Purification and Characterization of Mycobacterium tuberculosis Indole-3-glycerol Phosphate Synthase
Yanping Yang, Min Zhang, Hongmei Zhang, Jianqiang Lei, Ruiliang Jin, Shengfeng Xu, Jialing Bao, Lu Zhang, and Honghai Wang*
State Key Laboratory of Genetic Engineering, Institute of Genetics, School of Life Science, Fudan University, Shanghai 200433, PR China; fax: 86-21-65648376; E-mail: hhwang@fudan.edu.cn* To whom correspondence should be addressed.
Received November 15, 2004; Revision received January 13, 2005
Indole-3-glycerol phosphate synthase (IGPS) plays an important role in the survival of Mycobacterium tuberculosis. The trpC gene, encoding IGPS, is essential for the growth of M. tuberculosis. It was expressed at the transcriptional level in cultured M. tuberculosis. The recombinant IGPS with an added His-tag was purified. The His-tag was found to have a small effect on the biochemical properties of IGPS. IGPS is a monofunctional enzyme in M. tuberculosis. Recombinant IGPS has considerable beta-pleated sheet and is relatively compact. The enzyme activity is significantly inhibited by denaturants and antibiotics, suggesting that IGPS may be a novel potential drug target of M. tuberculosis.
KEY WORDS: Mycobacterium tuberculosis, indole-3-glycerol phosphate synthase, enzyme activity, monofunctional enzyme, secondary structure, drug targetDOI: 10.1134/S0006297906130062
Abbreviations: IGP) indole-3-glycerol phosphate; IGPS) indole-3-glycerol phosphate synthase; CdRP) 1-(o-carboxyphenylamino)-1-deoxyribulose-5´-phosphate; PRA) N-(5´-phosphoribosyl)anthranilate.
Mycobacterium tuberculosis, the primary etiologic agent
of tuberculosis, is a successful pathogen with the ability to persist
in the immunocompetent host macrophage and is one of the world's
leading causes of death, infecting 9 million and killing 2-3 million
people annually worldwide, as well as an opportunistic pathogen that
infects acquired immunodeficiency syndrome (AIDS) patients [1-3]. Worse still, the appearance
of multidrug resistant (MDR) strains of M. tuberculosis and the
AIDS epidemic worldwide have enormously reinforced the incidence rate
of tuberculosis (TB), and new optimal anti-tuberculosis drugs or drugs
with new acting mechanisms have not been developed in the past 30
years, relighting tuberculosis as a primary public health threat [4, 5]. The second meeting of Stop
TB Partners' Forum (March 24-25, 2004; New Delhi, India) and WHO's 2004
Global Tuberculosis Report both announced that tuberculosis is still
far from under control [3]. Thus, new therapeutic
strategies such as enhancing activity against MDR strains, reducing
toxicity, and shortening duration of therapy are urgently needed to
combat this disease.
The trpC gene, encoding indole-3-glycerol phosphate synthase (IGPS), was demonstrated to be an essential gene for the growth of M. tuberculosis and M. bovis BCG by Himar1-based transposon mutagenesis [6]. We could not find any M. tuberculosis IGPS homogenous protein or amino acid sequence from the protein database of Homo sapiens using the BLASTP program on the NCBI website (http://www.ncbi.nlm.nih.gov). Therefore, IGPS may be a good drug-design target site for more effective agents against tuberculosis.
IGPS is an enzyme catalyzing the fourth step reaction in the tryptophan biosynthesis pathway, in which the substrate 1-(o-carboxyphenylamino)-1-deoxyribulose-5´-phosphate (CdRP) is converted to the product indole-3-glycerol phosphate (IGP), undergoing a ring-closure reaction by releasing CO2 and H2O [7, 8]. IGPS functions as a separate monomeric enzyme in most microorganisms. Sometimes part of a bi- or multienzyme complex is fused to one or more other enzymes in the tryptophan biosynthesis pathway [9]. However, the IGPS gene trpC lies in a single operon in M. tuberculosis [10].
In this work we detected the expression of trpC at a transcriptional level in M. tuberculosis cultured in vitro, purified recombinant IGPS of M. tuberculosis expressed in E. coli, and determined the biochemical properties of the protein.
