dmitrieva, N., and Sverdlov, E. (2004) Mol. Genet.
Genomics, 271, 22-32.
65.Diatchenko, L., Lau, Y. F., Campbell, A. P.,
Chenchik, A., Moqadam, F., Huang, B., Lukyanov, S., Lukyanov, K.,
Gurskaya, N., Sverdlov, E. D., and Siebert, P. D. (1996) Proc. Natl.
Acad. Sci. USA, 93, 6025-6030.