Catalytically Important Amino Acid Residues in Endoglucanases from a Mutant Strain Trichoderma sp. M7
S. D. Petrova*, N. G. Bakalova, and D. N. Kolev
Department of Biochemistry, Faculty of Biology, University of Sofia “St. Kliment Ohridski”, Dragan Tsankov Blvd. 8, 1164 Sofia, Bulgaria; fax: +3592-8656641; E-mail: spetrova@biofac.uni-sofia.bg* To whom correspondence should be addressed.
Received October 19, 2004; Revision received December 16, 2004
Two endoglucanases, EG-III (49.7 kD) and EG-IV (47.5 kD), from a mutant strain Trichoderma sp. M7 were modified with several specific reagents. Water-soluble carbodiimide completely inactivated only one of the purified endoglucanases and kinetic analysis indicated that at least two molecules of carbodiimide bind to EG-IV for inactivation. The reaction followed pseudo-first-order kinetics with a second-order rate constant of 3.57*10-5 mM-1*min-1. Both endoglucanases were inhibited by iodoacetamide, but the absence of substrate protection excluded direct involvement of cysteine residues in the catalysis. N-Bromosuccinimide (NBS) showed a strong inhibitory effect on both endoglucanases, suggesting that tryptophan residues are essential for the activity and binding to the substrate, since the presence of substrates or analogs prior to NBS modification protected the enzymes against inactivation.
KEY WORDS: cellulase, chemical modification, endoglucanase, TrichodermaDOI: 10.1134/S0006297906130049
Trichoderma reesei secrets into its culture medium an enzyme system consisting of two exoglucanases (1,4-beta-D-glucan-4-cellobiohydrolase, EC 3.2.1.91) catalyzing the release of cellobiose from the chain ends of cellulose, several endoglucanases (1,4-beta-D-glucan-4-glucanohydrolase, EC 3.2.1.4) which randomly attack internal glucosidic bonds, and beta-glucosidases (beta-D-glucoside-glucohydrolase, EC 3.2.1.21) which finally convert the disaccharide cellobiose into glucose [1-3].
The mutant strain Trichoderma sp. M7 has proved to be one of the best in producing an optimal cellulase system with higher endoglucanase activity when grown on lignocellulosic materials and has been used commercially for the conversion of delignified waste-cellulose fibers from the paper industry [4]. The biotechnological applications of cellulases require a fundamental understanding of their mode of action, based on the large number of enzymes involved, displaying different preferences against cellulose regions.
The present work describes an attempt to identify some catalytically important amino acid residues in two endoglucanases from a mutant strain Trichoderma sp. M7 by chemical modification with specific reagents.
MATERIALS AND METHODS
Materials. Microcrystalline cellulose (Avicel PH 101) was purchased from Sigma (USA); carboxymethyl cellulose sodium salt, medium viscosity (CM-cellulose) was from Koch-Light Laboratories (UK); N-bromosuccinimide (NBS) and 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide hydrochloride (CMC) were from Merck (Germany); iodoacetamide (IAA), diethylpyrocarbonate (DEP), 5,5´-dithiobis-(2-nitrobenzoic acid) (DTNB), N-acetylimidazole (NAI), and 2-chloro-1,3,5-trinitrobenzene (TNBC) were purchased from Fluka (Switzerland). All the remaining chemicals and substrates used were of analytical grade.
Enzyme assays and protein determination. Endoglucanases from mutant strain Trichoderma sp. M7 were purified and characterized as previously reported [5]. Endoglucanase activity was assayed with CM-cellulose as a substrate in 0.05 M sodium acetate buffer (NaOAc), pH 4.8, at 50°C [6]. Enzyme activity towards Avicel PH 101 was determined by incubation at 50°C for 1 h in the same buffer. Endo- and exoglucanase activities were quantified by measuring of reducing sugars released [7, 8]. One unit (IU) of endo- or exoglucanase activity was defined as the amount of enzyme catalyzing the production of 1 µmol of reducing sugar equivalent per min under the conditions used.
Protein content was determined by the method of Lowry using bovine serum albumin (fraction V) as standard or by absorbance at 280 nm.
Adsorption of enzymes on cellulose. Avicel PH 101 was used as cellulose adsorbent. A 20-mg cellulose sample was suspended in 0.5 ml 0.05 M NaOAc buffer (pH 4.8) and preincubated at 4°C for 20 min. A 0.5-ml sample of an enzyme solution (0.1-1.0 mg/ml) was added and the reaction mixture was incubated at 4°C for 30 min (time sufficient to attain the adsorption equilibrium) with continuous mixing (hydrolysis of Avicel could be neglected under these conditions). After centrifugation (5000 rpm, 10 min), the concentration of the enzymes remaining in the supernatant was determined by the Lowry method. The bound enzyme concentration was calculated from the difference between the initial enzyme concentration and the free enzyme concentration in the supernatant.
