Affinity Modification of the Restriction Endonuclease SsoII by 2´-Aldehyde-Containing Double Stranded DNAs
A. E. Sud'ina1, T. S. Zatsepin2, V. Pingoud3, A. Pingoud3, T. S. Oretskaya1,2, and E. A. Kubareva1*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (7-095) 939-3181; E-mail: kubareva@belozersky.msu.ru2Faculty of Chemistry, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (7-095) 939-3181
3Institut für Biochemie, Justus-Liebig-Universität, Heinrich-Buff-Ring 58, D-35392 Giessen, Germany; fax: +49 (641) 99-35409
* To whom correspondence should be addressed.
Received February 7, 2005; Revision received March 25, 2005
Properties of 2´-aldehyde-containing double stranded DNAs (dsDNAs) have been studied for the first time as substrate analogs of the restriction endonuclease SsoII. These reactive oligonucleotides were successfully cross-linked to the restriction endonuclease SsoII by reductive amination, and conditions for DNA-protein conjugate trypsinolysis followed by the oligonucleotide-peptide conjugate purification were optimized. Use of MALDI-TOF mass spectrometry revealed that covalent linkage forms between the sugar moiety of the central pyrimidine nucleoside of the SsoII recognition site and Lys173 of the enzyme. The latter is probably involved in initial steps of enzyme-substrate recognition during dsDNA readout.
KEY WORDS: restriction endonuclease SsoII, DNA-protein interactions, modified double stranded DNAs, affinity modification
Abbreviations: dsDNAs) double stranded DNAs (DNA duplexes); Kdis) dissociation constant; U) 2´-O-(2-oxoethyl)uridine.
The discovery and intensive study of type II restriction endonucleases
represent significant progress in molecular biology and gene
engineering during the last decade. Type II restriction endonucleases
recognize specific (often palindromic) nucleotide sequence of a DNA
molecule and hydrolyze the DNA at certain sites inside (or near) this
sequence. These enzymes are widely used for construction of recombinant
DNA molecules and DNA mapping. Type II restriction endonucleases are
the only restriction enzymes for which results of numerous biochemical
studies can be compared with structural analysis data [1, 2]. This explains why type II
restriction endonucleases are often used as model systems for studies
of structural aspects of specific DNA-protein interactions and
mechanisms of Mg2+-dependent hydrolysis of phosphodiester
bonds in DNA molecules [3].
However, detailed mechanisms underlying specific enzyme-substrate recognition, regulation of these enzymes, and also evolutionary and structural relationship among various restriction endonucleases remain unclear. Subsequent study of type II restriction endonucleases and new information on structure and properties of complexes between these enzymes and DNA might not only clarify these important problems, but also allow directed modification or alteration of substrate specificity of the restriction endonucleases.
The method of affinity modification of proteins by DNA is often employed for determination of protein regions involved in the interaction of these proteins with DNA ligand [4-6]. This method consists of insertion of reactive group into one or both interacting biomolecules, and this results in covalent binding of protein to DNA. There are a few preconditions required for protein-nucleic acid conjugate formation: 1) reacting groups of a protein and DNA should locate in proximity to each other; 2) introduced modifications should not alter the structure of the protein or DNA and thereby influence the complex formation process.
The method of affinity protein modification by DNA is very useful for both localization of protein regions closely positioned to DNA during protein-nucleic acid complex formation and identification of amino acid residues directly involved in this interaction. The whole procedure includes hydrolysis of the protein component of such a conjugate using proteases to yield oligonucleotide-peptide conjugate. Subsequent amino acid sequencing of the resulting peptide (representing a part of active site or recognition domain) is carried out by mass spectrometry or by the Edman sequencing method [5, 6].
In the present study, we have investigated for the first time the interaction of 2´-aldehyde-containing double stranded DNAs (dsDNAs; DNA duplexes) with the type II restriction endonuclease SsoII. Reactive oligonucleotides were successfully cross-linked to the restriction endonuclease SsoII by reductive amination and the resulting DNA-protein conjugate was subjected to trypsinolysis. Use of MALDI-TOF mass spectrometry revealed involvement of Lys173 of the restriction endonuclease in covalent bond formation with the sugar moiety of the central pyrimidine nucleoside of the enzyme recognition site.
