Heterologous Expression, Purification, and Properties of a Potato Protein Inhibitor of Serine Proteinases
A. S. Speranskaya1, A. A. Krinitsina2, T. A. Revina1, N. G. Gerasimova1, Ya. S. Keruchen'ko1, A. B. Shevelev1, and T. A. Valueva1*
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: valueva@inbi.ras.ru2Faculty of Biology, Lomonosov Moscow State University, 119992 Moscow, Russia
* To whom correspondence should be addressed.
Received June 13, 2006; Revision received June 30, 2006
The gene PKPI-B10 [AF536175] encoding in potato (Solanum tuberosum L., cv. Istrinskii) a Kunitz-type protein inhibitor of proteinases (PKPI) has been cloned into the pET23a vector and then expressed in Escherichia coli. The recombinant protein PKPI-B10 obtained as inclusion bodies was denatured, separated from admixtures by ion-exchange fast protein liquid chromatography (FPLC) on MonoQ under denaturing conditions, and renatured. The native protein was additionally purified by ion-exchange FPLC on DEAE-Toyopearl. The PKPI-B10 protein effectively inhibits the activity of trypsin, significantly weaker suppresses the activity of chymotrypsin, and has no effect on other serine proteinases: human leukocyte elastase, subtilisin Carlsberg, and proteinase K, and also the plant cysteine proteinase papain.
KEY WORDS: Kunitz-type proteinase inhibitors, serine proteinases, potato, heterologous expressionDOI: 10.1134/S0006297906110022
Abbreviations: BAPA) Nalpha-benzoyl-L-arginyl p-nitroanilide; bp) base pairs; FPLC) ion-exchange fast protein liquid chromatography; HLE) human leukocyte elastase; PKPI) potato Kunitz-type proteinase inhibitors; Suc-AAV-pNa) N-succinyl-L-alanyl-L-alanyl-L-valyl p-nitroanilide; Suc-GGF-pNa) N-succinyl-glycyl-glycyl-L-phenylalanyl p-nitroanilide; Z-AAL-pNa) N-carbobenzoxy-L-alanyl-L-alanyl-L-leucyl p-nitroanilide.
Potato Kunitz-type proteinase inhibitors (PKPIs) are proteins with
molecular weight of 20-25 kD [1, 2]. Based on the structure of the N- and C-terminal
amino acid sequences, these proteins are divided into three structural
groups: A, B, and C [1]. The cultivated potato
genome contains genes encoding all these three groups, and each group
is probably encoded by multiple genes [2].
We have shown [3, 4] that the potato (Solanum tuberosum L., cv. Istrinskii) genome contains at least four genes encoding the group B proteins: PKPI-B1, PKPI-B2, PKPI-B9, and PKPI-B10. Analysis of the amino acid sequence, which was reconstructed based on the nucleotide sequence of the PKPI-B9 gene, showed that it encoded an isoform of the 21-kD protein, PSPI-21-6.3. The protein PSPI-21-6.3, an effective inhibitor of human leukocyte elastase (HLE), was isolated by us earlier from potato tubers of the same cultivar [5].
Nucleotide sequences of the genes PKPI-B1 and PKPI-B2 contained 99-100% of residues identical to the cDNA sequences isolated from the potato cultivar Provita and studied earlier [2]. The sequence of the gene PKPI-B10 was only 96% homologous to the known genomic nucleotide sequences and sequences of cDNA from different potato cultivars. And the amino acid sequence of the protein recovered based on the nucleotide sequence and encoded by the gene PKPI-B10 was found to contain unique substitutes [4].
Substitutions of residues in the amino acid sequences of PKPI inhibitors can change the specificity of its action on proteinases. Thus, three proteins of the B group prepared as a result of expression in Schizosaccharomyces pombe and only 97% homologous in the amino acid sequences varied in the action specificity to serine proteinases, trypsin, alpha-chymotrypsin, and also to cysteine proteinase papain [6]. The variability of the structure and properties of the PKPI proteins seems to be functionally important and to play a role in the plant defense against attack of pathogens [7-10]. In our works [11, 12] the potato tubers infected with the oomycete Phytophthora infestans and also subjected to mechanical damage were shown to accumulate proteinase inhibitors, mainly presented by the PKPI-B proteins. These proteins were highly toxic to the pathogen due to inhibition of the hyphae sprouting and acceleration of destruction of the zoomycete spores [11, 12].
