An Antiviral Protein Having Deoxyribonuclease and Ribonuclease Activity from Leaves of the Post-flowering Stage of Celosia cristata
M. Begam*, S. Narwal, S. Roy, S. Kumar, M. L. Lodha, and H. C. Kapoor
Division of Biochemistry, Indian Agricultural Research Institute, New Delhi-110012, India; E-mail: mb_bioc@yahoo.com* To whom correspondence should be addressed.
Received November 18, 2004; Revision received April 6, 2005
An antiviral protein named CCP-27 was purified from the leaves of Celosia cristata at the post-flowering stage by anion-exchange, cation-exchange, and gel-filtration chromatography. It exhibited resistance against sunnhemp rosette virus in its test host Cyamopsis tetragonoloba. It also exhibited deoxyribonuclease activity against supercoiled pBlueScript SK+ plasmid DNA. It was found to nick supercoiled DNA into nicked circular form at lower protein concentration followed by nicked to linear form conversion at higher protein concentration. CCP-27 also possesses strong ribonuclease activity against Torula yeast rRNA.
KEY WORDS: Celosia cristata, antiviral proteins, ribosome inactivating protein, DNase activity, RNase activityDOI: 10.1134/S0006297906130074
Abbreviations: AVPs) antiviral proteins; RIPs) ribose inactivating proteins; SRV) sunnhemp rosette virus.
The use of antiviral proteins (AVPs) from plants is one of the promising
approaches to control viral diseases. Some well-characterized AVPs are
from Phytolacca americana, Mirabilis jalapa, Dianthus
caryophyllus, Clerodendrum aculeatum, Amaranthus
viridis, and Trichosanthes kirilowii [1-6]. A definite mechanism whereby
these AVPs operate to inhibit viral infection remains unresolved. At
present, the most widely accepted mechanism of action is a direct
result of their ribosome specific N-glycosidase activity in
vitro [7]; such proteins are therefore known
as ribosome inactivating proteins (RIPs).
In addition to deadenylation activity, RIPs/AVPs are also known to possess DNA lyase activity [8-13]. The ribonucleolytic cleavage of naked rRNA by some RIPs has also been reported [14]. Celosia cristata, an ornamental plant of Amaranthaceae family is a potential candidate for the presence of strong antiviral proteins. Two potent growth stage dependent glycoproteins, CCP-25 and CCP-27 have been reported to be present in the leaves of this plant at flowering stage [15]. Here, we report the purification of CCP-27 from leaves of Celosia cristata at the post-flowering stage and its strong DNase and RNase activity against supercoiled plasmid DNA and Torula yeast rRNA, respectively.
MATERIALS AND METHODS
Virus inoculum. Sunnhemp rosette virus (SRV) was used for testing the virus inhibitory activities of leaf extract. The virus was maintained on its systemic host, Crotolaria juncea. The plants were grown in small plastic pots containing soil, FYM, sand, and vermiculite mixture in 2 : 1 : 1 : 0.5 ratios. The pots were kept in an insect free glass house. Virus inoculum was prepared by homogenizing the infected leaves with 20 mM sodium-potassium phosphate buffer, pH 7.0, with a sterilized pestle and mortar. The contents were squeezed through two layers of muslin cloth and the filtrate was centrifuged at 12,000g for 10 min. The clear supernatant was used as virus inoculum after suitable dilution with distilled water, so as to produce countable number of lesions on the test plant leaves.
Bioassay. Cyamopsis tetragonoloba was used as local lesion host for bioassay of the purified CCP-27. Test plants with same height, age, and vigor were selected for experiments. For each treatment, three plants with two-three leaves of equal size were used. The purified 27 kD protein from Celosia cristata leaves was applied on the test plant leaves. For controls, test plant leaves were treated with only buffer and virus inoculum. After 1 h, the protein treated leaves were washed with distilled water and gently blotted dry. The leaves were then sprinkled very lightly with 600 mesh carborundum powder and inoculated gently and uniformly with virus inoculum. Just after inoculation, leaves were washed with distilled water. Plants were observed for the development of lesions for 3-4 days. The inhibitory activity of the proteins was calculated in terms of percentage inhibition using the following formula: % inhibition = (C - T)/C × 100%, where, C is average number of lesions in control plants, and T is average number of lesions in treated plants.
Purification of antiviral protein CCP-27. Antiviral protein (CCP-27) was isolated and purified from the leaves of Celosia cristata at the post-flowering stage. For extraction, the dried leaves were homogenized with 4-5 volumes of 0.1 M sodium acetate buffer (pH 5.2) containing 12 mM beta-mercaptoethanol and 20 mg polyvinylpolypyrrolidone. The pulp was squeezed through two layers of muslin cloth, and filtrate was centrifuged at 12,000g for 10 min. The clear supernatant was used for purification of CCP-27.
Ammonium sulfate fractionation. The supernatant obtained from leaf extract was subjected to ammonium sulfate fractionation at 0-25 and 25-80% saturation, by slow addition of calculated quantities of solid ammonium sulfate at each step to the supernatant with constant stirring using a magnetic stirrer at 4°C. After obtaining the desired saturation of ammonium sulfate (0-25%), the suspension was kept at 4°C for 30 min to enable protein precipitation. The precipitated protein was pelleted by centrifugation at 12,000g for 10 min. The pellet was suspended in minimal amount of buffer A (20 mM sodium phosphate buffer, pH 6.2, containing 10 mM NaCl). The supernatant was used to precipitate the protein at subsequent higher saturation of ammonium sulfate, i.e. 25-80%. The pellet obtained at each step was suspended in buffer A and dialyzed against the same buffer at 4°C for 24 h. The proteins precipitated at 0-25% were not used for further characterization as they showed very much less antiviral activity. Therefore, the protein precipitated at 25-80% saturation showing high antiviral activity was used for further purification.