MATERIALS AND METHODS
Chemicals. The following chemicals were used in this study: poly(dI·dC) from ICN (USA); 10-200 kD protein molecular mass markers from MBI-Fermentas (Lithuania); Coomassie R-250 from Bio-Rad (USA); [gamma-32P]ATP from Izotop (Russia).
Enzymes and proteins. Bovine serum albumin (BSA) was from Sigma (USA); trypsin from Calbiochem (USA); T4 polynucleotide kinase from SibEnzyme (Russia). The plasmid pQESso9/pACMS7 carrying the SsoII gene was a generous gift from Dr. A. S. Karyagina (Institute of Agricultural Biotechnology, Russian Academy of Agricultural Sciences). The restriction endonuclease SsoII was expressed in E. coli JM109 as described in [7, 8]. A recombinant form of the restriction endonuclease SsoII contains the N-terminal sequence MetArgGlySer(His)6GlySerSerGln. The molecular mass of the recombinant enzyme is 37.2 kD.
DNA duplexes I-V were prepared as described in [9]. The DNA duplexes I, II, and V contained 5´-32P-label in both strands, whereas duplexes III and IV contained the label in the modified strand. Bands containing radioactivity were visualized in gels and analyzed using Molecular Dynamics PhosphorImager SI (Molecular Dynamics, USA) and the program Image Quantum 5.0.
Study of the restriction endonuclease SsoII binding to DNA duplexes I, II, and V. Complex formation between the restriction endonuclease SsoII and 100 nM dsDNAs was carried out in 20 µl buffer A (10 mM Tris-HCl, pH 8.5, containing 100 mM NaCl, 5 mM CaCl2, 0.1 mg/ml BSA, 10% glycerol) at 20°C for 30 min in the presence of 10 ng/µl poly(dI·dC). DNA-protein complex and free DNA duplex were separated by gel electrophoresis under non-denaturing conditions [10]. The dissociation constant (Kd) value for the restriction endonuclease SsoII complexes with dsDNAs corresponds to the enzyme dimer concentration at which 50% of DNA substrate is in the complex with the enzyme. Restriction endonuclease concentrations were varied from 1 to 2000 nM.
Determination of apparent rate constant (kapp) for hydrolysis of the DNA duplexes I-III and V by the restriction endonuclease SsoII. The reaction was carried out in the buffer B (10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 50 mM NaCl, 1 mM dithiothreitol (DTT), 0.1 mg/ml BSA) at 37°C for 0-120 min. The reaction was terminated by adding 10 µl of solution for oligonucleotide application to polyacrylamide gel (0.025% solution of bromophenol blue and xylene cyanol in formamide-water mixture, 4 : 1). Concentrations of DNA duplexes and the restriction endonuclease SsoII dimer were 100 and 12.5 nM, respectively. Reaction products were analyzed by electrophoresis in 20% polyacrylamide gel. The apparent rate constant was calculated using the following formula: kapp = delta * CS/CE min-1, where delta is a slope of the initial part of the kinetic curve, delta = Delta%/(Deltat * 100%); CS and CE are initial concentrations of dsDNA and enzyme, respectively.
Affinity modification of restriction endonuclease SsoII by DNA duplexes III and IV. The DNA duplexes III and IV were incubated with the restriction endonuclease SsoII in the buffer A at 20°C for 30 min or at 37°C for 15 min in the presence of 10 ng/µl poly(dI·dC). The concentration of the DNA duplexes was 100 nM and the concentrations of the restriction endonuclease SsoII (calculated per the enzyme dimer) were 25, 50, 100, 250, 500, and 1000 nM. After addition of NaBH3CN (final concentration 25 mM) the reaction mixture was incubated at 20°C for 2 h or at 37°C for 60 min. Reaction products were analyzed by electrophoresis in 4%/12% polyacrylamide gel using the Laemmli system [11].