The purpose of the present work was to obtain a highly purified recombinant protein PKPI-B10 and study its properties.
MATERIALS AND METHODS
To isolate the genomic DNA of potato (S. tuberosum L., cv. Istrinskii), the Escherichia coli strain BMH 71-19 (mutS), vectors pUK21 [13] and pGEM-T-Easy (Promega, USA) were used. The primers KEN1 and KEN2 were constructed as described in [3]. To perform heterologous expression, the E. coli strain BL21(DE3) and the vector pET23a (Promega) were used. Restrictases NdeI, SacI, and XhoI were from MBI Fermentas (Lithuania). Chromogenic substrates used were: N-succinyl-glycyl-glycyl-L-phenylalanyl p-nitroanilide (Suc-GGF-pNa), Nalpha-benzoyl-L-arginyl p-nitroanilide (BAPA), N-carbobenzoxy-L-alanyl-L-alanyl-L-leucyl p-nitroanilide (Z-AAL-pNa) (Calbiochem, USA); N-succinyl-L-alanyl-L-alanyl-L-valyl p-nitroanilide (Suc-AAV-pNa) (Bachem, Switzerland). alpha-Chymotrypsin (EC 3.4.21.1) was from Serva (Germany); trypsin (EC 3.4.21.4) from Spofa (Czechia) additionally recrystallized from MgSO4; subtilisin Carlsberg (EC 3.4.21.14) from Sigma (USA); proteinase K (EC 3.4.21.64) from Merck (Germany); papain (EC 3.4.22.1) from Fluka (Switzerland). Low-molecular-weight proteins for electrophoresis in SDS-polyacrylamide gel were from MBI Fermentas.
The isolation of potato genomic DNA, amplification, molecular cloning, isolation of the plasmid DNA, and restriction analysis were performed as described in [3]. The plasmid constructions were sequenced with an automated AbiPrism 310 sequencer (Applied Biosystems, USA) using the standard primers pUC/M13, T7-promotor, and SP6 (Promega).
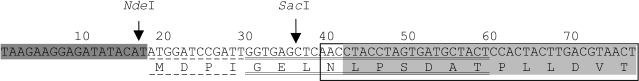
The product resulting by amplification of the potato genomic DNA with the KEN1 and KEN2 primers was cloned into the vector pUK21 linearized at the site Eco32.I. The recombinant plasmid pUK-10 was chosen which contained a DNA fragment of 587-bp size and included the part of the encoding region of the gene PKPI-B10, which corresponded to the mature protein PKPI-B10 possessing an additional N-terminal residue Asn. The plasmid pUK-10 was treated with restrictases NdeI and SacI, recognizable by the sites located, respectively, in the vector pUK21 polylinker and the primer KEN2. The resulting fragment of the plasmid was cloned in the expression vector pET23a degraded by the NdeI and XhoI restrictases. The resulting plasmid was called palphaK10.
Escherichia coli BL21(DE3) was transformed with the expression plasmid palphaK10 by the routine method [14]. The bacteria carrying the palphaK10 plasmid were cultured in dishes on agarized medium LB (peptone, 10 g/liter; yeast extract, 5 g/liter; NaCl, 10 g/liter) supplemented with ampicillin (100 mg/liter) and 0.4% glucose. The resulting bacterial colonies no older than 30 h after the transformation were washed off in 6 ml of fresh LB medium, and the resulting washing was planted into two flasks containing 100 ml of liquid LB medium (peptone, 10 g/liter; yeast extract, 5 g/liter; NaCl, 20 g/liter) supplemented with ampicillin (100 mg/liter). The fermentation was performed under conditions of intense aeration at 30°C for 24 in the absence of the inducer isopropyl thiogalactoside. After the fermentation, the grown biomass was collected by centrifugation and washed in saline to remove the medium completely. The cells were resuspended in 0.05 M Tris-HCl buffer (pH 8.0), cooled to 0°C, and disintegrated by ultrasonication on a Soniprep 150 (MSE-Sanyo, Great Britain-Japan). The resulting suspension was centrifuged at 1500g for 30 min to separate the insoluble cellular fraction containing inclusion bodies. The collected fraction of inclusion bodies was resuspended in deionized water and reprecipitated under the same conditions. The procedure was repeated two to five times.