Effect of group-specific reagents. The water-soluble carbodiimide (CMC) was used to modify the carboxylic groups by incubating the reagent (1-50 mM final concentration) with the purified endoglucanases (0.08 mg/ml) in 0.05 M MES/NaOH buffer (pH 6.0) at 25°C for 60 min [9]. At indicated times, 10 µl aliquots were withdrawn and diluted in 90 µl 0.05 M NaOAc buffer (pH 4.8) to quench the inactivation reaction. The remaining activity was determined under standard assay procedure with CM-cellulose as substrate and expressed as a percentage of a control (enzyme sample incubated without reagent).
The conversion of histidine residues in endoglucanases to N-carboethoxy derivatives assessed by treatment with DEP and oxidation of tryptophan residues by NBS were performed by the following procedure. Aliquots (10 µl) of freshly prepared DEP (0.1 M in ethanol) or NBS (0.5 mM in bidistilled water) were added successively to the enzyme sample (0.08 mg/ml in 0.05 M MES/NaOH buffer (pH 6.0) for DEP or in 0.05 M NaOAc buffer (pH 5.0) for NBS) directly into a quartz cuvette. After each addition, the increase in absorbance at 242 nm (in the case of DEP) or decrease in absorbance at 280 nm (in the case of NBS) was monitored and the residual activity was measured. The number of modified histidine or tryptophan residues was determined using the molar absorption coefficients of 3200 M-1*cm-1 for carboethoxylated histidine and 5500 M-1*cm-1 for tryptophan [10, 11]. Under the experimental conditions, the reactivity of NBS was restricted to the oxidation of Trp residues. After each addition of NBS samples were removed, stored on ice until the end of experiment, and then used to measure the adsorption on Avicel. The relative adsorption of the Trp oxidized endoglucanase onto Avicel was determined under the conditions used for the intact enzyme.
Endoglucanases (0.08 mg/ml in 0.05 M sodium-phosphate buffer, pH 7.8) were modified with IAA (5-50 mM) at 37°C for 60 min for assessment of the catalytically important thiol groups [12]. The number of free thiol groups was determined by titration of the enzymes (20 µM) in 5 M urea/50 mM Tris buffer (pH 8.0) with DTNB (1 mM final). The change in absorbance at 412 nm was continuously monitored and the number of free thiol groups was calculated using the epsilon412 value of 1.36*10-4 M-1*cm-1 for the released 5-thio-2-nitrobenzoic acid [9].
Chemical modifications with NAI (10-50 mM final concentration) and TNBC (10-50 mM) were performed in 0.05 mM sodium-phosphate buffer (pH 7.5) at 25°C for 1 h [13]. Aliquots of 10 µl were withdrawn at various time intervals and the residual enzyme activity was measured under standard conditions.
In all chemical modification reactions, the ability of the substrate to protect the endoglucanases from inactivation was assessed by incubating the enzymes with modifying reagents in the presence of various concentrations of the substrate under the same experimental conditions.
RESULTS AND DISCUSSION
Adsorption of the purified endoglucanases on Avicel PH 101. The adsorption phenomena of cellulase enzymes (a prerequisite step prior to hydrolysis) reflect their behavior on cellulose surface and synergistic interactions between the individual components of the system. Several investigators have suggested that there is a competitive adsorption between cellulase components and that an optimal initial endoglucanase/exoglucanase ratio is needed for the adsorption [14-16]. Kim et al. [15] supposed that endoglucanases were a “driving force” in the hydrolysis of cellulose because of the high degree of randomness and ability to provide a sufficient number of chain ends for exoglucanases to act on.