Isolation of oligonucleotide-peptide conjugate. The DNA duplex III (1 µM) was incubated with the restriction endonuclease SsoII (10 µM) in buffer C (10 mM Tris-HCl, pH 8.5, 100 mM NaCl, 5 mM CaCl2) at 37°C for 15 min. After addition of 100 mM aqueous solution of NaBH3CN to final concentration 25 mM, samples were incubated at 37°C for 1 h. Reaction products were separated by gel electrophoresis in the Laemmli system. Bands were visualized by autoradiography using Retina Roentgen film (Germany) and by Coomassie R-250 staining. Strips corresponding to the DNA-protein complex were excised and cut into pieces (1 × 1 mm) and each piece was placed into a 0.5 ml tube. Gel pieces were sequentially treated using the following protocol: 1) 150 µl 40% aqueous methanol containing 5% CH3COOH, 15 min; 2) 150 µl water, 5 min; 3) 150 µl 50% aqueous methanol containing 50 mM ammonium bicarbonate, 20 min at 56°C (this step was repeated if the dye was not completely removed); 4) 150 µl water, 5 min; 5) 150 µl 50 mM ammonium bicarbonate containing 5 mM DTT, 20 min at 56°C; 6) 150 µl freshly prepared 15 mM iodoacetamide in 50 mM ammonium bicarbonate, 15 min in the darkness (two times); 7) 150 µl water, 5 min (two times); 8) 150 µl acetonitrile, 10-15 min. Remaining acetonitrile was evaporated in vacuum (5-10 min). Gel pieces were then treated with 1.5 µl trypsin solution (10 ng/µl) in buffer D (20 mM ammonium bicarbonate, 50 µM DTT). Tubes were incubated on ice for 10 min and after addition of 1-1.5 µl water they were incubated at 37°C for 48 h. Every 12 h a new portion of trypsin was added. For extraction of proteolytic products of the oligonucleotide-peptide conjugate gel pieces were treated with 20 µl of 50 mM ammonium bicarbonate for 3 h at 45°C. After supernatant collection, the procedure was repeated again. The supernatant was evaporated to 10-15 µl, and after addition of 10 µl of the solution for oligonucleotide loading onto polyacrylamide gel, isolated products were analyzed by electrophoresis in 15% polyacrylamide gel in the presence of 2 M urea. After autoradiography, the band corresponding to the oligonucleotide-peptide conjugate was excised from the gel, placed in a 2 ml tube, and homogenized. After addition of 500 µl water and incubation at 20°C for 3-4 h, supernatant was collected and the procedure was repeated. The oligonucleotide-peptide conjugate was desalted using ultrafiltration on Microcon filters (YM-3, Millipore, USA) and 50 mM ammonium citrate as an elution buffer. Final concentration of the sample was 5-10 µM.
MALDI-TOF mass spectra were registered using a Voyager DE instrument (PerSeptive Biosystems, USA) using 2,4,6-trihydroxyacetophenone (20 mg/ml in acetonitrile) and aqueous ammonium citrate (20 mg/ml) (1 : 1 v/v). Comparison of experimentally obtained molecular masses of the oligonucleotide-peptide conjugate with theoretically possible peptides formed during trypsinolysis of the restriction endonuclease was carried out using the program PeptMass (http://cn.expasy.org/tools/peptide-mass.html) after subtraction of the oligonucleotide molecular mass.
RESULTS AND DISCUSSION
Endonuclease SsoII is a component of the restriction-modification system of Shigella sonnei 47 [12]. In double stranded DNA this enzyme recognizes a nucleotide sequence degenerated by a central nucleotide pair 5´-vCCNGG-3´ (where N = A, G, C, or T) and hydrolyzes phosphodiester bond marked with the arrow.
Enzyme contacts with a sugar-phosphate backbone of DNA mainly involve charged and polar amino acid residues, preferentially Arg and Lys [13]. In the present study, we have developed an approach for identification of amino acid residues closely positioned to the enzyme recognition site of DNA. This approach employs DNA ligands carrying a reactive group in 2´-position of the sugar moiety. Recent studies in our laboratory on a series of substrate analogs containing an aldehyde group in the 2´-position of the sugar moiety revealed their potential applicability for affinity modification of p50 subunit of human transcription factor NF-kappaB [9, 14].
