REVIEW: DNA Demethylation and Carcinogenesis#
N. P. Kisseljova* and F. L. Kisseljov
Institute of Carcinogenesis, Blokhin Russian Cancer Research Center, Kashirskoe Shosse 24, 115478 Moscow, Russia; fax: (7-095) 324-1205; E-mail: natalia_kis@crc.umos.ru* To whom correspondence should be addressed.
# This article was not published in the journal special issue devoted to the 70th anniversary of B. F. Vanyushin (Biochemistry (Moscow) (2005) 70, No. 5) because of the limiting volume of the journal.
Received November 12, 2004
DNA methylation plays an important role in the establishment and maintenance of the program of gene expression. Tumor cells are characterized by a paradoxical alteration of DNA methylation pattern: global DNA demethylation and local hypermethylation of certain genes. Hypermethylation and inactivation of tumor suppressor genes are well documented in tumors. The role of global genome demethylation in carcinogenesis is less studied. New data provide evidence for independence of DNA hypo- and hypermethylation processes in tumor cells. These processes alter expression of genes that have different functions in malignant transformation. Recent studies have demonstrated that global decrease in the level of DNA methylation is related to hypomethylation of repeated sequences, increase in genetic instability, hypomethylation and activation of certain genes that favor tumor growth, and increase in their metastatic and invasive potential. The recent data on the role of DNA demethylation in carcinogenesis are discussed in this review. The understanding of relationships between hypo- and hypermethylation in tumor cells is extremely important due to reversibility of DNA methylation and attempts to utilize for anti-tumor therapy the drugs that modify DNA methylation pattern.
KEY WORDS: DNA hypomethylation, DNA hypermethylation, gene expression, repeated DNA sequences, tumor suppressors, proto-oncogenes, genetic instability, metastasis
Alteration of expression of many genes occurs during tumor formation and progression. The epigenetic modifications, i.e. modifications that do not influence the genetic potential of genes, but lead to inherited alteration of their expression, play an important role in carcinogenesis. Two related components contribute to epigenetic modifications of gene expression: alteration of chromatin conformation (chromatin remodeling) and modification of DNA methylation pattern.
The methyl group is located in the fifth position of the ring in some cytosines within CpG dinucleotides in the genome of vertebrates [1]. The pattern of 5-methylcytosine distribution in the genome is unique for each cell type and is established in embryogenesis as a result of balance between DNA methylation and demethylation [2, 3]. In the cells of an adult organism the methylation pattern is maintained in relatively unmodified state, but may alter significantly with aging and during certain diseases including malignant neoplasias. Abnormal DNA methylation pattern was detected as a constant modification in tumor cells long ago. A considerable decrease in 5-methylcytosine content in comparison to corresponding normal tissues was observed in many human and animal neoplasias [4-8]. Together with global decrease in DNA methylation, a selective local hypermethylation of cytosine within CpG dinucleotides located in the 5´-regulatory region of genes is observed in tumor cells [9].
Hypermethylation and hypomethylation (demethylation) are terms that denote alteration of methylation status of a genome fragment in a tumor cell in comparison to a corresponding normal cell. Although aberrant local hypermethylation was detected later than the decrease in 5-methylcytosine content in neoplastic cells, actually the first effect is much better studied than the latter. It has been demonstrated in many studies that several suppressors of tumor growth, genes that regulate cellular motility, apoptosis, and angiogenesis and genes of the DNA repair system may be inactivated in tumors due to aberrant methylation of their 5´-regulatory regions [9]. Regulatory regions of these genes are located in the specific GC-rich sequences--CpG islands that are not methylated in all tissues of the adult organism. Aberrant methylation of CpG islands is accompanied by inhibition of transcription of the gene [10]. These numerous studies confirmed the suggestion that was made soon after the discovery of DNA methylation about a possible role of methylation in regulation of gene transcription [11].
The list of genes inactivated in tumors by methylation of their promoters is increasing constantly. In has been shown that the pattern of aberrantly methylated genes represents unique characteristics of a tumor and may be used for classification of a tumor or the stage of its progression. Attempts are being undertaken to use certain genes with high frequency of methylation as markers for diagnostics and determination of risk-groups for cancer development, prognosis, and monitoring of the disease. Inhibitors of methylation are under trial as anti-cancer drugs. The progress in these studies is summarized in numerous reviews [12-15].
Although DNA demethylation is widely spread within the genome and is clearly associated with tumor formation, its significance for tumor cells remained obscure until recently. Progress in the study of genome demethylation and its role in carcinogenesis was achieved only during recent years. The recent studies are reviewed below. The simultaneous presence of hyper- and hypomethylation in tumors seems to be paradoxical. Understanding of the role of these processes and interaction between them in carcinogenesis is of particular importance due to their reversibility and possibility for drug treatment of both processes.
HYPOMETHYLATED DNA SEQUENCES IN HUMAN TUMORS
It was already shown in early studies describing the decrease in the total content of 5-methylcytosine in different tumor forms that the level of general demethylation and frequency of demethylation increase with tumor progression [6, 8]. However, the establishment of demethylated sequences in neoplastic cells was necessary to understand what selective advantages are acquired by the cells with decreased level of DNA methylation. The majority of tumor cells possess the following properties that are obtained during progression and promote malignant growth: a) self-maintenance with growth signals; b) insensitivity to anti-growth signals; c) unlimited proliferative potency; d) block of apoptosis e) block of differentiation; f) stimulation of angiogenesis; g) ability for invasion and metastases; h) genetic instability. Actually many signal pathways, whose alteration in tumor cells favor the acquirement of these properties, are established. It appears to be confirmed that structural modifications of genome and local DNA hypermethylation are involved in alteration of functions or expression of many genes--components of these signal pathways. The role of DNA demethylation in acquiring of these properties by tumor cells is only beginning to be clarified.
It is evident that the targets for aberrant demethylation in tumors are represented by DNA sequences that are methylated in normal tissues. The following groups can be attributed to these DNA sequences: 1) repeated sequences that are methylated in all studied genome loci [16]; 2) unique DNA sequences encoding for genes whose suppression of expression in normal cells is achieved through methylation. Imprinted genes with mono-allelic expression that is maintained by methylation and certain tissue-specific genes are attributed to these genes [17-19].