The protein was dissolved in a denaturing buffer (0.05 M Tris-HCl buffer (pH 8.0) containing 7 M urea and 0.01 M dithiothreitol) at 65°C for 1 h, and then the solution was centrifuged at 10,000g for 30 min. The cleared supernatant (5 ml) was placed onto a Mono Q column (2.5 × 30 cm) (Pharmacia, Sweden) equilibrated with 0.05 M Tris-HCl buffer (pH 8.0) containing 7 M urea. The quantity of the purposeful protein in the preparation was from 10 to 50 mg. The sorbent-bound material was eluted in a linearly increasing gradient of NaCl concentration (from 0 to 1 M) in the starting buffer. The fractions containing the desired protein (1-2 mg/ml) were combined.
The denatured protein was renatured by dialysis against 0.05 M Tris-HCl buffer (pH 8.0) at 4°C for 96 h, with a single change of the buffer. Then the resulting suspension was centrifuged at 10,000g for 30 min. The supernatant containing the renatured protein PKPI-B10 was placed onto a column (2.5 × 30 cm) with DEAE-Toyopearl (Toyo Soda, Japan) equilibrated with 0.05 M Tris-HCl buffer (pH 8.0). The protein bound to the ion-exchanger was eluted in a linearly increasing gradient of NaCl concentration (from 0 to 1 M) in the starting buffer. The fractions containing protein PKPI-B10 were combined, desalted, and concentrated by ultrafiltration in an Amicon cell (USA) equipped with a UM-2 membrane.
Electrophoresis in the presence of SDS and beta-mercaptoethanol was performed in 15% polyacrylamide gel (SDS-PAGE) by the Laemmli method [15]. The gels were stained with 0.1% Coomassie R-250 solution in 25% ethanol supplemented with 5% formaldehyde.
The protein concentration was determined by the modified Bradford's method [16] with BSA as the standard.
The activity of recombinant protein PKPI-B10 was evaluated by suppression of activities of the corresponding enzymes (trypsin, chymotrypsin, HLE, subtilisin Carlsberg, proteinase K, and papain). The enzyme activities were determined by the rate of hydrolysis of chromogenic substrates: BAPA (for trypsin and papain), Suc-GGF-pNa (for chymotrypsin), Suc-AAV-pNa (for HLE), and Z-AAL-pNa (for subtilisin and proteinase K) [17-19]. Concentrations of the active enzymes were determined by titration of active sites: for trypsin with p-nitrophenyl-p´-guanidinobenzoate [20] and for chymotrypsin with N-trans-cinnamoylimidazole [21].
Suspension of macroconidia of the fungus Fusarium culmorum (2,000,000 spores per ml) was obtained by washing them off from the surface of 14-day-old mycelium grown at 25°C on oat-agar medium supplemented with potato thermostable proteins. To the suspension of macroconidia solution of protein PKPI-B10 was added at the ratio of 1 : 1. The mixture (0.1 ml) was placed onto an object glass, covered with a cover glass, and incubated in a humid chamber at 22°C for 18 h. The quantities of emerged and lysed macroconidia and the length of hyphae produced on their emergence were determined with a Laboval microscope (Germany) at the magnification ×120 (in three replicates for each concentration of the protein). A suspension of macroconidia without addition of protein PKPI-B10 was used as the control.