MATERIALS AND METHODS
Materials. All chemicals were purchased from Sigma-Aldrich (USA) unless otherwise specified. Polymerase, restriction endonucleases, and ligase were supplied by New England Biolabs (China). The pET30a vector and enterokinase cleavage capture kit were bought from Novagen (USA). The pUC18 vector was purchased from TaKaRa (China). Ni-NTA His*Binding resin and columns were supplied by Qiagen (Germany). M. tuberculosis H37Rv (ATCC 93009), streptomycin, geomycine, and kanamycin were provided by the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Custom oligonucleotide primers were commercially synthesized by Shanghai Sangon Biological Engineering and Technology Services Co., Ltd. (China). Gel extract, bacterial genome extract, and plasmid extract kits were bought from Watson Biotechnologies, Inc. (China).
Phylogenetic comparison of indole-3-glycerol phosphate synthase. The protein data banks were searched against M. tuberculosis IGPS using the BLASTP program (http://www.ncbi.nlm.nih.gov). The 12 most similar sequences matched with M. tuberculosis and E. coli IGPS were aligned by ALIGNX (Vector NTI Suite 6), and a tree was constructed by the neighbor-joining method with the best tree mode. The percentage of sequence identity between M. tuberculosis IGPS and the 12 other sequences varied from 100% (Mycobacterium bovis) to 49% (Bifidobacterium longum DJO10A). No amino acid sequence of Homo sapiens matched to M. tuberculosis IGPS.
Bacterial strains and culture conditions. E. coli strains DH5alpha and BL21 (DE3) were used for cloning and expression, respectively. E. coli strains were cultured in Luria-Bertani (LB) medium and on agar supplemented with 25 µg/ml kanamycin. M. tuberculosis was grown in Middlebrook 7H9 broth and on Middlebrook 7H10 agar supplemented with 10% (v/v) oleic acid-albumin-dextrose complex.
RNA and DNA extraction. RNA was isolated from M. tuberculosis cultured in 7H9 broth according to the procedure described previously with some modifications [11]. Genomic DNA was prepared according to the bacterial genome DNA extract kit manufacturer's instruction.
Primer design. The sequence of trpC (GenBank accession number NC_000962) taken from the published genome sequence was used to design primers for polymerase chain reaction (PCR) and reverse transcription-polymerase chain reaction (RT-PCR) amplification. The forward primer was: 5´-ATA GGA TCC ATG AGT CCG GCA ACC GTG CTC-3´, and the reversed primer was: 5´-ATA AAG CTT CTA GCG AGC CGG TTT CGG ACA-3´ containing BamHI and HindI restriction endonuclease sites, respectively.
Duplex RT-PCR. One microgram of RNA was added to 1 µl of random hexamer primer (0.2 µg), and water was added to make the volume of 12 µl. After incubation at 70°C for 5 min, the chilled mixture was added to 4 µl of 5× reaction buffer (Bio Basic Inc., Canada), 1 µl of RNase inhibitor (20 U), and 2 µl of dNTP mixture (10 mM). The samples were incubated at 25°C for 5 min, and 1 µl of AMV reverse transcriptase (20 U) (Bio Basic Inc.) was added. The mixture was incubated at 25°C for 10 min and subsequently at 42°C for 60 min, heated to 70°C for 10 min, and then chilled on ice. A control reaction to monitor DNA contamination was conducted with extracted RNA in a mock RT reaction without the presence of AMV reverse transcriptase.
The 50-µl PCR reaction mixture contained 5 µl of 10× reaction buffer, 5 µl of dNTP (2 mM, respectively), template nucleic acid from the appropriate RT reaction mixture, 1 µl of Taq polymerase, and 250 pmol primers. The reaction proceeded through 20 cycles of the following temperature profile: 94°C, 1 min; 63°C, 1 min; 72°C, 1.5 min.
Construction of trpC gene-containing plasmid. The 50-µl PCR reaction mixture contained 5 µl of 10× reaction buffer, 5 µl of dNTP (2 mM, respectively), 2.5 µl of M. tuberculosis H37Rv genomic DNA (50 ng/µl), 1 µl of Taq polymerase (2 U/µl), and 250 pmol primers. The reaction was performed using the same temperature profile as RT-PCR, but the reaction proceeded through 34 cycles instead of 20.