Demethylation of Repeated DNA Sequences
It was demonstrated in one of the first studies of DNA demethylation in animal tumors that a significant decrease in 5-methylcytosine content (by 30-40%) occurs in moderately and frequently repeated DNA sequences [20]. The classes of repeated sequences with tumor-specific demethylation were characterized much later. Endogenous retrotransposons are attributed to these DNA sequences. These elements of the human genome are methylated in all studied genomic loci. Many transposons have strong promoters; methylation of the latter correlates with suppression of transcription activity and apparently prevents their further expansion throughout the genome [16, 21]. Hypomethylation of long (several thousands nucleotides) dispersed retrotransposons LINE-1 in tumor cells in comparison to normal tissues was demonstrated for many tumors [22-28]. In some cases, demethylation of LINE-1 was accompanied by transcription. The authors suggest that the decrease in the level of methylation of these genomic elements may activate their transcription and provides them competence for transposition and recombination. In reality, exogenous expression of LINE-1 in transformed cells is accompanied by LINE-1 transpositions that are connected with genome deletions and inversions [29]. However, the recombination with involvement of LINE-1 in tumors has not been described yet.
The decrease in the level of methylation of another group of retro-elements, the endogenous retroviruses from the HERV-K family, was also shown in certain tumors. The human genome contains approximately 50 full-length copies of these viruses and about several hundred fragments of the viral genome located in all chromosomes. Deleted copies contain regulator regions--LTR (long terminal repeats) [30]. Hypomethylation of HERV-K observed in bladder carcinomas was not accompanied by transcription [23]. Correlation of hypomethylation of the sequences of the viral gene GAG with expression of the protein was detected in primary testicular tumors [31]. The data on demethylation of these retro-elements are not sufficient yet for understanding of the role of this process in carcinogenesis. It is known that HERV-K LTRs can direct transcription in both orientations in vitro and that some of them are located in gene introns in the antisense orientation with respect to these genes, i.e. potentially they may be involved in the production of antisense RNA [32, 33]. It is not yet known whether LTRs of these endogenous viruses are involved in destabilization of transcription in tumor cells.
In addition to retrotransposons, satellite DNA representing approximately 10% of the genome is another target for demethylation. Satellite repeats are relatively short (up to 200 bp) highly repetitive non-coding DNA sequences that form tandem blocks located in heterochromatin regions of chromosomes (pericentromeric and telomeric regions). Heterochromatin is transcriptionally inactive chromatin characterized by histone deacetylation, DNA methylation, and compactization of nucleosomes. Hypomethylation of several satellite repeats (Satalpha, Sat2, Sat3) has been observed in different tumor forms with high frequency (40-90%) in comparison to related normal tissues [34-40]. Hypomethylation of satellite DNA increased with progression of ovarian tumors, thus it can be considered as a marker of poor prognosis for this form of tumors [38, 39]. It has also been demonstrated that hypomethylation of satellite DNA does not correlate with hypermethylation of the promoter regions of several genes--suppressors of tumor growth in the same tumors, thus representing an independent alteration of DNA methylation pattern [34, 38]. In addition, demethylation of satellite DNA correlates with the decrease in 5-methylcytosine content in DNA of tumor cells [39]. Taking into account this coincidence together with the fact that satellite DNA represents 10% of the genome, it can be suggested that demethylation of the satellite repeats contributes heavily in global genome demethylation in tumors.
What is the role of hypomethylation of the repeats in carcinogenesis? It is known that homologous recombinations between repeats can lead to chromosomal rearrangements [41, 42]. It was suggested that methylation of the repeats restricts homologous recombination [16, 43]. The following facts favor this hypothesis. It was shown that methylation of the DNA fragment that overlaps the hot spot of meiotic recombination leads to more than 100-fold decrease in the frequency of crossing-over within this locus in fungi [44]. Translocations that occur as a result of exchange between decondensed and demethylated satellite repeats and multibranched structures formed by the long arms of chromosomes 1 and 16 on which these repeats are located are often observed in patients with a rare genetic disorder, ICF syndrome (immunodeficiency, centromeric heterochromatin instability and facial abnormalities). The analogous chromosomal anomalies are observed in normal B-cells after their treatment with demethylating agents [45, 46]. The absence of the functional gene DNMT1 (one of the DNA-methyltrasferases) in mice embryonic cells ES leads to DNA hypomethylation followed by the increase in mutations of both the endogenous gene (HPRT) and transgenes. The mutations of these loci were mainly represented by deletions caused by mitotic recombinations or chromosomal losses with subsequent duplication of the remaining chromosome [47]. In mice with hypomorphic DNMT1 allele the expression of DNA-methyltransferase 1 was reduced by 90% in comparison to the wild-type mice. This reduction was accompanied by decrease in the global level of genome methylation and centromeric repeats in all studied tissues. These mice developed aggressive T-cell lymphomas that displayed trisomy of chromosome 15 in 80% of cases [48]. The authors managed to demonstrate without using genotoxic demethylating agents that first, genome hypomethylation may induce tumors in animals, and second, these tumors are characterized by increased chromosomal instability. Finally, it should be noted that in human tumors with demethylated repeats Satalpha and Sat2 located in the pericentromeric region of chromosome 1 rearrangements of these regions are often observed [36, 37].
The abovementioned data clearly indicate that hypomethylation of repeats may lead to the development of genetic instability that is a significant peculiarity of tumor cells.
Demethylation of Unique DNA Sequences
Imprinted genes. It is known that DNA methylation is involved in the maintenance of mono-allelic expression of imprinted genes. Genome imprinting is a gamete-specific epigenetic genome modification that maintains the unequal expression of the maternal and paternal alleles of a gene in the tissues of an adult organism. A relatively small number of genes undergo imprinting in mammals. Imprinted genes are often located in clusters with coordinated regulation of gene expression within such clusters. The genes that are transcribed from either the maternal or paternal alleles can be located in the same cluster. The specific CG-rich regulating regions DMR (differentially methylated regions) or ICR (imprinting control regions) are located within the imprinted loci and can influence the gene imprinting at distances of several hundred thousand base pairs. DMRs are normally highly methylated only on one allele and are partly or fully demethylated on the other allele. DMRs in hypomethylated state seem to function as insulators or silencers [17, 49]. Not only deletions in imprinted loci, but also the disruption of imprinting due to deregulation of DNA methylation are observed in tumor cells [50, 51]. Both hypermethylation and hypomethylation of distinct regulatory DNA regions may occur in an imprinted locus. Demethylation of the DMR allele that is methylated in normal cells may differently modulate expression of genes controlled by this DMR leading to: a) appearance of bi-allelic gene expression (relaxation of imprinting); b) silencing of both alleles. Examples of DMR hypomethylation and variation of expression of certain genes in human tumors are listed below.