RESULTS
Clones of the potato genomic DNA were analyzed, and a recombinant plasmid pUK-10 was chosen which contained a DNA fragment 587 bp in size. This fragment included a part of the encoding region of the gene PKPI-B10 [AF536175], which corresponded to the mature protein PKPI-B10 containing an additional Asn residue at the N-terminus. The Asn residue is a constituent of the sequence of the conservative peptide of the precursor protein encoding the vacuole-distributing signal [1, 22]. Plasmid pUK-10 was used for producing the expression plasmid palphaK10 encoding the protein containing additionally seven amino acid residues in the N-terminal region (Fig. 1).
The expected product of palphaK10 expression, which is the recombinant protein PKPI-B10, was mainly detected in inclusion bodies and in lesser amount in the soluble fraction of the E. coli strain BL21(DE3) cell proteins. The molecular weight of the resulting PKPI-B10 protein determined by SDS-PAGE was about 22 kD and coincided with the calculated value of 21.7 kD [4]. No protein product with such molecular weight was detected in the control bacteria transformed by the insertion-free pET23b vector.Fig. 1. Scheme of production of recombinant plasmid palphaK10 in the beginning of the translated region. The nucleotide sequence (and the amino acid residues encoded by them) of the vector pET23a is shaded darkly, that of the N-terminal region of the mature protein PKPI-B10 is shaded lightly; the nucleotide sequence of the polylinker pUK21 fragment is underlined with a dashed line, and that of the KEN1 primer with a double solid line; the N-terminal region of the gene PKPI-B10 is framed. The arrows indicate the action sites of NdeI and SacI restrictases.
The insoluble fraction containing inclusion bodies of E. coli was dissolved in denaturing buffer. It is known that during the in vitro renaturation of proteins their molecules can be folded both correctly and incorrectly and also aggregate due to intermolecular interactions [23]. Although foreign admixtures are sometimes reported to virtually have no influence on the folding of the desired protein [23], preliminary experiments have shown that non-protein admixtures affected the protein PKPI-B10 renaturation. The absence of protein renaturation under these conditions can be also caused by aggregation of its molecules. Therefore, the denatured inclusion bodies were purified from protein and non-protein admixtures.
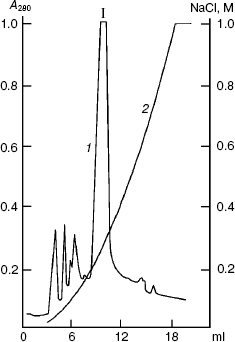
Figure 2 shows results of the sought protein isolation by anion-exchange FPLC on a column with Mono Q at pH 8.0 in the presence of 7 M urea. By data of SDS-PAGE, component I mainly contained protein PKPI-B10 and was virtually separated from the admixtures that were present in the lysate of E. coli cells. The yield of the denatured protein PKPI-B10 was 2 mg per 0.4 g biomass.
Refolding of solubilized proteins of E. coli inclusion bodies is known to occur either by fast dilution of the solution or by dialysis, which provides a smooth decrease in the concentration of the denaturing agent. The correct folding of the protein, in the absence of intermolecular aggregation, can occur under either conditions and depends on the properties of the protein [23]. This can only be determined experimentally. Preliminary experiments showed that PKPI-B10 is folded with a high efficiency under conditions of both fast dilution of the denaturing solution and dialysis. The bulk of the PKPI-B10 protein was renatured by dialysis against 0.05 M Tris-HCl buffer (pH 8.0).Fig. 2. Ion-exchange FPLC on a Mono Q column of the proteins produced on denaturation of the inclusion bodies. I is the component containing the denatured recombinant protein PKPI-B10; 1) A280; 2) NaCl.
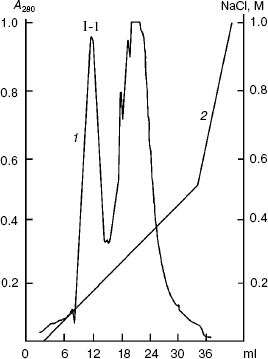
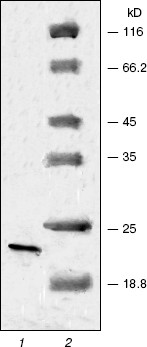
The renatured PKPI-B10 was additionally purified to homogeneity by anion-exchange FPLC on DEAE-Toyopearl at pH 8.0 (Fig. 3). As shown by data of SDS-PAGE, the component I-1 eluted with 0.15 M NaCl contained the homogenous protein PKPI-B10 (Fig. 4). It was also concluded that the recombinant protein PKPI-B10 molecule consisted of one polypeptide chain.