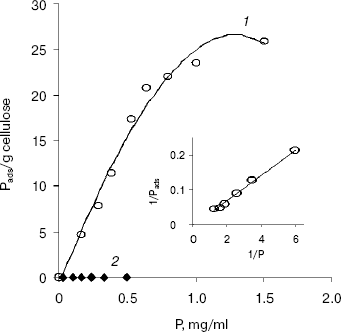
The adsorption isotherm of EG-III from Trichoderma sp. M7 on Avicel PH 101 showed a rapid increase in the amount of adsorbed enzyme onto cellulose when the concentration of enzyme in solution was increased from 0 to 1 mg/ml. At enzyme concentrations higher than 1 mg/ml, an adsorption plateau is reached (Fig. 1). The values of the adsorption association constant (Ka) and available binding sites (Pads,m) for EG-III calculated from a double-reciprocal plot of the Langmuir adsorption equation were 0.18 ml/mg and 151.17 mg/g cellulose, respectively. The relative association constant (Kr = Pads,m * Ka) defined by Gilkes et al. [17] (roughly comparing the “affinities” of related cellulase ligands for a given substrate) was 27.21 ml/g. EG-IV did not absorb on Avicel under the conditions of the reaction, suggesting a lack of substrate binding domain. The observed difference in the binding capacities of the two purified endoglucanases suggests their different roles in the degradation of cellulose.
Modification of amino acid residues with group-specific reagents. The retaining glycosyl hydrolases utilize a double-displacement mechanism of catalysis involving a glycosyl-enzyme intermediate, whereas for the inverting glycosidases a direct displacement of the leaving group by water was proposed. Both classes operate via transition states with substantial oxocarbenium ion character [18].Fig. 1. Adsorption isotherms of EG-III (1) and EG-IV (2) on Avicel PH 101 at 4°C. Inset: double reciprocal plot of the adsorption isotherm of EG-III on Avicel PH 101. P, free enzyme concentration in mg/ml; Pads, concentration of the bound enzyme (calculated from the difference between the initial enzyme concentration and the free enzyme concentration in the supernatant after centrifugation).
The pH optima of the purified EG-III and EG-IV (5.0 and 5.2, respectively) and the plots log v versus pH indicated that two groups with apparent pKe values of 5.0 and 5.5 for EG-III and 4.6 and 5.0 for EG-IV are involved in the catalysis [5]. These values suggest that residues with carboxylic side chain (aspartate/glutamate) or histidine are responsible for the catalytic activity. On the other side, the slopes of Dixon plots were not ±1, which is indicative of a complex mechanistic pathway for this endoglucanase-CM-cellulose system (the substrate with pKa about 3.65 contributed to the acidic region of the curve).
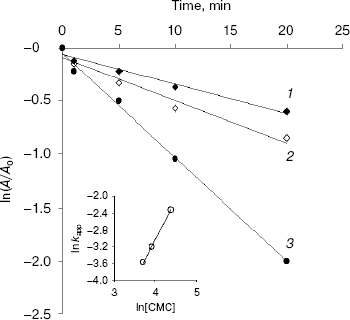
Semi-logarithmic plots of endoglucanase activity as a function of time at different CMC concentrations were linear for EG-IV (Fig. 2) indicating that the inactivation followed pseudo-first-order kinetics. The double logarithmic plot of pseudo-first-order rate constants (kapp) against CMC concentration gave a slope of 1.82, showing that at least two carbodiimide molecules are needed to inhibit one molecule of endoglucanase EG-IV. The intercept of the plot gave a second-order rate constant (k) of 3.57*10-5 mM-1*min-1 (Fig. 2, inset). The protection of the enzyme with CM-cellulose as a substrate was difficult to follow because of the reaction between the substrate and carbodiimide. The protection with cellobiose as an inhibitor of the endoglucanases was only partial.
Surprisingly, EG-III was not inactivated by CMC at different pH values (between 4.0 and 6.5). Slight inactivation was observed during the first 5 min of the reaction, and after that the activity was restored to the initial values. The reversible inactivation probably reflects structural changes induced by the modification of some exposed groups (carbodiimides are also capable of modifying Tyr and Cys residues). Alcalde et al. [19] demonstrated that modification of 4-5 carboxylic groups with carbodiimide in cyclodextrin glucanotransferase (CGTase) from Thermoanaerobacter sp. 501 kept residual activities up to 80% for hydrolysis and 50% for coupling and cyclization reactions. Carboxymethylcellulase from Aspergillus niger was thermally stabilized by chemical modification of the carboxylic groups with carbodiimide [20].Fig. 2. Inactivation of EG-IV by water-soluble carbodiimide (CMC) at various concentrations (mM): 40 (1); 50 (2); 80 (3). Inset: plot of logarithm of the observed pseudo-first-order kapp constants against ln[CMC]. kapp values were calculated from the slopes of the semi-log plots. The value of specific activity for EG-IV corresponding to 100% is 26.7 µmol/min per mg.