The gene IGF2 located in the well-known imprinted locus of chromosome 11p15 encodes for embryonic growth factor. Deletions within this locus are typical for many tumor types. In colorectal cancer, relaxation of imprinting of the IGF2 gene was accompanied by hypomethylation of IGF2 DMR located between the second and the third exons of this gene [52].
The gene CDKN1C encodes for the inhibitor of cyclin-dependent kinases p57KIP2 and is located in the same imprinted locus as IGF2 (11p15.5). Reduction in CDKN1C gene expression correlated with hypomethylation of DMR-LIT1 (KvDMR1) in esophageal cancer cell lines. DMR-LIT1 is located in the intron of another gene from the same locus, KvLQT1, and determines imprinting of several genes in this locus [53]. Hypomethylation of DMR-LIT1 was accompanied by modification of histones within this region. The decrease in expression of the CDKN1C gene did not correlate either with methylation of CpG island in its own promoter or with histone modification in the region of the gene promoter [54]. Hypomethylation of the maternal allele KvDMR1 has also been detected in breast, gastric, cervical, and liver carcinomas [55, 56]. However the authors did not investigate the expression of which genes within this locus was altered in the majority of these tumor types. The correlation between hypomethylation of the maternal allele KvDMR1 with the absence or decrease in expression of both the CDKN1C and IGF2 genes located in the same locus at significant distance from the KvLQT1 gene was observed only in liver tumors (see above) [56].
Thus, DNA demethylation observed in tumor cells may disrupt imprinting leading to significant alteration of expression of imprinted genes.
Tissue-specific genes. Tissue-specific genes appear to represent potential targets for demethylation. These genes are expressed in only a few normal tissues of an adult organism. About half of the tissue-specific genes possess CpG islands in regulatory regions. However, CpG islands of many studied genes were not methylated in all normal tissues independently of their expression. Other tissue-specific genes contain individual CpG dinucleotides within promoters and coding regions of genes. The majority of CpG dinucleotides within these genes are methylated in those tissues, where these genes are not expressed [10]. However, there are no data that provide evidence that observed demethylation of specific CpG sites of these genes determines their transcription in the normal tissues where they are expressed. In this connection, the role of methylation in regulation of expression of tissue-specific genes in normal tissues has been intensively discussed. In was demonstrated only recently that methylation is involved in the control of expression of certain genes in normal tissues [19].
For several tissue-specific genes with regulatory regions methylated in normal tissues, it was shown that they are demethylated and ectopically expressed in neoplastic cells. Some proto-oncogenes, the genes that determine the metastatic potential of tumor cells, and a number of genes with undetermined functions in normal cells are among these genes (table) [57-74]. Brief information about the genes for which activation with involvement of demethylation in tumor cells is confirmed is presented below.
Cancer-associated tissue-specific genes whose
sequences are hypomethylated in human tumors
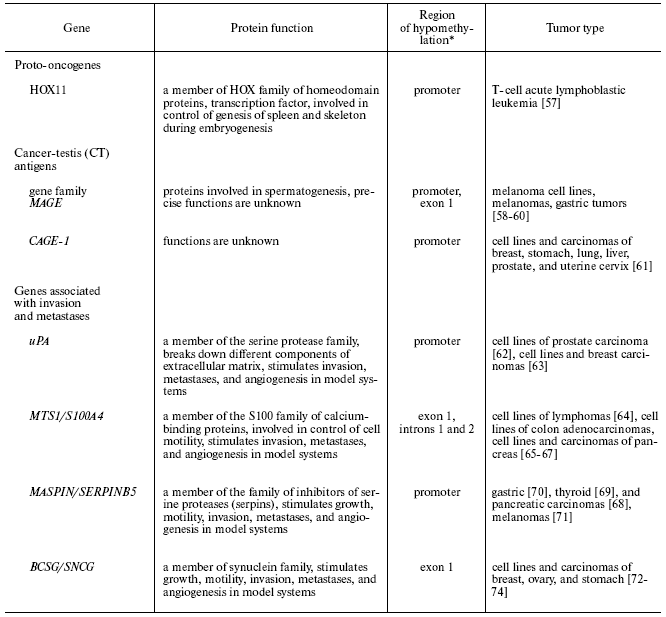
*The hypomethylation of this region is accompanied by
transcription.
Proto-oncogenes. HOX11 is a transcriptional factor, proto-oncogene, and member of the family of homeodomain proteins HOX. The gene is expressed only in embryogenesis where its protein product is necessary for formation of spleen and skeleton [75, 76]. Aberrant expression of the gene is observed in T-cell acute lymphoblastic leukemia. Gene expression is accompanied by promoter demethylation. It was demonstrated that in normal T-cells, the promoter is methylated, and that methylation of the promoter in vitro is sufficient to silence the gene promoter [57].
Demethylation of individual CpG dinucleotides located in C-MYC, Ha RAS, and ERB-A1 proto-oncogenes was revealed in human tumors long ago. However, the data providing evidence that this site-specific demethylation results in activation of these genes are still lacking [77-80].
Cancer-testis antigens. Cancer-testis antigens (CT antigens: MAGE, GAGE, LAGE, and others) are specifically expressed only in male germinal cells at the definite stage of spermatogenesis and also in various tumor cells [58-61]. Protein products of these genes cannot be presented on the surface of germinal cells due to the absence of expression of MHC first class molecules in them. Hence, these proteins are tumor-specific antigens and may induce immune response in the host organism. These properties provide the possibility to use them in immunotherapy [81, 82]. It is suggested that CT antigens share a common mechanism for regulation of expression, in which methylation plays a significant role. However, experimental data favoring this hypothesis are actually obtained only for a few genes. Demethylation of the promoter and the first exon of MAGE-A1 gene (melanoma antigen A1) strongly correlate with its expression in male germinal cells, certain melanoma cell lines, and in melanoma metastasis to skin and lymph nodes. In other normal cells of the adult organism, the gene is methylated and not expressed [58, 59]. Hypomethylation of the promoters and aberrant expression of MAGE-A1 and five another genes of the MAGE family have been observed in gastric tumors [60]. Hypomethylation of the promoter of CAGE-1 (cancer-associated gene-1), a new cancer-testis antigen, strongly correlates with its expression in many tumor types. In the studied normal tissues, the promoter is methylated [61, 83]. Methylation of the promoters of MAGE-A1 and CAGE-1 in vitro inhibits binding of the transcription factors and transcription of these genes [61, 84]. It was also demonstrated that the expression of genes GAGE, LAGE, and NY-ESO-1 can be induced in normal and tumor cells that do not express these genes by treatment of cells with the demethylating agent 5-azacytidine [85-87]. The role of these genes in carcinogenesis is not yet established.