For selection of the position for modified nucleoside insertion into DNA substrate, we have investigated the interaction of restriction endonuclease SsoII with 32P-labeled DNA duplexes I and II containing AT or GC base pairs in the central position of the recognition site. The nature of the central nucleoside pair was not essential for binding effectiveness and hydrolysis of DNA substrate by the restriction endonuclease SsoII (table). Heterocyclic base removal and preservation of intact sugar fragment of these nucleoside residues were also not critical for the enzyme functioning. Substitution of (2R,3S)-2-oxomethyltetrahydrofuranol-3 residue, a structural analog of 2-deoxy-D-ribofuranose, for 1,3-propanediol did not reduce effectiveness of DNA hydrolysis (compared with the non-modified duplex) [15]. Nevertheless, introduction of a pyrophosphate group instead of the phosphodiester bond in the central position of the recognition site resulted in blockade of catalytic activity of restriction endonuclease SsoII [16].
Parameters of interaction of restriction endonuclease SsoII with
non-modified and 2´-aldehyde-containing DNA duplexes I-V
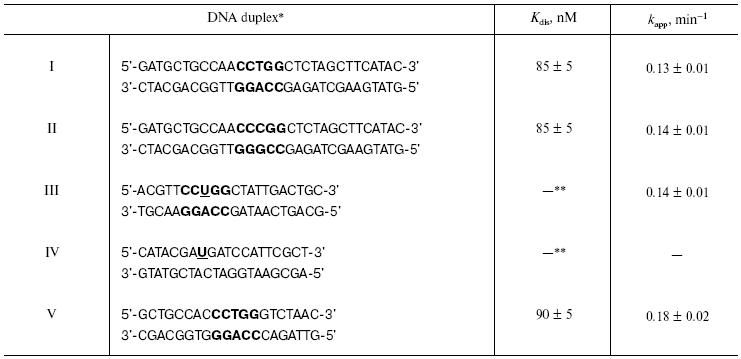
*Recognition site of restriction endonuclease SsoII is
shown in bold.
**Kdis cannot be determined because the process of
complex formation is accompanied by covalent bond formation between
enzyme and substrate.
Note: U is 2´-O-(2-oxoethyl)uridine
residue.
Taking into consideration these data, we suggest that recognition of central nucleotide pair by the restriction endonuclease involves contacts between sugar-phosphate backbone and the enzyme during DNA-protein complex formation known as indirect DNA readout [2].
For experimental verification of this hypothesis, we prepared 32P-labeled DNA duplex III containing the 32P-label into the modified strand. In this duplex a 2´-aldehyde group was introduced into the sugar moiety of the central pyrimidine nucleotide of the recognition site. Initially we investigated the effect of the 2´-aldehyde group introduction into DNA on the enzyme binding and catalytic activity (with DNA duplex III as substrate). The modified DNA duplex IV lacking the enzyme recognition site was used as control.
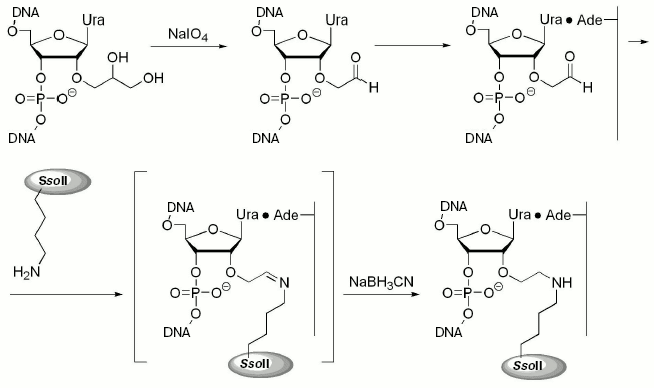
The 2´-aldehyde group was generated by oxidation of the corresponding 1,2-diol with sodium periodate (Fig. 1). Excess of the oxidizing agent was removed by oligonucleotide precipitation with alcohol and DNA duplexes were then formed. Interaction of the epsilon-amino group of Lys residues (in proteins) with 2´-aldehyde group (in DNA) resulted in formation of a Schiff base, which was then reduced to a secondary amine with NaBH3CN (Fig. 1).
