Identification of beta-Lactamase Inhibitory Peptide Using Yeast Two-Hybrid System
Wei Sun1*, Youjia Hu2, Jiawei Gong2, Chunbao Zhu2, and Baoquan Zhu2
1School of Medicine, Tongji University, 1239 Siping Road, Shanghai 200092, China; E-mail: wayofsun1@yahoo.com.cn2Department of Bio-Pharmaceutics, Shanghai Institute of Pharmaceutical Industry, 1320 West Beijing Road, Shanghai 200040, China
* To whom correspondence should be addressed.
Received February 12, 2005; Revision received February 25, 2005
Random oligonucleotide fragments were designed and amplified by PCR and fused with the activating domain of pGAD424 to construct a random peptide library. The DNA fragment encoding beta-lactamase was fused with the binding domain of pGBT9 (+2). Subsequently, using yeast two-hybrid system we found two positive clones encoding peptides P1 and P2 that have the ability to bind beta-lactamase in vivo. The genes encoding P1 and P2 were cloned into pGEX-4T-1. GST-peptide fusion proteins were expressed in Escherichia coli and isolated by glutathione-Sepharose 4B affinity chromatography. Finally, P1 and P2 were cleaved from the fusion protein with thrombin and purified by ultrafiltration. Inhibition assay of peptides with beta-lactamase in vitro indicated that only P1 has the ability to inhibit beta-lactamase.
KEY WORDS: beta-lactamase, yeast two-hybrid system, beta-lactamase inhibitory peptide, GST-peptide fusion system
Since the introduction of beta-lactam antibiotics, bacterial resistance to these agents has become an increasing problem. The production of the enzyme beta-lactamase by both Gram-positive and Gram-negative bacteria is the most common mechanism of resistance to beta-lactam antibiotics [1]. beta-Lactamases hydrolyze the amide bond of the beta-lactam antibiotics to create an ineffective antimicrobial agent; the enzymes have been grouped into four classes (A, B, C, and D) based on amino acid sequence homology [2, 3]. The most prevalent plasmid-mediated beta-lactamase in Gram-negative bacteria is the TEM-1 beta-lactamase from class A [4]. Like other class A beta-lactamases, TEM-1 beta-lactamase is capable of hydrolyzing both penicillins and cephalosporins [2]. To overcome the drug resistance mediated by TEM-1 beta-lactamase, extended-spectrum antibiotics including aztreonam, cefotaxime, and ceftazidime were developed. However, soon after their introduction, selective pressure resulted in the emergence of variant beta-lactamases capable of hydrolyzing these antibiotics [5].
An additional strategy that has been employed to combat antimicrobial drug resistance is the use of beta-lactamase inhibitors such as clavulanic acid, sulbactam, and tazobactam. Although not capable of antimicrobial activity themselves, these agents are used in conjunction with various beta-lactam antibiotics to bind beta-lactamase and prevent the hydrolysis of the antibiotics, thereby restoring the therapeutic value to the antimicrobial agent. Unfortunately, beta-lactamase variants have been identified that are resistant to these enzyme inhibitors while still retaining the ability to hydrolyze beta-lactam antibiotics [6].
Clavulanic acid was initially purified from the soil bacterium Streptomyces clavuligerus, which also produces a protein inhibitor of beta-lactamase called beta-lactamase inhibitor protein (BLIP) [7]. BLIP is a 165 amino acid protein composed of two domains of ~78 residues each and is a protein inhibitor of TEM-1 beta-lactamase (Ki = 0.1-0.6 nM) [8-10]. In addition, BLIP inhibits beta-lactamases from both Gram-positive and Gram-negative bacteria to varying degrees and also inhibits the cell-wall transpeptidase PBP5 from Enterococcus faecallis [8]. The cocrystal structure of TEM-1 beta-lactamase and BLIP indicates that residues involved in binding and inhibition of beta-lactamase, such as Asp49 and Phe142, lie in different domains [9], and that residues 46 to 51 (AAGDYY) of BLIP make critical interactions with the active site of TEM-1 beta-lactamase [11].
These findings provide an important basis for the design and development of new drugs that could inhibit beta-lactamase, which is the common target for antibiotic resistance in both Gram-positive and Gram-negative bacteria. In this paper, we report the screening of a library of peptide-based potential inhibitors of the TEM-1 beta-lactamase by the yeast two-hybrid system. Peptides gained can provide the basis for further development of more potent peptide derivatives against beta-lactamase activity.