Both the purified PCR product and pUC18 vector were digested with BamHI and HindI, extracted with gel extract, and then ligated together using T4 DNA ligase overnight at 4°C. The ligation mix was transformed into E. coli strain DH5alpha cells and screened on LB plates containing 100 µg/ml ampicillin. One recombinant clone was selected and sequenced by automated DNA sequencing, which confirmed that trpC was in the proper configuration for expression and no mutations had occurred during the PCR amplification. The recombinant plasmid DNA was extracted and digested with BamHI and HindI to isolate the DNA fragment containing trpC. Then the fragment was purified and cloned into the pET30a vector predigested with the same restriction endonucleases.
Expression and purification of recombinant indole-3-glycerol phosphate synthase in E. coli. Being propagated in E. coli strain DH5alpha, the construct containing trpC coding sequence in pET30a vector was purified and transformed into E. coli strain BL21 (DE3) for expression to produce IGPS protein. Several transformants were cultured in 50 ml LB medium supplemented with kanamycin (25 µg/ml) at 37°C until the absorbance (A600) reached 0.4-0.6. Isopropyl-D-thiogalactopyranoside (IPTG) was added to the final concentration of 1 mM for induction and the bacteria were incubated continually for 10 more hours at 25°C to reach an absorbance value of 1.2. Cells were harvested and washed with cold 5 mM Tris-HCl, pH 7.9. The expression of the recombinant protein was then checked by SDS-PAGE in the whole bacterial extract and pellet, and also in supernatant fraction after sonication. The molecular weight of the recombinant IGPS was estimated by gel electrophoresis calibrated with molecular weight standards.
Recombinant IGPS was purified by Ni-NTA His*Binding resin affinity chromatography according to the manufacturer's protocol. All purification steps were performed with pH 8.0 buffers and at 4°C unless otherwise noted. The bacterial extract from 100 ml culture was suspended at 5 ml binding buffer containing 10 mM imidazole and broken by sonication. The lysate was centrifuged to discard the bacterial debris and insoluble material, and the supernatant was gently removed and used for purification. SDS-PAGE homogeneous purified recombinant IGPS was dialyzed against 5 mM Tris-HCl buffer (pH 7.9) and saved. The recombinant IGPS was cleaved by enterokinase at 4°C for 24 h. The enterokinase was removed according to the manufacturer's protocol, and the IGPS without the His-tag was obtained by binding the His-tag fragment to the Ni-NTA His*Binding resin.
Enzyme activity assay. The concentration of the protein was determined with the Bio-Rad method [12] using bovine serum albumin (BSA) as the standard. CdRP and N-(5´-phosphoribosyl)anthranilate (PRA) were chemically synthesized, with a yield of 30 mM [13]. Ten microliters of 30 mM CdRP and 10 µl of 1.24 µM IGPS were added into 480 µl of 5 mM Tris-HCl (pH 7.0) and incubated at 37°C for 20 min. The enzyme activity was measured with a spectrophotometer by following the absorbance increase of the solution at 280 nm due to the conversion of CdRP to IGP [14, 15]. Complete conversion of 0.1 mM CdRP to IGP led to an increase in the absorbance at 280 nm of 0.448 [16]. The amount of the enzyme catalyzing formation of 1 µmol IGP per min at 37°C was defined as one unit.
Two hundred microliters of freshly prepared stock solution of PRA (approximately 0.75 mM in 0.1 M triethanolamine-HCl buffer, pH 8.6, 0°C) was diluted with 0.79 ml of 0.1 M Tris-HCl buffer, pH 7.6, containing 5 mM EDTA, 1 mM dithiothreitol, and 0.05 mg/ml BSA for stabilizing the enzyme and kept at 25°C. The reaction was initiated by rapidly adding 10 µl of 1.24 µM IGPS, and the absorbance of the solution at 350 nm due to the conversion of PRA to CdRP was recorded [13]. Control reactions were done to eliminate the influence of optical density value increase caused by the substrate and IGPS.