The table shows rate constants for substrate analog hydrolysis by restriction endonuclease SsoII in comparison with parameters of the enzyme interaction with non-modified duplex V. Introduction of the 2´-aldehyde group into the DNA insignificantly influenced catalytic activity of the restriction endonuclease SsoII assayed with such substrates. This implies that the 2´-aldehyde containing DNA duplex III is an effective reagent for affinity modification of the restriction endonuclease SsoII.Fig. 1. Scheme of preparation of 2´-aldehyde containing DNA duplexes and their interaction with restriction endonuclease SsoII.
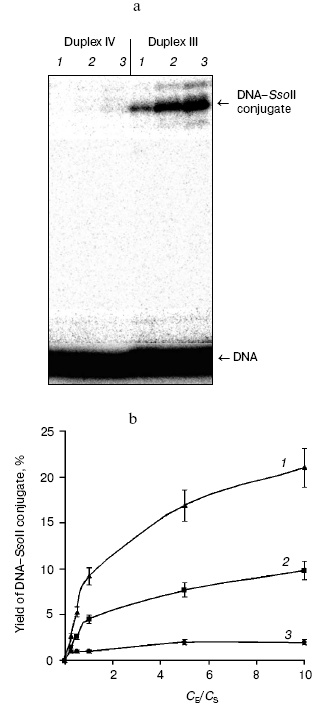
Optimization of conditions for preparation of cross-linked complex between the restriction endonuclease SsoII with DNA also employed DNA duplexes III (containing the enzyme recognition site) and IV (lacking the enzyme recognition site). It was important to elucidate the dependence of cross-linking on protein/DNA ratio. Figure 2 shows results of such experiments. The highest effectiveness of the interaction between the restriction endonuclease SsoII and DNA duplex III was observed at 5-fold excess of the enzyme over substrate. The cross-linked conjugate formation was observed both with specific substrate, DNA duplex III, and nonspecific substrate, DNA duplex IV. However, in the case of the specific substrate (duplex III) yield of reaction products was several times higher than in the case of duplex IV. Increase in reaction temperature from 20 to 37°C was accompanied by 2-fold increase in yield of the DNA-protein conjugate (Fig. 2b). The high yield of the conjugate (~20%) suggests that contact formation with sugar moiety of the central nucleoside of the enzyme recognition site involves a Lys residue of the restriction endonuclease SsoII.
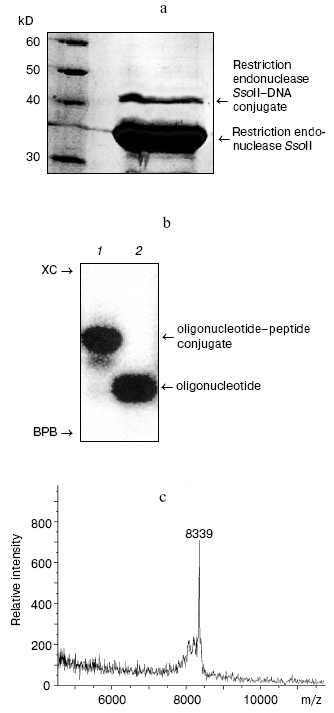
During the next step, we have developed an approach for determination of the amino acid residue of the enzyme interacting with the 2´-aldehyde group of DNA duplex III. Oligonucleotide-peptide conjugate was isolated by gel electrophoresis using the Laemmli system (Fig. 3a). Comparison of electrophoretic mobility of the SsoII-DNA complex with molecular masses of protein markers revealed that molecular mass of the enzyme-substrate conjugate was within 42-43 kD. Since molecular masses of the restriction endonuclease SsoII and the modified oligonucleotide are 37.2 and 6.5 kD, respectively, it is reasonable to suggest that the oligonucleotide-peptide conjugate corresponds to a cross-linked product of protein monomer and one molecule of the modified oligonucleotide of the duplex used.Fig. 2. a) Radioautograph of separation of products of restriction endonuclease SsoII modification by 2´-aldehyde-containing DNA duplexes III and IV by gel electrophoresis in the Laemmli system. Lanes 1-3: ratios of the restriction endonuclease SsoII concentration (CE) to concentration of DNA duplex (CS = 100 nM) were 1 : 2, 5 : 1, and 10 : 1, respectively. The enzyme concentration was calculated as a dimer. b) The effect of component concentration ratio in reaction mixture and temperature on effectiveness of the restriction endonuclease SsoII conjugate formation with DNA duplex III (at 37°C (1); at 20°C (2)) and IV (at 20°C (3)).