Genes associated with invasion and metastases. uPA (urokinase-type plasminogen activator). The protein product of the gene is a serine protease that is involved in transformation of inactive plasminogen into the active form and cleaves certain components of extracellular matrix: laminin, fibronectin, and collagen. Gene expression is repressed in epithelial prostate and breast cells, where the gene promoter is hypermethylated. In several cell lines of carcinomas of prostate and breast and in primary breast tumors transcription of the gene is activated in correlation with promoter demethylation [62, 63, 88]. Gene demethylation and expression in breast tumors correlates with invasive phenotype, presence of metastasis, and high mortality [89]. Restoration of the hypermethylated status of the promoter by treatment of tumor cells with S-adenosyl methionine inhibits gene expression and represses invasive and metastatic potential of the cells [90].
MTS1/S100A4 (metastasin 1). This gene encodes for a small protein that binds calcium ions and is a member of S100 family. The gene is specifically expressed only in several types of normal cells in the adult organism (T-lymphocytes, neutrophils, macrophages). The gene product is involved in the control of mobility of these cell types [91]. The gene is also expressed in many human tumors, where its expression correlates with invasive and metastatic phenotype of the tumor. Exogenous expression of the gene results in appearance of the metastatic phenotype in non-metastatic tumor cells, while the expression of the antisense RNA to MTS1 represses cellular metastatic potential [92, 93]. Correlation of gene expression with hypomethylation of certain CpG sites in the first exon and two first introns was demonstrated for normal blood cells, several carcinoma cell lines, and primary carcinomas (table) [61-64]. MTS1 expression may be induced in tumor cells that do not express this gene by their treatment with demethylating agents [64].
MASPIN (SERPINB5). The gene encodes for a multifunctional protein, a member of the family of serpins (inhibitors of serine proteases) [94]. It has been demonstrated in many studies that in culture of tumor cells and after injection of tumor cells into animals, expression of exogenous maspin inhibits growth, motility of cells and their ability for invasion, suppresses angiogenesis in tumors, and stimulates apoptosis [95, 96]. Gene expression is observed only in certain types of epithelial cells of the adult organism. The gene promoter is methylated in those tissues and epithelial cells, where its expression is repressed, and is hypomethylated in those types of epithelial cells, where its mRNA is present [97]. In primary human tumors derived from epithelial cells that do not express maspin, hypomethylation of the gene promoter that correlates with its re-expression is often observed (table) [68-70].
Maspin expression in different tumor types (without analysis of methylation status in the majority of cases) is intensively studies. The results obtained in some studies contradict the hypothesis that maspin is a tumor suppressor. This suggestion was based on maspin properties studied in cultured cells. It was demonstrated in some studies that maspin expression correlates with tumor progression, presence of metastasis, with shorter remission duration and decreased survival of patients [98-100]. However, in some type of tumors maspin expression decreased with tumor progression [101, 102]. Contradictory results of these studies may be related to the fact that localization of the protein (nuclear or cytoplasmic) and protein phosphorylation that are apparently important for manifestation of maspin functions were not taken into account in all studies [103, 104]. The role of maspin expression for tumors remains obscure.
BCSG1 (synuclein gamma, SNCG). BCSG1 protein is a chaperon associated with heat-shock proteins, a member of a family of small proteins that are expressed in neurons. Since alteration of tissue-specific expression of BCSG1 was primarily detected in breast tumors, the gene was designated as BCSG (breast cancer specific gene). BCSG1 expression stimulates growth and motility of tumor cells in vitro, increases their metastatic and invasive potential in vivo, and favors the development of resistance to some drugs utilized in cancer therapy [105-107]. Gene expression in tumors correlates with demethylation of a CpG island in the first exon of the gene (table) [72-74]. Treatment of carcinoma cells not expressing BCSG1 with demethylating agents activates its transcription [73].
The new methods that provide the possibility to study variations in expression of gene panels have revealed a large number of genes whose expression is activated in tumor cells. It can be suggested that aberrant demethylation as the mechanism of activation of gene expression in tumor cells is connected with a larger number of genes than is established to date.
Thus, total DNA demethylation may be involved in acquiring specific properties of tumor cells through activation of proto-oncogenes, genes whose expression increases metastatic and invasive potential of cells, by favoring genome instability and also through inhibition of expression of tumor suppressor genes by destruction of their imprinting (p57/KIP2).
MECHANISMS OF DEMETHYLATION
Hypermethylation of certain DNA regions and hypomethylation of others may be observed in the same tumors. On the other hand, neither positive nor negative correlations between hyper- and hypomethylation of DNA in tumors are yet established [26, 38, 108, 109]. This observation provides evidence to conclude that the mechanisms responsible for these two alterations of methylation pattern are independent from each other. Apparently, in normal cells different enzymes are responsible for methylation and demethylation, and in tumor cells the alterations of functioning of these different enzymes are not coordinated. Several DNA-methylating enzymes, DNA methyltransferases (DNMTs), are known. They are components of multiple protein complexes that are involved in DNA replication, transcription control, and chromatin remodeling [110]. The enzymes that are responsible for the processes of global DNA demethylation in embryogenesis and carcinogenesis are not yet identified. Hence it was suggested that DNA demethylation it tumor cells is a passive process occurring due to the lack of the activity of DNMT1 or the lack of the donor of methyl groups S-adenosylmethionine. (DNMT1 is the major methyltransferase that methylates the newly synthesized DNA chain in the replication complex.) However, this suggestion does not explain the observed co-existence of hyper- and hypomethylated DNA regions in the same tumor and the fact that the level of DNMT1 in tumors is not only decreased, but is often elevated and does not correlate either with total hypomethylation or with hypomethylation of repeats [40, 109].
When the methylation status is analyzed, it is very difficult to discriminate between the possible causes of observed demethylation: inhibition of methylation during DNA synthesis or elimination of methyl groups from DNA without replication. Confirmations of the existence of an active (independent from replication) DNA demethylation were obtained recently. It was shown that the mouse male genome is subjected to active demethylation a few hours after fertilization before the beginning of the first round of DNA replication [111-113]. Demethylation of the promoter of the tissue-specific gene (PGK2) is observed in mouse spermatogenesis. Demethylation precedes gene activation and occurs without DNA replication [114].
Demethylation of the promoter region of interleukin-2 gene was observed in human T-lymphocytes 20 min after activation, and it was impossible to block this demethylation by inhibitors of DNA replication [115]. The treatment of human embryonic kidney cells HEK293 with inhibitors of histone deacetylases led to the induction of histone acetylation, demethylation of plasmid DNA that was methylated in vitro, and plasmid expression in the absence of replication [116, 117].