Fig. 3. Ion-exchange FPLC on a column with DEAE-Toyopearl of component I. I-1 is the component containing recombinant protein PKPI-B10; 1) A280; 2) NaCl.
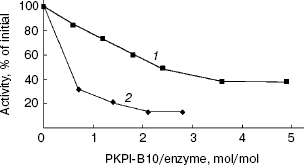
The effects of protein PKPI-B10 on activities of trypsin and alpha-chymotrypsin are shown in Fig. 5. PKPI-B10 protein effectively suppressed the activity of trypsin and less markedly the activity of chymotrypsin. The suppression of the trypsin activity depended linearly on the inhibitor quantity up to 80% inhibition (Fig. 5, curve 1). The calculations showed that 1 mol PKPI-B10 interacted stoichiometrically with 1 mol trypsin. But the inhibition of chymotrypsin did not depend on the PKPI-B10 quantity stoichiometrically, and more than a threefold excess of the inhibitor was required to reach the 50% inhibition (Fig. 5, curve 2).Fig. 4. SDS-PAGE of component I-1 resulting after ion-exchange chromatography: 1) protein PKPI-B10; 2) marker proteins (from top down): beta-galactosidase, BSA, ovalbumin, lactate dehydrogenase, endonuclease BSP981, beta-lactoglobulin (on the right, molecular weights of marker proteins in kD).
Protein PKPI-B10 had no effect on HLE, subtilisin Carlsberg, and proteinase K (a thiol-dependent subtilisin-like enzyme from Tritirachium album), as well the plant cysteine proteinase papain.Fig. 5. Influence of protein PKPI-B10 on the activities of target proteinases: 1) chymotrypsin; 2) trypsin. To determine the enzymatic activity BAPA (1) and Suc-GGF-pNa (2) were used as substrates.
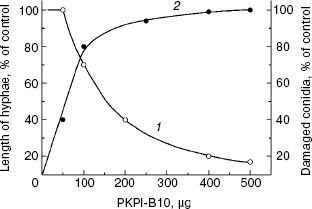
The effect of recombinant protein PKPI-B10 was studied on the growth and development of the phytopathogenic fungus F. culmorum, which is an agent of a most dangerous disease of potato, so-called vine wilt [24]. The addition of 160 µg PKPI-B10 to the suspension of the fungus macroconidia decreased by 50% the length of the emerging hyphae as compared to the control, and the addition of 500 µg virtually completely suppressed emergence of the fungus macroconidia (Fig. 6, curve 1), and all the conidia were damaged (curve 2). Thus, protein PKPI-B10 was highly toxic for the phytopathogen. It not only suppressed the emergence of hyphae, but also accelerated destruction of the fungal macroconidia.
Fig. 6. Influence of recombinant protein PKPI-B10 on the emergence of hyphae and lysis of macroconidia of the fungus F. culmorum: 1) size of hyphae; 2) number of lysed conidia; mean values of three independent experiments are presented with standard error from 2 to 10%.
DISCUSSION
Heterologous expression in E. coli cells transformed by plasmid palphaK10 resulted in synthesis of the 22-kD protein PKPI-B10 with a molecule consisting of one polypeptide chain. Protein PKPI-B10 effectively inhibited trypsin, weakly suppressed chymotrypsin activity, but suppressed the growth and development of the phytopathogenic fungus F. culmorum. Therefore, protein PKPI-B10 could be a constituent of the potato defense system.