The large molecular mass of the conjugate complicated its effective isolation from polyacrylamide gel and the method of trypsinolysis in the gel developed by Shevchenko et al. [17] was employed for proteolysis of the peptide fragment.
According to literature data, cross-linking of DNA to proteins significantly increases resistance of proteins to proteases [18]. So, trypsinolysis was carried out at 37°C for 48 h. Proteolytic treatment significantly increased effectiveness of isolation of the oligonucleotide-peptide conjugate to 90-95%. The conjugate was further purified by gel electrophoresis in polyacrylamide gel under denaturing conditions (Fig. 3b). After the isolation from the gel, the oligonucleotide-peptide conjugate was desalted by ultrafiltration. Employment of an alternative desalting method using StageTips reversed-phase microcolumns (Proxeon, Switzerland) or ZipTip (Millipore, USA) was much less effective because losses of the oligonucleotide-peptide conjugate exceeded 50%.
For determination of the Lys residue responsible for contact formation of restriction endonuclease SsoII with DNA duplex III, the resulting oligonucleotide-peptide conjugate was analyzed by MALDI-TOF mass spectrometry (time of flight mass spectrometry with matrix-assisted laser desorption/ionization). The MALDI-TOF mass spectrum of the oligonucleotide-peptide conjugate contained a peak corresponding to molecular mass of 8339 daltons (Fig. 3c). Subtraction of the molecular mass of the oligonucleotide part (6425 daltons) yielded molecular mass of attached peptide (1914 daltons). This molecular mass corresponded (within experimental error) to the fragment 158LVDLVMPGVVQYTSNKR174 (1919 daltons) of the restriction endonuclease SsoII. Since cross-linking of 2´-aldehyde-containing ligand to proteins selectively involves Lys residues, this means that covalent bonding of the DNA duplex III involves Lys173 of the restriction endonuclease SsoII.Fig. 3. a) Separation of reaction products of cross-linking of restriction endonuclease SsoII with DNA duplex III. Reaction products were analyzed by gel electrophoresis in the Laemmli system followed by subsequent staining with Coomassie R-250. Protein molecular mass markers are shown on the left. b) Radioautograph of separation of products of dsDNA III-restriction endonuclease conjugate trypsinolysis in 15% polyacrylamide gel under denaturing conditions (lane 1). Lane 2: dsDNA III. XC, xylene cyanol; BPB, bromophenol blue. c) MALDI-TOF mass-spectrum of the oligonucleotide-peptide conjugate.
Attempts to confirm peptide structure by the method of tandem mass-spectrometry (MALDI-TOF or ESI MS/MS) were unsuccessful due to fragmentation of the oligonucleotide component. We plan to develop a method for determination of primary structure of the peptide after hydrolysis of the oligonucleotide component. Such variant would be very useful for simultaneous analysis of several DNA-protein conjugates.