Thus, methylation is a reversible modification like many other biochemical modifications: phosphorylation and dephosphorylation of proteins, acetylation and deacetylation of histones. However, the search for DNA demethylating enzymes has led to controversial results.
The protein MBD2 (methyl-CpG-binding domain protein 2) appears to be the most likely candidate for the enzyme responsible for global DNA demethylation. It was demonstrated that MBD2 is a processive DNA demethylase that is able to recognize methylated CpG dinucleotides and to catalyze the cleavage of methyl group from 5-methylcytosine without DNA destruction [118]. However, MBD2 was found to be present in protein complexes that repress transcription, and its demethylating activity was not confirmed in other studies [119, 120]. At the same time it was shown that exogenous expression of MBD2 in human embryonic kidney cells HEK293 stimulates DNA demethylation and induces transcriptional activity of several promoters that are methylated in vitro [116, 121]. It can be suggested that MBD2 is a polyfunctional protein that interacts with different proteins to exhibit various functions. It is also possible that MBD2 acts as a coactivator that is essential for activity of an yet unknown DNA demethylase. Apparently, MBD2 can be considered as a protein associated with demethylating activity. The role of MBD2 in DNA demethylation should be clarified in further experiments. Thus, the enzymes responsible for global demethylation in normal and tumor cells are not identified yet.
Few data are accumulated about the alterations of signal pathways that lead to activation of DNA demethylation in tumor cells. It has been demonstrated that exogenous expression of v-Ha-ras oncogene in mice embryonic cells is accompanied by increase in demethylating activity and demethylation of certain genes that are methylated in these cells [122]. Apparently, the Ras signal pathway controls at least the promoter-specific demethylation. It is known that DNMT1 is also effecter of Ras in mouse cells [123]. Activation of both methylation and demethylation by the signal pathway that is often altered in tumors is in accordance with simultaneous presence of hyper- and hypomethylated sequences in tumors.
Expression of Set/TAF-1beta is increased in many tumor forms [124]. Set/TAF-1beta is a subunit of the protein complex INHAT (inhibitor of histone acetyltransferases) that inhibits histone acetylation [125]. Exogenous expression of SET/TAF-1beta gene in normal human embryonic kidney cells inhibits demethylation of in vitro methylated DNA. Mutant SET/TAF-1beta gene that is incapable of inhibition of histone acetylation is not able to inhibit DNA demethylation too [124]. These data [124, 125] indicate the existence of links between epigenetic modifications of DNA and histones and possible common pathways of their regulation. The following questions are not yet solved: alteration of which signal pathways in tumor cells activate SET/TAF-1beta expression; whether this activation inhibits DNA demethylation in tumors in the same manner as in normal cells and how it occurs.
It is still unknown how the balance between methylation and demethylation in normal cells is maintained and how the mechanisms of maintenance of the balance are altered in tumor cells. In the accepted model of maintenance of methylation pattern in offspring cells after division the main role is played by DNA methyltransferases. According to this model, DNMT1 methylates only those CpG dinucleotides in the newly synthesized DNA chain that were methylated in the maternal DNA chain. This model does not explain the relative instability of methylation pattern in normal cells, in particular the detection of CpG dinucleotides methylated only in one DNA chain, or alleles that differ in internal distribution of methylated CpG dinucleotides [126]. Apparently, the pattern of DNA methylation represents the result of dynamic equilibrium between DNA methylation and opposite processes. It remains obscure whether active DNA demethylation is involved in maintenance of the methylation pattern or the relative instability is maintained by other mechanisms.
Reversibility of DNA methylation, in contrast to structural modifications of the genome, provides the theoretical possibility of treating tumor cells with agents that modify DNA methylation pattern in order to restore normal gene expression. DNA methyltransferases were thought to be the most suitable targets for such treatment until recently. Inhibitors of DNA methyltransferase-1 (antisense oligonucleotides and nucleoside cytidine analogs) demonstrate anti-tumor effect on laboratory models in some cases and are under the first stages of clinical trial [127, 128]. However, recent studies indicate that not only local hypermethylation but global demethylation is also involved in carcinogenesis. These processes coexist in tumor cells and alter gene expression with opposite roles in malignant transformation, inactivate tumor suppressor genes, and activate proto-oncogenes and genes associated with metastases. These data indicate that the carcinogenic effect of DNA demethylation should be taken into account for design and trial of agents for anti-tumor therapy.
We thank Dr. G. M. Volgareva for critical reading of the manuscript.
This work was supported in part by the Russian Foundations for Basic Research (grant 04-04-49-074) and for Scientific Schools (grant 1877).
REFERENCES
1.Vanyushin, B. F., Tkacheva, S. G., and Belozersky,
A. N. (1970) Nature, 225, 948-949.
2.Razin, A., and Szyf, M. (1984) Science,
210, 604-610.
3.Razin, A., and Shemer, R. (1995) Human Mol.
Genet., 4, 1751-1755.
4.Burtzeva, N. N., Asisof, U. M., Itkin, B. Z., and
Vanyushin, B. F. (1977) Biokhimiya, 42, 1296-1302.
5.Burtzeva, N. N., Asisof, U. M., and Vanyushin, B.
F. (1979) Biokhimiya, 44, 1690-1696.
6.Gamma-Sosa, M. A., Slagel, V. A., Trewyn, R. W.,
Oxenhandler, R., Kuo, K. C., Gehrke, C. W., and Ehrlich, M. (1983)
Nucleic Acids Res., 11, 6883-6894.
7.Feinberg, A. P., and Vogelstein, B. (1983)
Nature, 301, 89-92.
8.Kim, Y. I., Giuliano, A., Hatch, K. D., Sshneider,
A., Nour, M. A., Dallal, G. E., Selhub, J., and Mason, J. B. (1994)
Cancer, 74, 893-899.
9.Baylin, S. B., Herman, J. G., Graff, J. R.,
Vertino, P. M., and Issa, J. P. (1998) Adv. Cancer Res.,
72, 141-198.
10.Antiquera, F., and Bird, A. (1993) in DNA
Methylation: Molecular Biology and Biological Significance (Jost,
J. P., and Saluz, H. P., eds.) Brikhauser Verlag, Basel, pp.
169-185.
11.Razin, A., and Riggs, A. D. (1980)
Science, 210, 604-610.
12.Jones, P. A., and Baylin, S. B. (2002) Nat.
Rev. Genet., 3, 415-428.
13.Laird, P. W. (2003) Nat. Rev. Cancer,
3, 253-266.