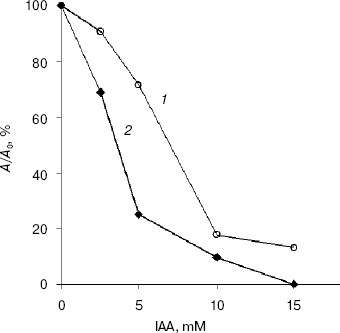
At pH values greater than 7.5, IAA modifies greatly the side group of Cys, where as at pH values between 5.0 and 7.0 the side group of His is principally affected [12]. At pH 7.85 and IAA concentrations higher than 10 mM, EG-IV completely lost the activity, whereas EG-III lost 80% of its activity after 15 min of incubation at 50°C (Fig. 3). The inclusion of the substrate in the reaction mixture before the addition of 15 mM IAA prevented the inactivation only to 20% of the initial activity. These low values of substrate protection against the inactivation with IAA might exclude the involvement of cysteine residues in the catalysis. Skoubas and Georgatsos [13] observed similar patterns of IAA inactivation of barley beta-glucosidase and explained the loss of activity and the absence of substrate protection with conformational changes in the enzymes caused by acylation of other than cysteine residues. Second-order rate constants calculated from the plots of lnkapp versus ln[IAA] were 4.16*10-2 and 5.36*10-4 mM-1*min-1 for EG-III and EG-IV, respectively [21]. The inactivation reaction order, determined from the slope of the secondary plots, was 1.0 for EG-III and 1.83 for EG-IV, whereas the reaction with Ellman's reagent under denaturing conditions revealed the presence of 20.5 and 11.8 mol free thiol groups per mol EG-III and EG-IV, respectively. This indicates that one or two thiol groups are exposed and react very rapidly, accompanied by the loss of activity up to 50% of the initial, whereas the others react more slowly, followed by complete inactivation of the endoglucanases.
The modification of enzymes with DEP (25 mM), NAI (50 mM), and TNBC (20 mM) over an extended period of 3 h did not result in any loss of their activity indicating that His, Tyr, and Lys residues are not involved in the catalysis.Fig. 3. Inactivation of EG-III (1) and EG-IV (2) by iodoacetamide (10 min incubation).
The effect of treatment of the endoglucanases with NBS was dependent upon the time and reagent concentration. After 10 min of incubation, the purified EG-III was strongly inhibited and lost up to 90% of the initial activity when incubated with 5 µM NBS, while EG-IV was more stable and retained 17% of its activity at 50 µM NBS (Fig. 4a). Semi-logarithmic plots of the residual activity as a function of time were non-linear (Fig. 4c) indicating that the modification of one Trp residue may alter the rates at which the other groups are modified. The reaction with NBS can be resolved into two phases--a rapid phase completed in less than 5 min and a slower phase lasting up to 15 min. Extrapolation of the linear portion of the curves at longer times back to the ordinate allowed for the calculation of the faster rate constant [22]. Second-order rate constants of inactivation determined from the plot of lnkapp versus ln[NBS] were 0.097 µM-1*min-1 (EG-III) and 0.008 µM-1*min-1 (EG-IV) (Fig. 4d). The double logarithmic plot gave a slope of 0.9 corresponding to the equivalent of one oxidized Trp residue for EG-IV. The presence of 0.5% CM-cellulose in the reaction mixture partially inhibited the modification with NBS indicating that some of the Trp residues remained exposed to the reagent. The treatment of EG-III with NBS affected both the catalysis and substrate binding, as the modification changed the adsorption behavior of the enzyme. The relative binding of the modified endoglucanase to Avicel was reduced by 40% compared to the adsorption of the intact enzyme at the same concentrations. The number of oxidized Trp residues was established to be 3.4 (1.3%) per mol EG-III and 0.9 (0.7%) per mol EG-IV (Fig. 4b) under the conditions given. Since, the modification of EG-IV requires higher concentrations of NBS and no substrate protection was demonstrated, it might be concluded that Trp residues are not involved directly in the catalysis. On the other hand, EG-IV did not adsorb on Avicel, which explained the results and confirmed the role of Trp residues in the substrate binding. In the presence of 6 mM Gdn-HCl (unfolding the protein), 2.9 Trp residues per mol EG-IV were modified. The role of Trp residues in the substrate binding and catalytic properties of other glycosyl hydrolases (CBH and EG-III from Trichoderma sp. [23, 24], glucanase-xylanase from Cellulomonas fimi [25], xylanases from Bacillus sp., Chainia sp., Fibrobacter succinogenes, Thermomyces lanuginosus [26-28]) have been investigated.