Thus, in the present study we have investigated for the first time the properties of the synthetic 2´-aldehyde-containing DNA duplex as substrate analog for restriction endonuclease SsoII. Results of this study indicate that the presence of the modified sugar moiety insignificantly influenced binding efficacy and catalytic activity of restriction endonuclease SsoII with such substrate. This property makes the 2´-aldehyde substrates perspective reagents for affinity modification of restriction endonuclease SsoII and other DNA-recognizing proteins. We have optimized conditions for trypsinolysis of the restriction endonuclease SsoII-DNA conjugate and isolation of the oligonucleotide-peptide conjugate. Use of MALDI-TOF mass spectrometry revealed that formation of covalent bond between sugar moiety of the central pyrimidine nucleotide of the enzyme recognition site and the restriction endonuclease SsoII involves Lys173. The central nucleoside pair is recognized by restriction endonuclease SsoII during readout of secondary structure of the DNA duplex; this implies formation of contacts with the sugar-phosphate backbone. The data suggest that Lys173 participates in the process of the indirect readout during recognition and binding of DNA substrate.
Isolation of DNA-protein conjugate in gel is especially attractive in the cases of formation of several conjugates during cross-linking of proteins to DNA. This helps better characterization of each of the forming products and determination of protein regions (or amino acid residues) involved in their formation.
This work was supported by grants from the Russian Foundation for Basic Research (Nos. 04-04-48714 and 03-04-48957) and the program “Russian Universities” (No. 07.02.567).
REFERENCES
1.Aggarwal, A. K. (1995) Curr. Opin. Struct.
Biol., 5, 11-19.
2.Pingoud, A., and Jeltsch, A. (2001) Nucleic
Acids Res., 29, 3705-3727.
3.Kovall, R. A., and Matthews, B. W. (1999) Curr.
Opin. Chem. Biol., 3, 578-583.
4.Meisenheimer, K. M., and Koch, T. H. (1997)
Crit. Rev. Biochem. Mol. Biol., 32, 101-140.
5.Steen, H., and Jensen, O. N. (2002) Mass
Spectrom. Rev., 21, 163-182.
6.Zatsepin, T. S., Dolinnaya, N. G., Kubareva, E. A.,
Ivanovskaya, M. G., Metelev, V. G., and Oretskaya, T. S. (2005) Rus.
Chem. Rev., 74, 77-95.
7.Sheflyan, G., Kubareva, E. A., Kuznetsova, S. A.,
Karyagina, A. S., Nikolskaya, I. I., Gromova, E. S., and Shabarova, Z.
A. (1996) FEBS Lett., 390, 307-310.
8.Pingoud, V., Kubareva, E., Stengel, G., Friedhoff,
P., Bujnicki, J. M., Urbanke, C., Sudina, A., and Pingoud, A. (2002)
J. Biol. Chem., 277,
14306-14314.
9.Romanenkov, A. S., Ustyugov, A. A., Zatsepin, T.
S., Nikulova, A. A., Kolesnikov, I. V., Metelev, V. G., Oretskaya, T.
S., and Kubareva, E. A. (2005) Biochemistry (Moscow), in
press.
10.Carey, J. (1991) Meth. Enzymol.,
208, 103-117.
11.Laemmli, U. K. (1970) Nature, 227,
680-685.
12.Uporova, T. M., Kartashova, I. M., Skripkin, E.
A., Lopareva, E., and Nikolskaya, I. I. (1985) Vopr. Med. Khim.,
31, 131-136.
13.Gromiha, M., Siebers, J. G., Selvaraj, S., Kono,
H., and Sarai, A. (2004) J. Mol. Biol., 337, 285-294.
14.Turutin, D. V., Zatsepin, T. S., Timchenko, M.
A., Kubareva, E. A., and Oretskaya, T. S. (2002) Mol. Biol.
(Moscow), 36, 877-879.
15.Kubareva, E. A., Petrauskene, O. V., Karyagina,
A. S., Tashlitsky, V. N., Nikolskaya, I. I., and Gromova, E. S. (1992)
Nucleic Acids Res., 20, 4533-4538.
16.Gromova, E. S., and Shabarova, Z. A. (1990)
Progr. Nucleic Acids Res. Mol. Biol., 39, 1-47.
17.Shevchenko, A., Wilm, M., Vorm, O., and Mann, M.
(1996) Analyt. Chem., 68, 850-858.
18.Steen, H., Petersen, J., Mann, M., and Jensen, O.
N. (2001) Protein Sci., 10, 1989-2001.