MATERIALS AND METHODS
Strains, reagents, and plasmid constructs. All strains and plasmids utilized and constructed in this study are described in the table.

Note: SIPI) Shanghai Institute of Pharmaceutical Industry;
Apr) ampicillin resistance.
The following random oligonucleotide fragments were synthesized by Invitrogen (USA): 5´-ACGAATTCACTATCCACTGCNNYNNYNNYGCTGCAGGTGACTACTACNNYNNYNNYNNYNNYNNYGGCACCTCTTTCTAGGATCCGTGC-3´, in which N is A, T, C or G, while Y is C or G. There were EcoRI (5´ end) and BamHI (3´ end) sites in the fragment, respectively. The fragments were amplified by PCR with the sense primer (5´-ACGAATTCACTATCCACT-3´) and anti-sense primer (5´-GCACGGATCCTAGAAAGA-3´) synthesized by Shanghai Sangon Biological Engineering Technology and Service Co., Ltd (China).
Various restriction endonucleases, T4 DNA ligase, and Taq DNA polymerase were purchased from TakaRa Biotechnology Co., Ltd (Japan). Zymolyase-20T was purchased from the Kirin Brewery Co., Ltd (Japan). Sephadex G-75 gel-filtration chromatography columns and glutathione-Sepharose 4B affinity chromatography columns were purchased from Pharmacia (USA).
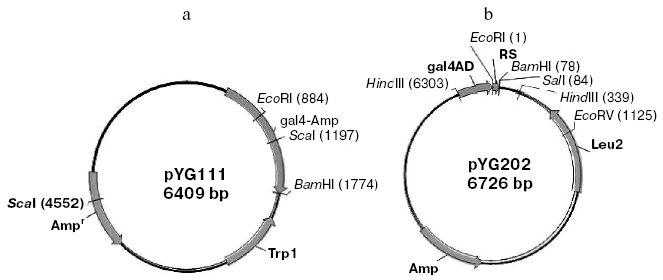
Construction of bait plasmid and random peptide library. The beta-lactamase gene was amplified from pBR322 using the sense primer with a BamHI site (5´-CGGGATCCTTACCAATGCTTAAT-3´) and the anti-sense primer with an EcoRI site (5´-CGGAATTCAGTATGAGTATTCA-3´). The PCR product was fused with the binding domain of pGBT9 (+2) to generate bait plasmid pYG111 (Fig. 1a).
The random oligonucleotide fragments were amplified by PCR. The PCR products were digested by EcoRI and BamHI and cloned into the same sites of the plasmid pGAD424 to generate random peptide library (designated pYG202) (Fig. 1b). Fragments encoding random peptides were fused with the activating domain of pAGD424. pYG202 was transformed into E. coli DH5alpha, and to form the random peptide library all transformants were collected.Fig. 1. Physical map of bait plasmid pYG111 (a) and prey plasmid library pYG202 (b).
Screening of yeast two-hybrid system. For the two-hybrid screening, S. cerevisiae Y153 (100 ml, A600 = 0.68) was transformed with 100 µg of pYG111 and 100 µg of pYG202. Then we made two dilutions of transformation reactions (100 µl of 1 : 100, 1 : 1000) and spread on SD-Leu-Trp-His plates to determine the total number of transformants. To identify the optimal spreading density for His+ transformants, aliquots of the cell suspension (25, 50, and 100 µl) were spread on SD-Leu-Trp-His contained 25 mM 3-AT (60 plates in total). The plates were incubated at 30°C for six days. The remainder of the transformation solution was kept at 4°C for five days. Once it was established that 50 µl was the optimal spreading density, 50 µl aliquots were spread on 40 SD-Leu-Trp-His plates containing 25 mM 3-AT. These plates were incubated at 30°C for six days.
beta-Galactosidase assays. His+ transformants were screened out by the expression of beta-galactosidase using the colony lift assay with 5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside (X-gal) as the substrate or the liquid assay with o-nitrophenyl-beta-D-galactoside (ONPG) as the substrate (CLONTECH Yeast Protocols Handbook). The liquid assay gives a quantitative measure of beta-galactosidase expression in Miller units: Miller units = 1000A420/(VtA600), where V is the volume of cell suspension, and t is the time for color development (CLONTECH Yeast Protocols Handbook).