14.Lichtenstein, A. V., and Kisseljova, N. P. (2001)
Biochemistry (Moscow), 66, 235-255.
15.Szyf, M. (2000) Curr. Drug. Targets,
1, 101-118.
16.Yoder, J. A., Walsh, C. P., and Bestor, T. H.
(1997) Trends Genet., 8, 335-340.
17.Brannan, C. I., and Bartolomei, M. S. (1999)
Curr. Opin. Genet. Devel., 9, 164-170.
18.Feinberg, A. P., and Tycko, B. (2004) Nature
Rev. Cancer, 4, 143-153.
19.Ehrilch, M. (2003) J. Cell. Biochem.,
88, 899-910.
20.Burtzeva, N. N., Romanov, G. A., Asisof, U. M.,
and Vanyushin, B. F. (1979) Biokhimiya, 44,
2066-2072.
21.Bender, J. (1998) Trends Biol. Sci.,
23, 252-256.
22.Jurgens, B., Schmitz-Drager, B. J., and Schulz,
W. A. (1996) Cancer Res., 56, 5698-5703.
23.Flrol, A. R., Lower, R., Schmitz-Drager, B. J.,
and Schulz, W. A. (1999) Br. J. Cancer, 80,
1312-1321.
24.Takai, D., Yagi, Y., Habib, N., and Sigimura, T.
(2000) Jpn. J. Clin. Oncol., 30, 306-309.
25.Santourlidis, S., Flrol, A. R., Ackermann, R.,
Wirtz, H. C., and Schulz, W. A. (1999) Prostate, 39,
166-174.
26.Flrol, A. R., Steinhoff, C., Muller, M., Seifert,
H. H., Hander, C., Engers, R., Ackermann, R., and Schulz, W. A. (2004)
Br. J. Cancer, 91, 985-994.
27.Dante, R., Dante-Paire, J., Rigal, D., and
Roizes, G. (1992) Anticancer Res., 12, 559-563.
28.Menendez, L., Benigno, B. B., and McDonald, J. F.
(2004) Mol. Cancer, 3, 12.
29.Kazazian, H. H., Jr., and Goodier, J. L. (2002)
Cell, 110, 277-280.
30.Leib-Mosch, C., Haltmeier, M., Werner, T., Geigl,
E. M., Brack-Werner, R., Francke, U., Erfle, V., and Hehlmann, R.
(1993) Genomics, 18, 261-269.
31.Gotzinger, N., Sauter, M., Roemer, K., and
Mueller-Lantzsch, N. (1996) J. Gen. Virol., 77,
2983-2990.
32.Domansky, A. N., Kopantzev, E. P., Snezhkov, E.
V., Lebedev, Y. B., Leib-Mosch, C., and Sverdlov, E. D. (2000) FEBS
Lett., 472, 191-195.
33.Buzdin, A. A., Lebedev, Y. B., and Sverdlov, E.
D. (2003) Bioorg. Khim., 29, 103-106.
34.Narayan, A., Ji, W., Zhang, X. Y., Marrogi, A.,
Graff, J. R., Baylin, S. B., and Ehrlich, M. (1998) Int. J.
Cancer, 77, 833-838.
35.Qu, G., Grundy, P. E., Narayan, A., and Ehrlich,
M. (1999) Cancer Genet. Cytogenet., 109, 34-39.
36.Ehrlich, M., Hopkins, N. E., Jiang, G., Dome, J.
S., Yu, M. C., Woods, C. B., Tomlinson, G. E., Chintagumpala, M.,
Champagne, M., Dillerg, L., Parham, D. M., and Sawyer, J. (2003)
Cancer Genet. Cytogenet., 141, 97-105.
37.Wong, N., Lam, W. C., Lai, P. B., Pang, E., Lau,
W. Y., and Johnson, P. J. (2001) Am. J. Pathol., 159,
465-471.
38.Widschwendter, M., Jiang, G., Woods, C., Muller,
H. M., Fiegl, H., Goebel, G., Marth, C., Muller-Holzner, E., Zeimet, A.
G., Laird, P. W., and Ehrlich, M. (2004) Cancer Res., 64,
4472-4480.
39.Qu, G., Dubeau, L., Narayan, A., Yu, M. C., and
Ehrlich, M. (1999) Mutat. Res., 423, 91-101.
40.Saito, Y., Kanai, Y., Sakamoto, M., Saito, H.,
Ishii, H., and Hirohashi, S. (2001) Hepatology, 33,
561-568.
41.Rouyer, F. M., Simmler, D., Page, D., and
Weissenbach, J. (1987) Cell, 51, 417-425.
42.Small, K. J., Iber, J., and Warren, S. T. (1997)
Nature Genet., 16, 96-99.
43.Rossignol, J. L., and Fauferon, G. (1994)
Experientia, 15, 307-317.
44.Maloisel, L., and Rossingol, J. L. (1998)
Genes Devel., 12, 1381-1389.
45.Ji, W., Hernandez, R., Zhang, X. Y., Qu, G.,
Frady, A., Varela, M., and Ehrlich, M. (1997) Mutant. Res.,
379, 33-41.
46.Mimiou, P. M., Jeanpierre, V., Blanquet, V.,
Sibella, D., Bonneau, C., Herbelin, C., Fischer, A., Niveleau, A., and
Viegas-Pejignot, E. (1994) Hum. Mol. Genet., 3,
2093-2102.
47.Chen, R. Z., Petterson, U., Beard, C.,
Jackson-Grusby, L., and Jaenisch, R. (1998) Nature, 395,
89-93.
48.Gaudet, F., Hodgson, J. G., Eden, A.,
Jackson-Grusby, L., Dausman, J., Gray, J. W., Leonhardt, H., and
Jaenisch, R. (2003) Science, 300, 489-492.
49.Delaval, K., and Feil, R. (2004) Curr. Opin.
Genet. Devel., 14, 188-195.
50.Jirtle, R. L. (1999) Exp. Cell Res.,
248, 18-24.
51.Feinberg, A. P. (2004) Semin. Cancer
Biol., 14, 427-432.
52.Cui, H., Onyahgo, P., Brandenburg, S., Wu, Y.,
Hsieh, C. L., and Feinberg, A. P. (2002) Cancer Res., 62,
6442-6446.
53.Horike, S., Mitsuya, K., Meguro, M., Kotobuki,
N., Kashiwagi, A., Notsu, T., Schulz, T. C., Shirayoshi, Y., and
Oshimura, M. (2000) Hum. Mol. Genet., 9, 2575-2583.