Fig. 4. Inactivation of EG-III (1) and EG-IV (2) by NBS (5 min incubation) and substrate protection of EG-III (3) (a). NBS (5-40 µM) oxidation of EG-III (1) and EG-IV (2) (enzymes (EG) (1.5 µM) in 0.05 M sodium acetate buffer (pH 5.0) were titrated with 0.5 mM NBS to provide the molar excess as indicated) (b). Kinetics of EG-IV inactivation by NBS at various concentrations (µM) (12.5 (1), 25 (2), 50 (3)) (c) and plot of lnkapp versus ln[NBS] for determination of second-order rate constant for EG-IV (d).

REFERENCES
1.Teeri, T. T. (1997) TIBTECH, 15,
160-167.
2.Schülein, M. (1988) Meth. Enzymol.,
160, 234-242.
3.Chen, H., Hayn, M., and Esterbauer, H. (1992)
Biochim. Biophys. Acta, 1121, 54-60.
4.Nikolov, T., Bakalova, N., Petrova, S., Benadova,
R., Spasov, S., and Kolev, D. (2000) Biores. Technol.,
71, 1-4.
5.Petrova, S. D., Bakalova, N. G., and Kolev, D. N.
(2006) Biores. Technol., in press.
6.Wood, T. M., and Bhat, K. M. (1988) Meth.
Enzymol., 160, 87-112.
7.Somogyi, M. (1952) J. Biol. Chem.,
119, 19-23.
8.Nelson, N. (1944) J. Biol. Chem.,
153, 375-378.
9.Bray, M. R., and Clarke, A. G. (1990) Biochem.
J., 270, 91-96.
10.Mata, I., Castillón, M. P., Domínguez,
J. M., Macarrón, R., and Acebal, C. (1993) J. Biochem.,
114, 754-759.
11.Spande, T. F., and Witkop, B. (1967) Meth.
Enzymol., 11, 498-506.
12.Means, G. E., and Feeney, R. E. (1971) in
Chemical Modification of Proteins, Holden-Day Inc., San
Francisco, pp. 105-110.
13.Skoubas, A., and Georgatsos, J. G. (1997)
Phytochem., 46, 997-1003.
14.Kim, D. W., Kim, T. S., Jeong, Y. K., and Lee, J.
K. (1992) J. Ferment. Bioeng., 73, 461-466.
15.Kim, D. W., Kim, T. S., Jeong, Y. K., and Lee, J.
K. (1994) Enzyme Microb. Technol., 16, 649-658.
16.Nidetzky, B., Steiner, W., and Claeyssens, M.
(1994) Biochem. J., 303, 817-823.
17.Gilkes, N. R., Jervis, E., Henrissat, B., Tekant,
B., Miller, R. C., Jr., Warren, R. A., and Kilburn, D. G. (1992) J.
Biol. Chem., 267, 6743-6749.
18.Rye, C. S., and Withers, S. G. (2000) Curr.
Opin. Chem. Biol., 4, 573-576.
19.Alcalde, M., Plou, F. J., Perez-Boada,
Garcia-Arellano, M. H., Valdes, I., Mendez, E., and Ballesteros, A.
(2003) J. Mol. Catal. B: Enzym., 26, 57-67.
20.Siddiqui, K. S., Saqib, A. N., Rashid, M. H., and
Rajoka, M. I. (1997) Biotechnol. Lett., 10, 325-329.
21.Levy, H. M., Leber, P. H., and Ryan, E. M. (1963)
J. Biol. Chem., 238, 3654-3659.
22.Dixon, M., and Webb, E. C. (1979) in
Enzymes, Academic Press, New York, pp. 332-467.
23.Macarrón, R., Henrissat, B., and Claeyssens,
M. (1995) Biochim. Biophys. Acta, 1245, 187-190.
24.Koivula, A., Kinnari, T., Harjunpaa, V.,
Ruohonen, L., Teleman, A., Drakenberg, T., Rouvinen, J., Jones, A., and
Teeri, T. T. (1998) FEBS Lett., 429, 341-345.
25.Bray, M. R., Johnson, P. E., Gikes, N. R.,
McIntosh, L. P., Kilburn, D. G., and Warren, A. J. (1996) Protein
Sci., 5, 2311-2318.
26.Deshpande, V., Hinge, J., and Rao, M. (1990)
Biochim. Biophys. Acta, 1041, 172-177.
27.McAllister, K. A., Marrone, L., and Clarke, A. J.
(2000) Biochim. Biophys. Acta, 1480, 342-352.
28.Bakalova, N. G., Petrova, S. D., Atev, A. P.,
Bhat, M. K., and Kolev, D. N. (2002) Biotechnol. Lett.,
24, 1167-1172.