Segregation of peptide-containing plasmids from S. cerevisiae transformants by shuttling through E. coli C600. His+ LacZ+ transformants were grown in 5 ml of the appropriate media for 1-2 days at 30°C with shaking. The cells were pelleted and resuspended in 200 µl of yeast zymolyase buffer (containing 30 mM DTT) placed for about 15 min at room temperature. The cells were pelleted and resuspended in 500 µl of yeast zymolyase buffer, with 1.5 mg zymolyase T-20 added. Then the cells were placed for 2 h in a 37°C constant temperature water bath and agitated gently. Protoplasts were prepared through enzyme lysing of the yeast cell wall. Plasmid DNA was extracted by alkaline lysis, then the plasmid DNA was transformed to competent cell of E. coli C600 (CLONTECH Yeast Protocols Handbook). E. coli transformants were selected on M9 + Pro + Trp-Leu plates containing 100 µg/ml ampicillin. Plasmid was prepared from ampicillin resistant transformants using the high pure plasmid isolation kit from TakaRa.
Peptide purification. Peptides were purified using the protein fusion and purification system according to the manufacture's instructions. EcoRI-BamHI fragments containing the peptide genes were obtained by PCR in pYG202 and subcloned into the same sites in pGEX-4T-1, then expressed as a fusion protein with the glutathione-S-transferase protein (GST) in E. coli DH5alpha. The fusion protein was purified on a glutathione-Sepharose 4B affinity chromatography column by eluting with 10 mM glutathione, and the peptide was subsequently released from GST by cleavage with thrombin. The peptides were separated from GST by filtration through Microcon Ultracel-YM 5000 membrane (Millipore).
Expression and purification of beta-lactamase. The beta-lactamase gene was subcloned into plasmid pLY5 to construct plasmid pLY5BL. The beta-lactamase was expressed in the form of inclusion bodies. The inclusion bodies were washed, dissolved, and refolded. Finally, beta-lactamase was purified to ~90% homogeneity with Sephadex G-75 gel-filtration chromatography (data not shown).
In vitro inhibition assay. Inhibition assays were performed as described previously [9]. Briefly, various concentrations of peptides or control peptide (insulin) were incubated with TEM-1 beta-lactamase for 2 h at 25°C to establish equilibrium. Enzyme assays were performed in 0.05 M phosphate buffer (pH 7.0) containing 1 mg/ml BSA. After incubation, 100 µl of ampicillin solution (10 mg/ml) was added, and hydrolysis of the substrate was monitored at 235 nm. The final volume of the reaction was 3 ml.
RESULTS
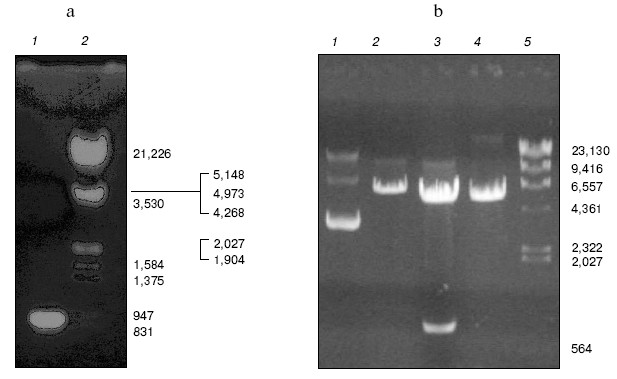
Construction of bait plasmid and prey plasmid library. The TEM-1 beta-lactamase gene was amplified by PCR with primers containing EcoRI and BamHI sites, respectively. The size of PCR products (Fig. 2a) and sequencing result of the gene both matches the expectation very well. Then the TEM-1 beta-lactamase gene was fused with the binding domain of pGBT9 (+2) to generate bait plasmid, pYG111, the structure of which was tested by digestion with restriction endonucleases EcoRI and BamHI (Fig. 2b).