The optimal pH and temperature values for IGPS catalysis were determined in 5 mM Tris-HCl buffers. To determine the Michaelis constant, concentrations of CdRP from 0.1 to 0.7 mM were used.
Circular dichroism (CD) measurements. The CD spectrum was obtained in a 0.1 cm cell of a Jasco-715 spectrometer (Jasco, Japan) equipped with RTE bath/circulator (NESLAB RTE-111; NESLAB, Japan). After a 30 min N2 purge, the spectra were recorded from 190 to 300 nm with a resolution of 0.2 nm and accumulated for five scans at 25-95°C. Recombinant IGPS concentration was 7.47 µM. Secondary structure parameters were estimated by the computer program PROSEC developed by Yang et al. [17].
RESULTS AND DISCUSSION
Phylogenetic comparison of indole-3-glycerol phosphate synthase. Phylogenetic analysis of the IGPS proteins (Fig. 1) revealed that the mycobacterial IGPS proteins composed an outstanding cluster close to the highly GC rich bacterium Thermobifida fusca. The IGPSs of the mycobacteria that cause disease in humans are remarkably similar to the avian strain. The broad evolutionary distance between the IGPS of mycobacteria and E. coli and human shows their inherent dissimilitude and would provide an appropriate target for selective inhibition of the enzyme.
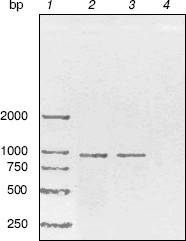
RT-PCR analysis. To monitor trpC expression at a transcriptional level in M. tuberculosis cultured in vitro, mRNA of M. tuberculosis H37Rv in logarithmic stage was detected by RT-PCR (Fig. 2). The size of the PCR products of trpC was 819 bp. No amplicons appeared in the absence of reverse transcriptase, excluding the contamination of genomic DNA. RT-PCR analysis certificated that trpC was transcribed in M. tuberculosis cultured in vitro. However, we only used cultured organisms for experiment, and expression may be different in vivo.Fig. 1. Phylogenetic comparison of indole-3-glycerol phosphate synthases. The numbers shown in the figure represent the evolutionary distances between M. tuberculosis IGPS and other sequences analyzed. The distances are related to the degree of divergence between the sequences. Both sequence alignment and phylogenetic tree construction were made using ALIGNX (Vector NTI Suite 6).

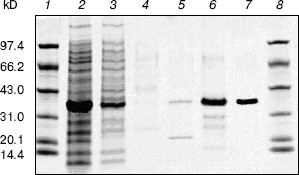
Cloning, expression, purification, and concentration of indole-3-glycerol phosphate synthase. The trpC gene of M. tuberculosis was obtained by PCR using M. tuberculosis H37Rv genomic DNA as template and cloned into pET30a plasmid. IGPS was expressed in E. coli BL21 (DE3) from the pET30a T7 promoter. About 30% of the recombinant enzyme was present in the soluble fraction. The recombinant IGPS expressed in E. coli produced a protein of approximately of 33.5-kD molecular weight. The solubilized fraction was purified to homogeneity by Ni-NTA affinity chromatography (Fig. 3). Being washed with 10 mM (lane 4), 50 mM (lane 5), and 100 mM (lane 6) imidazole successively, the protein was eluted as a single band with 200 mM imidazole (lane 7). The eluted protein band migrated slightly slower than the 31-kD protein standard and had appreciable IGPS activity. The purification yield was about 600 µg per 100 ml culture of transformed E. coli cells.Fig. 2. RT-PCR analysis of TrpC expression in Mycobacterium tuberculosis cultured in vitro. The size of the PCR product is 819 bp. Lanes: 1) DNA marker DL2000 (Takara); 2) positive PCR control: the genomic DNA of M. tuberculosis H37Rv was amplified using primers specific to trpC; 3) RNA samples from M. tuberculosis H37Rv at logarithmic stage was reverse-transcribed and PCR amplified using primers specific to trpC; 4) RNA samples from M. tuberculosis H37Rv (same as lane 3) amplified without reverse transcriptase using primers specific to trpC.