Figure 7 presents amino acid sequences of PKPI proteins of group B, which act as proteinase inhibitors. The amino acid sequences of these proteins are very similar. The primary structure of protein PKPI-B10 is most like the structure of proteins PSPI-21-6.3 [5], P1H5 [6], and P4B1 [6]. In fact, these proteins contain 94, 93, and 92% of identical amino acid residues, respectively. However, protein PKPI-B10 is different from protein PSPI-21-6.3 [5] not only by the absence of the double-chained structure, but also by the specificity of its effects on enzymes. Thus, unlike PKPI-B10 protein, protein PSPI-21-6.3 is a powerful inhibitor of HLE, trypsin, and chymotrypsin [5]. Contrasting to protein P1H5 capable of suppressing the activities of subtilisin and papain and also those of trypsin and chymotrypsin, PKPI-B10 protein does not influence subtilisin and papain [6]. Proteins PKPI-B10 and P4B1 are similar in the specificity of the effects on proteinases--they display an effective inhibition of trypsin and a poor suppression of chymotrypsin [6]. The amino acid sequences of the proteins PKPI-B10 and P1D4 are 23% identical (39 different residues). Moreover, protein P1D4 effectively inhibits the activity of papain, although its effects on trypsin and chymotrypsin are similar to those of PKPI-B10 [6]. Thus, all known protein inhibitors PKPI-B from potato are effective inhibitors of trypsin, but are different in the specificity of action on chymotrypsin, subtilisin, and the cysteine proteinase papain.
The PKPI proteins are so-called “canonical” inhibitors of serine proteinases and interact with the enzymes via a substrate-like mechanism [25]. Molecules of such type inhibitors have on their surface a specific structure, the “binding loop” which includes the peptide bond P1-P1´ forming the active site of the inhibitor [26]. In fact, the amino acid sequences of all PKPI-B inhibitors include a conservative region surrounding the peptide bond Arg68-Phe69, which is inside the peptide loop between residues Cys47-Cys96 (Fig. 7). This region of the inhibitor molecule is the first active site responsible for trypsin binding [27].Fig. 7. Comparison of the amino acid sequences of recombinant protein PKPI-B10 and PKPI proteins of the B group: 1) PKPI-B10 reconstructed sequence; 2) protein PSPI-21-6.3 [5]; 3, 4, 5) proteins P1D4, P1H5, and P4B1, respectively, from potato cv. Provita [6]. Numbers of the amino acid residues correspond to protein PKPI-B10 sequence; different residues are dashed with gray; the arrows indicate residues which are components of the active sites; and disulfide bonds are shown by * and **.

We concluded in work [5] that in the molecule of the double-chained protein PSPI-21-6.3 the peptide bond between residues Met114-Leu115 is a component of the second active site responsible for binding of HLE (or chymotrypsin) (Fig. 7). It has also been suggested that the disulfide bond between residues Cys145-Cys162 connecting the A- and B-chains of protein PSPI-21-6.3 produces the “canonical” binding loop, which includes the peptide bond Met114-Leu115. Molecules of all five inhibitory proteins PKPI-B have similar amino acid sequences in this region between residues Asp106-Gln124 (Fig. 7). However, protein P1D4, which acts on chymotrypsin 5-6-fold weaker than PSP-21-6.3, lacks the Cys145 residue, and the second peptide loop cannot be produced. The presence in the protein PKPI-B10 sequence of another disulfide bond (Cys153-Cys156) in the region between the Cys145-Cys162 residues also can prevent formation of the “canonical” binding loop (Fig. 7). Protein P1H5, which effectively inhibits chymotrypsin, has this loop and is capable of producing the second active site with residues Met151-Thr152 in the P1-P1´ position is an exception [6]. Note that protein P4B1 lacked the residue Met151. It was suggested that deletion in the P4B1 gene resulted in the removal from the protein molecule of the Met151 residue, which probably produced the second active site responsible for chymotrypsin binding. Just such a substitution led to a considerable decrease in the effect of protein P4B1 on this enzyme [6]. It is highly probable that proteins PKBI-B10, P1D4, and P4B1 poorly bind chymotrypsin without stoichiometry by the trypsin-binding active site.
It should be noted that variable amino acid residues are mainly located in the region between Cys145 and Cys162 of molecules of the above-mentioned proteins. Thus, substitutions of certain amino acid residues, as well as deletions in the primary structure of the proteins, are very likely to determine the specificity of protein effects as inhibitors of serine proteinases.