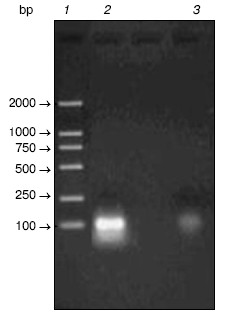
Random oligonucleotides were PCR amplified. PCR products were analyzed by agarose gel electrophoresis (Fig. 3). Then PCR products were subcloned into pGAD424 to construct prey plasmid library, designated pYG202. Finally, E. coli DH5alpha was transformed with pYG202, and transformation mixtures were spread on 100 plates of LB (containing 100 µg/ml ampicillin) and incubated for 16 h at 37°C. With amount of 107, all transformants were collected.Fig. 2. a) Amplification of beta-lactamase gene using PCR: 1) PCR product; 2) lambda_DNA/HindIII + EcoRI marker. b) Electrophoresis analysis of pYG111: 1) pYG111 (no digest); 2) pYG111/EcoRI; 3) pYG111/EcoRI + BamHI; 4) pGBT9(+2)/EcoRI + BamHI; 5) lambda_DNA/HindIII + EcoRI marker.
Screening of random peptide library. Bait plasmid pYG111 and random peptide library (plasmid pYG202) were co-transformed into S. cerevisiae Y153, and the transformants were screened for HIS3 expression. More than 10 million transformants were screened, and about 6000 His+ transformants were obtained. In addition to HIS3, Y153 has a second reporter gene, lacZ. When the His+ transformants were screened for lacZ expression, twenty transformants were His+ LacZ+ phenotype in the primary screening.Fig. 3. Amplification of single oligonucleotide chain by PCR: 1) marker; 2) amplified oligonucleotides; 3) negative control (without templates).
The GAL4AD-peptide fusion plasmids from the His+ LacZ+ transformants were segregated by shuttling through E. coli C600. Then these plasmids were transformed back into Y153 containing pYG111. Transformed cells were plated onto SD-Trp-Leu as well as SD-Trp-Leu-His + 3-AT media. Growth on SD-Trp-Leu confirms the presence of both the BD (bait) and AD (peptide) plasmids. Colony formation on SD-Trp-Leu-His + 3-AT demonstrates activation of the HIS3 reporter gene. These His+ colonies were tested for activation of lacZ reporter again. Through re-screening, two positive clones were obtained finally (Fig. 4). Single and co-transformants were respectively plated on nonselective (YPD) synthetic dextrose medium lacking tryptophan and histidine (SD-Trp-His), or leucine and histidine (SD-Leu-His), or tryptophan, leucine, and histidine (SD-Trp-Leu-His), but containing 25 mM 3-AT to select for clones in which the HIS3 gene was activated (Fig. 4). Two positive clones were transferred to nitrocellulose filter for the beta-galactosidase filter assay, on which they turned blue.
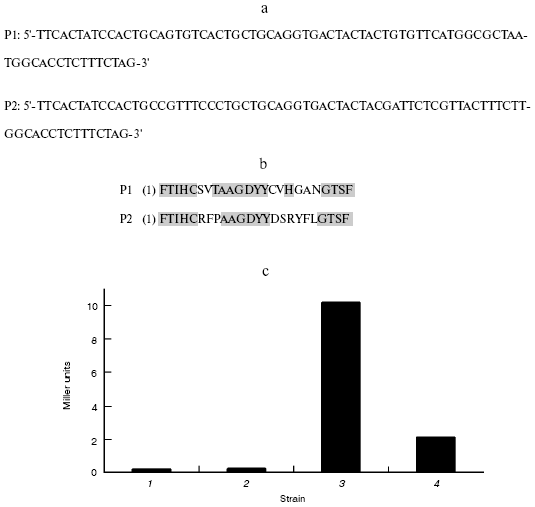
Alignment of sequences and assay of binding activity in vivo. DNA sequencing of these two clones revealed that they correspond to two unique peptide-encoding genes (Fig. 5a). They were designated P1 and P2, respectively. Sequence alignment (Fig. 5b) indicated that the sequences of P1 and P2 had a great deal of difference in the random region of the designed fragment.Fig. 4. Two-hybrid results showing interactions of peptide and beta-lactamase. a) Template for (b) through (f); b-e) growth on YPD, SD-Trp-His, SD-Leu-His, and SD-Trp-Leu-His + 3-AT, respectively; f) beta-galactosidase filter assay. BD, binding domain; BD/lac, beta-lactamase fused to binding domain; AD, activating domain; AD/oligo, random peptide fused to activating domain.