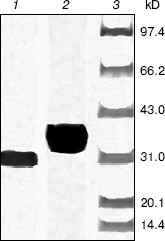
After cleavage of the His-tag using enterokinase, the molecular weight decreased to about 28 kD (Fig. 4).Fig. 3. Purification of recombinant indole-3-glycerol phosphate synthase on a Ni-NTA column. Lanes: 1, 8) protein markers; 2) crude E. coli supernatant fraction before application to column; 3) fraction passing through Ni-NTA column; 4) fraction eluted with 10 mM imidazole; 5) fraction eluted with 50 mM imidazole; 6) fraction eluted with 100 mM imidazole; 7) fraction eluted with 200 mM imidazole. Proteins were visualized with Coomassie blue stain.
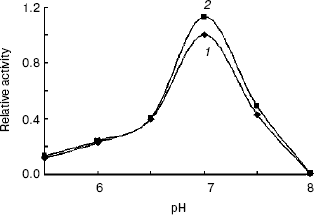
Enzymatic properties of indole-3-glycerol phosphate synthases. IGPS, both with or without the His-tag, was characterized using CdRP as the substrate. The pH optimum of the IGPS was 7.0 in buffers ranging from pH 5.5 to 8.0. The enzyme had almost no activity at pH 8.0 and slight activity at pH 5.0 (Fig. 5). The optimal incubation temperature was 37°C. After a 20 min incubation period, the activity of IGPS leveled off indicating equilibrium was attained.Fig. 4. Removal of His-tag from recombinant indole-3-glycerol phosphate synthase. Lanes: 1) IGPS after enterokinase treatment; 2) IGPS before enterokinase treatment; 3) protein markers.
The IGPS activity was significantly affected by various metal ions. Their optimal concentrations were about 0.4-2.0 mM (Fig. 6). Na+ and Ca2+ seemed to be much more effective than Mg2+ and Mn2+. However, the IGPSs, both with or without His-tag, showed no absolute requirement for the cations.Fig. 5. Effect of pH on indole-3-glycerol phosphate synthase activity (the activity of recombinant IGPS at pH 7.0 was taken as 1.0): 1) recombinant IGPS with His-tag; 2) IGPS without His-tag obtained after enterokinase treatment.
Thus, the enzyme activity assay was performed in pH 7.0 buffers at 37°C, after preincubating the enzyme for 20 min. The recombinant IGPS exhibited a specific activity of about 4 U/µg under these conditions. The Km of the recombinant enzyme for CdRP was determined to be about 0.5 mM.Fig. 6. Effect of cation concentration on indole-3-glycerol phosphate synthase activity. Assay mixtures were incubated in pH 7.0 buffers at 37°C for 20 min with various amounts of metal chlorides. The average of enzyme activity measured in the absence of metal salts was taken as 1.0. Dashed lines refer to recombinant IGPS with His-tag; solid lines refer to IGPS without His-tag. Cations used were as following: Na+ (squares); Mg2+ (circles); Ca2+ (triangles); Mn2+ (diamonds).
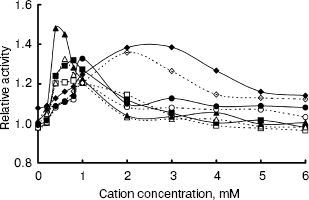
Since IGPS was found to be a bifunctional enzyme in E. coli, we estimated IGPS activity against PRA. The results showed no PRA conversion to CdRP, suggesting that IGPS is a monofunctional enzyme in M. tuberculosis.
Stability of the recombinant indole-3-glycerol phosphate synthase. The purified recombinant IGPS was stable if frozen and retained its activity for at least two months at -70°C. It was also quite stable to heating, and there was little loss of activity after a 10 min heating in a water bath at temperatures from 40 to 50°C. Actually, even at 70°C the enzyme maintained notable activity for several minutes and was gradually inactivated afterwards. This behavior indicates that the protein is tightly folded and quite resistant to denaturation.
The enzyme activity was rapidly lost in the presence of H2O2. Even 0.001% H2O2 inactivated the enzyme almost completely. Methanol was less toxic than H2O2. Enzyme activity was gradually lost in the presence of increasing concentrations of methanol and completely lost at 65%.