54.Soejima, H., Nakagawachi, T., Zhao, W.,
Higashimoto, K., Urano, T., Matsukura, S., Kitajima, Y., Takeuchi, M.,
Nakayama, M., Oshimura, M., Miyazaki, K., Joh, K., and Mukai, T. (2004)
Oncogene, 25, 4380-4388.
55.Scelfo, R. A. M., Schwienbacher, C., Veronese,
A., Gramantieri, L., Bolondi, L., Querzoli, P., Nenci, I., Calin, G.
A., Angioni, A., Barbanti-Brodano, G., and Negrini, M. (2002)
Oncogene, 21, 2564-2572.
56.Schwienbacher, C., Gramantieri, L., Scelfo, R.,
Veronese, A., Calin, G. A., Croce, C. V., Barbanti-Brodano, G., and
Negrini, M. (2000) Proc. Natl. Acad. Sci. USA, 97,
5445-5449.
57.Watt, P. M., Kumar, R., and Kees, U. R. (2000)
Genes Chrom. Cancer, 29, 371-377.
58.DeSmet, C., Lurquin, C., Lethe, B., Martelange,
V., and Boon, T. (1999) Mol. Cell Biol., 19,
7327-7335.
59.DeSmet, C., Loriot, A., and Boon, T. (2004)
Mol. Cell Biol., 24, 4781-4790.
60.Kaneda, A., Tsukamoto, T., Takamura-Enya, T.,
Watanabe, N., Kaminishi, M., Sugimura, T., Tatematsu, M., and Ushijima,
T. (2004) Cancer Sci., 95, 58-64.
61.Cho, B., Lee, H., Jeong, S., Bang, Y. J., Lee, H.
J., Hwang, K. S., Kim, H. Y., Lee, Y. S., Kang, G. H., and Jeoung, D.
I. (2003) Biochem. Biophys. Res. Commun., 307, 52-63.
62.Pakneshan, P., Xing, R. H., and Rabbani, S.
(2003) FASEB J., 17, 1081-1088.
63.Pakneshan, P., Tetu, B., and Rabbani, S. (2004)
Clin. Cancer Res., 10, 3035-3041.
64.Tulchinsky, E., Grigorian, M., Tkatch, T.,
Georgiev, G., and Lukanidin, E. (1995) Biochim. Biophys. Acta,
1261, 243-248.
65.Nakamura, N., and Takenaga, K. (1998) Clin.
Exp. Metastasis, 16, 471-479.
66.Rosty, C., Ueki, T., Argani, P., Jansen, M., Yeo,
C. J., Cameron, J. L., Hruban, R. H., and Goggins, M. (2002) Am. J.
Pathol., 160, 45-50.
67.Sato, N., Maitra, A., Fukushima, M., van Heek, N.
T., Matsubayashi, H., Iacobuzio-Donahue, C. A., Rosty, C., and Goggins,
M. (2003) Cancer Res., 63, 4158-4166.
68.Sato, N., Fukushima, N., Matsubayashi, H., and
Goggins, M. (2004) Oncogene, 23, 1531-1538.
69.Ogasawara, S., Maesawa, C., Yamamoto, M.,
Akiyama, W., Wada, K., Fujisawa, K., Higuchi, T., Tomisawa, Y., Sato,
N., Endo, S., Saito, K., and Musuda, T. (2004) Oncogene,
23, 1117-1124.
70.Akiyama, W., Maesawa, C., Ogasawara, S.,
Terashima, M., and Musuda, T. (2003) Am. J. Pathol., 163,
1911-1919.
71.Wada, K., Maesawa, C., Akasaka, T., and Musuda,
T. (2004) J. Invest. Dermatol., 122, 805-811.
72.Lu, A., Gupta, A., Li, C., Ahlborn, T. E., Ma,
Y., Shi, E. Y., and Lui, J. (2001) Oncogene, 20,
5173-5185.
73.Gupta, A., Godwin, A. K., Vanderveer, L., Lu, A.,
and Lui, J. (2003) Cancer Res., 63, 664-673.
74.Ynanagawa, N., Tamura, G., Honda, T., Endoh, M.,
Nishizuka, S., and Motoyama, T. (2004) Clin. Cancer Res.,
10, 2447-2451.
75.Roberts, C. W., Shutter, C. W., and Korsmeyer, S.
J. (1994) Nature, 368, 747-749.
76.Wellik, D. M., and Capecchi, M. R. (2003)
Science, 301, 363-367.
77.Cheah, M. S., Wallace, C. D., and Hoffman, R. M.
(1984) J. Natl. Cancer Inst., 73, 1057-1065.
78.Fienberg, A. P., and Vogelstein, B. (1983)
Biochem. Biophys. Res. Commun., 111, 47-54.
79.Lipsanen, V., Leinonen, P., Alhonen, L., and
Janne, J. (1988) Blood, 72, 2042-2044.
80.Borrello, M. G., Pierotti, M. A., Tamborini, E.,
Biassoni, D., Rizzetti, M. G., Pilotti, S., and Porta, G. D. (1992)
Oncogene, 7, 269-275.
81.Old, L. (2001) Cancer Immun., 1,
1.
82.Sahin, U., Tureci, O., Chen, Y. T., Seitz, G.,
Villena-Heinsen, C., Old, L. J., and Pfreundchuh, M. (1998) Int. J.
Cancer, 78, 387-389.
83.Park, S., Lim, D., Cho, B., Bang, Y. J., Sung,
S., Kim, H. Y., Kim, D. K., Lee, Y. S., Song, Y., and Jeoung, D. I.
(2003) Biochim. Biophys. Acta, 1652, 173-182.
84.De Smet, C., De Backer, O., Faraoni, I., Lurquin,
C., Brasseur, F., and Boon, T. (1996) Proc. Natl. Acad. Sci.
USA, 93, 7149-7153.
85.Lethe, B., Lukas, S., Michaux, L., DeSmet, C.,
Godelaine, D., Serrano, A., DePlaen, E., and Boon, T. (1998) Int. J.
Cancer, 76, 903-908.
86.DeBacker, O., Arden, K. C., Boreti, M., Vantomme,
V., DeSmet, C., Czekay, S., Viars, C. S., DePlaen, E., Brasseur, F.,
Chomez, P., van den Eynde, B., Boon, T., and van der Bruggen, P. (1999)
Cancer Res., 59, 3157-3165.
87.Lee, L., Wang, R. F., Wang, X., Mixon, A.,
Johnson, B. E., Rosenberg, S. A., and Schrump, D. S. (1999) Cancer
J. Sci. Am., 5, 20-25.
88.Xing, R. H., and Rabbani, S. A. (1999) Int. J.