In S. cerevisiae Y153, the interaction of bait and prey leads to the expression of beta-galactosidase, which is used to measure the strength and stability of the bait-prey interaction. To ascertain the strength of the interaction between the peptides and beta-lactamase, the activity of beta-galactosidase in cell extract prepared from the various strains was measured by monitoring the formation of o-nitrophenol at 420 nm. P1 demonstrated stronger interaction with beta-lactamase with 10.2 Miller units, while P2 was approximately fivefold less with 2.1 Miller units. The control indicated that there was no interaction in yeast (Fig. 5c). The results can be interpreted as showing the effect of some amino acid residues in peptides on its interaction with beta-lactamase.Fig. 5. a) DNA sequences of the peptides that interact with beta-lactamase; b) alignment of P1 and P2; c) the strength of the beta-lactamase-peptide interaction: 1) Y153; 2) BD + AD/Y153; 3) BD-lac + AD-P1/Y153; 4) BD-lac + AD-P2/Y153. (The sequence of P1 is pending a patent of China.)
Expression and purification of peptides, and assay of inhibitory activity in vitro. To investigate whether the peptides inhibited the activity of beta-lactamase in vitro, they were expressed as GST fusion protein in E. coli and purified by affinity chromatography (Fig. 6a). The peptides were purified and separated from GST by cleavage with thrombin and ultrafiltration (Fig. 6b). The yields of purified peptides were 710 µg of P1 and 750 µg of P2.
The peptides were assayed for their ability to inhibit the activity of beta-lactamase as described above. The results of this assay indicated that P1 can inhibit the activity of beta-lactamase, but P2 cannot do so (Figs. 7 and 8). In addition, P1 inhibits the activity of beta-lactamase in a dose-dependent manner.Fig. 6. SDS-PAGE map of GST fusion protein and target peptide. a) Lanes: 1) elution; 2) flow through; 3) cytoplasmic extracts of E. coli expression GST fusion protein; 4) protein molecular weight marker. b) Lanes: M) protein marker (composed of cytochrome c, 13,000 daltons; insulin, 5733 daltons; and bacitracin, 1422 daltons); 1) P1; 2) P2.
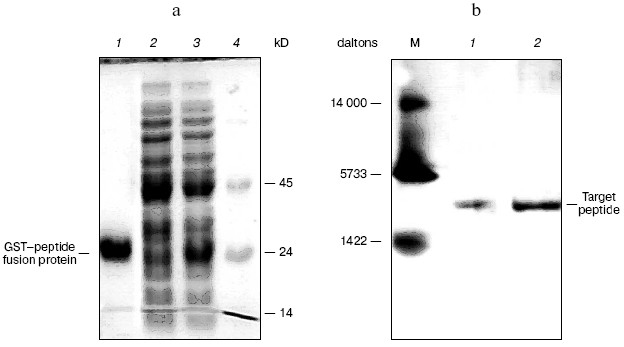
Fig. 7. Effects of P1 and P2 on the time-courses of beta-lactamase reaction: 1) without peptides; 2) P1 (50 µM); 3) P3 (50 µM).
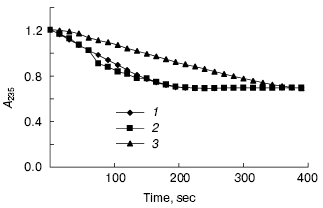
Fig. 8. Inhibitory effect of peptides on beta-lactamase: 1) P1; 2) P2; 3) insulin.
DISCUSSION
Protein-protein interactions have a key role in most biological processes and are important targets for drug design [12]. Over the past decade, there has been increased interest in the design of small peptide molecules that mimic functional epitopes of various proteins. The yeast two-hybrid system is one of the effective methods for analysis of large randomized libraries of specific proteins or peptides that bind a target protein with high affinity and sensitivity. The yeast two-hybrid system was originally devised by Fields and Song [13] as a genetic method for detecting interactions between proteins in vivo. The basis of this system is that certain eukaryotic transcriptional activators, which are composed of a DNA-binding domain and an activating domain, can be reconstructed in vivo from their separately expressed domains [14]. Thus, if a “bait” protein expressed as a fusion with the DNA-binding domain interacts with a “prey” protein expressed as a fusion with the activating domain, a functional transcriptional activator is generated that initiates expression of a “reporter” gene. It has been subsequently improved to allow screening of cDNA fusion libraries or random peptide libraries to identify partners of specific baits.