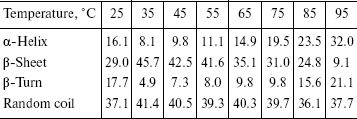
The recombinant IGPS was subjected to circular dichroism measurements. The data presented in the table indicated about 45% beta-pleated sheet at 35°C. The beta-sheet content decreased and alpha-helix and beta-turn content increased gradually as the temperature was raised from 35 to 95°C. The beta-sheet content was low at 25°C.
Percentage of secondary structure elements of the recombinant IGPS
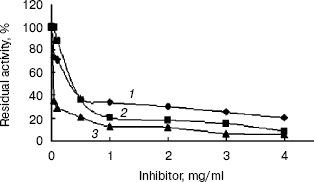
Inhibition of the activity of the recombinant indole-3-glycerol phosphate synthase with His-tag. The activity of recombinant enzyme with His-tag was significantly inhibited by streptomycin and kanamycin or geomycine at a concentration of 0.5 mg/ml (Fig. 7). It is interesting that these antibiotics are aminoglycosides, which are mainly used to treat infectious diseases caused by gram-negative aerobes or facultative aerobes in clinical management [18]. Streptomycin and kanamycin are also currently used in treatment of tuberculosis. Therefore, IGPS may serve as a novel potential target for the antibiotics to exterminate the M. tuberculosis pathogen. Designing of experiments to screen for highly potent and effective anti-tuberculosis drugs by a high-throughput method based on the results described in this paper is underway.
This work was supported by grants from both the National Basic Research Program of China (973 Program) (No. 2002CB512804) and the Ph. D. Programs Foundation of Ministry of Education of China (No. 20020246019).Fig. 7. Inhibition of the recombinant indole-3-glycerol phosphate synthase with His-tag by antibiotics: 1) streptomycin; 2) kanamycin; 3) geomycine. Assay mixtures were incubated in pH 7.0 buffers at 37°C for 20 min containing various concentrations of the antibiotics.
REFERENCES
1.Dye, C., Scheele, S., Dolin, P., and Pathania, V.
(1999) JAMA, 282, 677-686.
2.Dolin, P. J., Raviglione, M. C., and Kochi, A.
(1994) Bull. World Health Organ., 72, 213-220.
3.Sharma and Dinesh, C. (2004) The Lancet,
363, 1122.
4.Harries, A. D., and Mahler, D. (1996) TB/HIV, a
Clinical Manual Published by the World Health Organization,
Stabilimento Tipografico Ferrero s. r. l.-Romano Canavese (TO),
Italy.
5.Floyd, K., Blanc, L., Raviglione, M., and Lee,
J.-W. (2002) Science, 295, 2040.
6.Sassetti, C. M., Boyd, D. H., and Rubin, E. J.
(2003) Mol. Microbiol., 48, 77-84.
7.Parry, R. J. (1972) in The Chemistry of
Heterocyclic Compounds in Indoles (Houlihan, W. J., ed.) Pt. II,
Wiley-Interscience, New York, pp. 1-64.
8.Creighton, T. E., and Yanofsky, C. (1966) J.
Biol. Chem., 241, 4616-4624.
9.Nichols, B. P. (1996) in Escherichia coli
and Salmonella (Neidhardt, F. C., ed.) ASM Press, Washington,
DC, pp. 2638-2648.
10.Yu Zheng and Szustakowski, J. D. (2002) Genome
Res., 12, 1221-1230.
11.Mangan, J. A., Sole, K. M., Mitchison, D. A., et
al. (1997) Nucleic Acids Res., 25, 675-676.
12.Bradford, M. M., McRorie, R. A., and Williams, W.
L. (1976) Analyt. Biochem., 72, 248-254.
13.Kirschner, K., Szadkowski, H., and Jardetzky, T.
S. (1987) Meth. Enzymol., 142, 386-397.
14.Giuseppina, A. M., Vittoria, C., and Michela, D.
P. (1997) Biochem. J., 323, 259-264.
15.Manuel, M. S., and Alan, R. F. (1997)
Biochem., 36, 5560-5565.
16.Creigthon, T. E., and Yanofsky, C. (1970)
Meth. Enzymol., 17A, 365-380.
17.Yang, J. T., Wu, C. H. C., and Martinez, H. M.
(1986) Meth. Enzymol., 130, 208-269.