Cancer, 81, 443-450.
89.Xing, R. H., and Rabbani, S. A. (1996) Int. J.
Cancer, 67, 423-429.
90.Pakneshan, P., Szyf, M., Farias-Eisner, R., and
Rabbani, S. A. (2004) J. Biol. Chem., 279,
31735-31744.
91.Grigorian, M., Tulchinsky, E., Burrone, O.,
Tarabukina, S., Georgiev, G., and Lukanidin, E. (1994)
Electrophoresis, 15, 463-468.
92.Grigorian, M., Zain, S., Georgiev, G. P., and
Lukanidin, E. M. (1993) Gene, 135, 229-238.
93.Takenaga, K., Nakamura, Y., and Sakiyama, S.
(1997) Oncogene, 14, 331-337.
94.Zou, Z., Anisowicz, A., Hendrix, M. J., Thor, A.,
Naveu, M., Sheng, S., Rafidi, K., Seftor, E., and Sager, R. (1994)
Science, 263, 526-529.
95.Schaefer, J. S., and Zhang, M. (2003) Curr.
Mol. Med., 3, 653-658.
96.Liu, J., Yin, S., Reddy, N., and Sheng, S. (2004)
Cancer Res., 64, 1703-1711.
97.Futscher, B, W., Oshiro, M. M., Wozniak, R. J.,
Holtan, N., Hanigan, C. L., Duan, H., and Domann, F. E. (2002)
Nature Genet., 31, 175-179.
98.Sood, A. K., Fletcher, M. S., Gruman, L. M.,
Coffin, J. E., Jabbari, S., and Hendrix, M. J. (2002) Clin. Cancer
Res., 8, 2924-2932.
99.Maas, N., Hojo, T., Ueding, M., Luttges, J.,
Kloppel, G., Jonat, W., and Nagasaki, K. (2001) Clin. Cancer
Res., 7, 812-817.
100.Sugimoto, S., Maass, N., Takimoto, Y., Sato,
K., Minei, S., Zhang, M., Hoshikawa, Y., and Nagasaki, K. (2004)
Cancer Lett., 203, 209-215.
101.Pierson, C. R., MeGowen, R., Grignon, D., Sark,
W., Dey, J., and Sheng, S. (2002) Prostate, 53,
255-262.
102.Xia, W., Lau, Y. K., Li, L., Johnston, D. A.,
Sheng, S., El-Naggar, A., and Hang, M. C. (2000) Oncogene,
19, 2398-2403.
103.Mohsin, S. K., Zhang, M., Clark, G. M., and
Craig Allred, D. (2003) J. Pathol., 199, 432-435.
104.Odero-Marah, V. A., Khalknali-Ellis, Z.,
Schneider, G. B., Seftor, R. E., Koland, G. J., and Hendrix, M. J.
(2002) Biochem. Biophys. Res. Commun., 295, 800-805.
105.Jia, T., Liu, J., and Shi, Y. E. (1999)
Cancer Res., 59, 742-747.
106.Liu, J., Spence, M. J., Zhang, Y. L., Jiang,
Y., Liu, Y. E., and Shi, Y. E. (2000) Breast Cancer Res. Treat.,
62, 99-107.
107.Pan, Z. Z., Bruening, W., Giasson, B. I., Lee,
V. M., and Godwin, A. K. (2002) J. Biol. Chem., 277,
35050-35060.
108.Pini, J. T., Franchina, M., Taylor, J. M., and
Kay, P. H. (2004) Oncol. Res., 14, 399-405.
109.Ehrlich, M., Jiang, G., Fiala, E., Dome, J. S.,
Yu, M. C., Long, T. I., Youn, B., Sohn, O. S., Widschwendter, M.,
Tomlinson, G. E., Chintagumpala, M., Parham, D., Liang, G., Malik, K.,
and Laird, P. W. (2002) Oncogene, 21, 6694-6702.
110.Robertson, K. D. (2001) Oncogene,
20, 3139-3155.
111.Mayer, W., Niveleau, A., Walter, J., Fundele,
R., and Haaf, T. (2000) Nature, 203, 501-502.
112.Oswald, J., Engemann, S., Lane, N., Mayer, W.,
Olek, A., Fundele, R., Reik, W., Dean, W., and Walter, J. (2000)
Curr. Biol., 10, 475-478.
113.Santos, F., and Hendrich, B. (2002) Dev.
Biol., 241, 172-182.
114.Geyer, C. B., Kiefer, C. M., Yang, T. P., and
McCarrey, J. R. (2004) Biol. Reprod., 71, 837-844.
115.Bruniquel, D., and Schwartz, R. H. (2003)
Nat. Immunol., 4, 235-240.
116.Chervoni, N., and Szyf, M. (2001) J. Biol.
Chem., 276, 40778-40787.
117.Detich, N., Bovenzi, V., and Szyf, M. (2003)
J. Biol. Chem., 278, 27586-27592.
118.Bhattacharya, S. K., Ramchandani, S., Chervoni,
N., and Szyf, M. (1999) Nature, 397, 579-583.
119.Zhang, Y., Ng, H. H., Erdjument-Bromage, H.,
Tempst, P., Bird, A., and Reinberg, D. (1999) Genes Dev.,
13, 1924-1935.
120.Ng, H. H., Zhang, Y., Hendrich, B., Johnson, C.
A., Turner, B. M., Erdjument-Bromage, H., Tempst, P., Reinberg, D., and
Bird, A. (1999) Nat. Genet., 23, 58-61.
121.Detich, N., Theberge, J., and Szyf, M. (2002)
J. Biol. Chem., 277, 35791-35794.
122.Szyf, M., Theberge, J., and Bozovic, V. (1995)
J. Biol. Chem., 270, 12690-12696.
123.MacLeod, A. R., Rouleou, J., and Szyf, M.
(1995) J. Biol. Chem., 270, 11327-11337.
124.Cervoni, N., Detich, N., Seo, S. B.,
Chakravarti, D., and Szyf, M. (2002) J. Biol. Chem., 277,
25026-25031.
125.Seo, S. B., McNamara, P., Heo, S., Turner, A.,
Lane, W. S., and Chakravarti, D. (2001) Cell, 104,
119-130.
126.Turker, M. S. (1999) Semin. Cancer
Biol., 9, 329-337.
127.Goffin, J., and Eisenhauer, E. (2002) Ann.
Oncol., 13, 1699-1716.
128.Issa, J. P. (2003) Curr. Opin. Oncol.,
15, 446-451.