This work was supported by the Russian Foundation for Basic Research (project Nos. 04-04-48644 and 04-04-48694).
REFERENCES
1.Ishikawa, A., Ohta, S., Matsuoka, K., Hattori, T.,
and Nakamura, K. (1994) Plant Cell Physiol., 35,
303-312.
2.Heibges, A., Glaczinski, H., Ballvora, A.,
Salamini, F., and Gebhardt, C. (2003) Mol. Gen. Genomics,
269, 526-534.
3.Speranskaya, A. S., Dorokhin, A. V., Novikova, S.
I., Shevelev, A. B., and Valueva, T. A. (2003) Genetika,
39, 1490-1497.
4.Speranskaya, A. S., Krinitsina, A. A., Poltronieri,
P., Fasano, L., Santino, A., Shevelev, A. B., and Valueva, T. A. (2005)
Biochemistry (Moscow), 70, 292-299.
5.Valueva, T. A., Revina, T. A., Mosolov, V. V., and
Mentele, R. (2000) Biol. Chem. Hoppe-Seyler, 381,
1215-1221.
6.Heibges, A., Glaczinski, H., Ballvora, A.,
Salamini, F., and Gebhardt, C. (2003) Mol. Gen. Genomics,
269, 535-541.
7.Jongsma, M. A., and Bolter, C. (1997) J. Insect.
Physiol., 43, 885-895.
8.Valueva, T. A., and Mosolov, V. V. (2004)
Biochemistry (Moscow), 69, 1305-1309.
9.Cristeller, J. T. (2005) FEBS J.,
272, 5710-5722.
10.Dunaevskii, Ya. B., Elpidina, E. N., Vinokurov,
K. S., and Belozerskii, M. A. (2005) Mol. Biol. (Moscow),
39, 608-613.
11.Valueva, T. A., Revina, T. A., Kladnitskaya, G.
V., and Mosolov, V. V. (1998) FEBS Lett., 426,
131-134.
12.Valueva, T. A., Revina, T. A., Gvozdeva, E. L.,
Gerasimova, N. G., and Ozeretskovskaya, O. L. (2003) Bioorg.
Khim., 29, 499-504.
13.Vieira, J., and Messin, J. (1991) Gene,
100, 184-194.
14.Hanahana, D. (1983) J. Mol. Biol.,
5, 557-580.
15.Laemmli, U. K. (1970) Nature, 227,
680-685.
16.Appenroth, K. J., and Angsten, H. (1987)
Biochem. Physiol. Pflanzen, 182, 85-89.
17.Kakade, M. L., Simons, N., and Liener, I. E.
(1969) Cereal Chem., 46, 518-526.
18.Wenzel, H. R., Engelbrecht, S., Reich, H.,
Mondry, W., and Tschesche, H. (1980) Hoppe-Seyler's Z. Physiol.
Chem., 361, 1413-1416.
19.Lyublinskaya, L. A., Yakusheva, L. D., ans
Stepanov, V. M. (1977) Bioorg. Khim., 3, 273-279.
20.Chase, T., and Shaw, E. (1970) Meth.
Enzymol., 19, 20-27.
21.Shonbaum, G. R., Lerner, B., and Bender, M. Z.
(1961) J. Biol. Chem., 236, 2930-2935.
22.Matsouka, K., Matsumoto, S., Hattori, T.,
Machida, Y., and Nakamura, K. (1990) J. Biol. Chem., 265,
19750-19757.
23.Rainer, R., and Hauke, L. (1996) FASEB J.,
10, 49-56.
24.Popkova, K. V., Shneider, Yu. I., Volovik, A. S.,
and Shmyglya, V. A. (1980) Potato Diseases [in Russian], Kolos,
Moscow.
25.Laskowski, M., Jr., and Kato, I. (1980) Annu.
Rev. Biochem., 49, 593-626.
26.Bode, W., and Huber, R. (1992) Eur. J.
Biochem., 204, 433-451.
27.Valueva, T. A., Revina, T. A., and Mosolov, V. V.
(1999) Biochemistry (Moscow), 64, 1074-1078.