In this work, the yeast two-hybrid system was used to identify peptides that can interact with beta-lactamase in a random library. There are nine NNY (Y stands for C or T) in the random oligonucleotide fragment. The aim of design is that amino acids encoded by NNY distribute evenly in this fragment. Additionally, gene sequence (GCTGCAGGTGACTACTAC) was also designed into the random sequence as core sequence, because amino acids (AAGDYY) encoded by this gene sequence make critical interactions with the active site of TEM-1 beta-lactamase [11]. This design may improve inhibitory activity of peptides and facilitates screening. Theoretically, adequate screening would require a minimum of 1010 transformants. However, E. coli and yeast transformation efficiency could hardly exceed 1010, thus limiting the integrality of the library two-hybrid screening. This limitation caused by the transformation efficiency may be partially overcome by screening the peptide library repeatedly.
Both P1 and P2 can interact with beta-lactamase in vivo, but the inhibition assay in vitro indicated that only P1 can inhibit beta-lactamase. Although the core sequence of amino acids (AAGDYY) makes critical interactions with the active site of TEM-1 beta-lactamase [11], our work indicates that certain amino acids in both flanks of the core sequence affect peptide interacting with the active site of TEM-1 beta-lactamase. We presume that these amino acids control peptide conformation, with some conformations unable to fit the active site of TEM-1 beta-lactamase. More work is needed to understand the inhibition mechanism of P1. We also expect stronger inhibitory peptides to be found by some other techniques, such as point mutation.
Peptide inhibitors have been used to prevent the function of proteins associated with diseases [15]. Recently, Thompson et al. obtained two peptides that inhibited activity of exotoxin A from Pseudomonas aeruginosa, using the yeast two-hybrid system [16]. So this strategy could be used to screen peptide inhibitors of TEM-1 beta-lactamase, which is the target of bacterial resistance.
This work was supported by the National Natural Science Foundation of China (No. 30171120).
REFERENCES
1.Livermore, D. M. (1995) Clin. Microbiol.
Rev., 8, 557-584.
2.Bush, K., Jacoby, G. A., and Medeiros, A. A. (1995)
Antimicrob. Agents Chemother., 39, 1211-1233.
3.Ghuysen, J. M. (1991) Annu. Rev. Microbiol.,
45, 37-67.
4.Wiedemann, B., Kliebe, C., and Kresken, M. (1989)
J. Antimicrob. Chemother., 24 (Suppl. B), 1-22.
5.Petrosino, J., Cantu, C., III, and Palzkill, T.
(1998) Trends Microbiol., 6, 323-327.
6.Bush, K. (2002) Curr. Opin. Invest. Drugs,
3, 1284-1290.
7.Doran, J. L., Leskiw, B. K., Aippersbach, S., and
Jensen, S. E. (1990) J. Bacteriol., 172, 4909-4918.
8.Strynadka, N. C., Jensen, S. E., Johns, K.,
Blanchard, H., Page, M., Matagne, A., Frere, J. M., and James, M. N.
(1994) Nature, 368, 657-660.
9.Petrosino, J., Rudgers, G., Gillbert, H., and
Palzkill, T. (1999) J. Biol. Chem., 274, 2394-2400.
10.Rudgers, G. W., and Palzkill, T. (1999) J.
Biol. Chem., 274, 6963-6971.
11.Rudgers, G. W., Huang, W., and Palzkill, T.
(2001) Antimicrob. Agents Chemother., 45, 3279-3286.
12.Vidal, M., and Endoh, H. (1999) Trends
Biotechnol., 17, 374-381.
13.Fields, S., and Song, O. (1989) Nature,
340, 245-246.
14.Brent, R., and Ptashne, M. (1985) Cell,
43, 729-736.
15.Ferrer, M., and Harrison, S. C. (1999) J.
Virol., 73, 579-582.
16.Thompson, C., Merrill, A. R., and Mangroo, D.
(2003) FEMS Microbiol. Lett., 218, 85-92.