Ion-exchange chromatography. Both anion- and cation-exchange chromatography were performed for further purification of the antiviral protein obtained in ammonium sulfate fraction 25-80%.
DEAE-cellulose chromatography. After equilibrating the column by passing several volumes of buffer A, the active 25-80% ammonium sulfate dialyzed protein fraction was loaded on top of the column. The unbound fraction (having antiviral activity) was collected and subjected to cation-exchange chromatography.
CM-Sepharose chromatography. The DEAE-cellulose unadsorbed fraction was loaded onto the CM-Sepharose column (35 ×_1.0 cm) equilibrated with buffer B (20 mM sodium acetate buffer, pH 5.2, containing 10 mM NaCl and 0.01% sodium azide) and washed with three-four bed volumes of equilibration buffer. The adsorbed protein was then eluted with a discontinuous gradient of NaCl (50, 100, 150, and 200 mM, three bed volumes each), which was prepared in equilibration buffer. The eluate was collected in fractions using a Pharmacia-LKB fraction collector system (Sweden), at a flow rate of 30 ml/h. The eluate was simultaneously monitored for absorbance at 280 nm. The protein fractions eluting at each NaCl concentration were separately pooled and tested for the antiviral activity.
Gel filtration chromatography. Active protein fractions were concentrated in a lyophilizer and loaded onto a Superose-12 column (45 ×_1.0 cm) equilibrated with buffer C (20 mM sodium acetate, pH 5.2, containing 0.01% sodium azide). Elution was done with the same buffer at a flow rate of 15 ml/h and 30 ml fractions of 1.6 ml each were collected. The eluate was simultaneously monitored for absorbance at 280 nm. The protein fractions eluting at each NaCl concentration were separately pooled and tested for the antiviral activity.
Protein estimation. Protein concentration at each step of purification was determined by the method of Lowry et al. [16] with bovine serum albumin as standard.
SDS-polyacrylamide gel electrophoresis. Discontinuous SDS-PAGE was carried out in 12% separating gel with a 5% stacking gel according to Laemmli [17]. The proteins were visualized by staining with 0.1% Coomassie brilliant blue R-250.
DNase activity. pBlueScript SK+ plasmid DNA was isolated by the alkaline hydrolysis mini-preparation method of Ahn et al. [18]. An agarose (1%) gel electrophoresis was done to determine the quality, quantity, and size of the plasmid DNA. For this, 1 µg of plasmid DNA was incubated with serially increasing amount of purified CCP-27 in a final volume of 25 µl. The reaction mixture was incubated at 37°C for 4 h. At the end of the incubation period, 5 µl loading solution (30% Ficoll, 200 mM EDTA, 0.25% bromophenol blue, and 0.25% xylene cyanole FF) was added. Electrophoresis was carried out under non-denaturing condition in a 1% agarose gel. DNA bands were visualized by staining with ethidium bromide.
RNase activity. To determine if CCP-27 could degrade RNA, a method for substrate-based RNase activity assay was adapted [19] using Torula yeast RNA. CCP-27 samples were prepared in 50 mM Tris-HCl, pH 6.8, containing 0.1% SDS and 0.01% bromophenol blue without the addition of reducing agents. As a positive control for RNase activity, 2 µg RNase A was prepared in the same buffer. The sample and control were separated using 12% SDS-PAGE with 2 mg/ml Torula yeast RNA (Sigma, USA) in the resolving gel. Following electrophoresis, the gel was washed with 25% isopropanol in 10 mM Tris-HCl, pH 7.0, to remove SDS. The isopropanol was removed by further washing in 10 mM Tris-HCl, pH 7.0, 2 µM ZnCl2. Protein in the gel was renatured by incubation in 100 mM Tris-HCl, pH 7.0, at 50°C for 1 h. The gel was stained with 0.2% Toluidine blue O in 10 mM Tris-HCl, pH 7.0, for 10 min and destained in distilled water until transparent bands appeared. Lanes containing protein markers and CCP-27 were cut from the gel and stained with Coomassie blue to indicate the location of CCP-27.
RESULTS AND DISCUSSION
Purification of antiviral protein. CCP-27 was purified to homogeneity using conventional biochemical techniques. The leaf material was extracted with sodium acetate buffer, pH 5.2, and subjected to ammonium sulfate fractionation at 0-25 and 25-80% saturation, respectively. The majority of antiviral activity was recovered from 25-80% (NH4)2SO4 saturated fraction. This fraction was subjected to anion-exchange chromatography on DEAE-cellulose column for further purification. The column was eluted with 20 mM sodium phosphate buffer (pH 6.2) containing 10 mM NaCl. Most of the antiviral activity was extracted in unadsorbed fraction indicating the basic nature of the protein. The active unadsorbed fractions were pooled and subjected to cation-exchange chromatography on CM-Sepharose column and adsorbed proteins were eluted with a discontinuous NaCl gradient (50, 100, 150, and 200 mM, three bed volumes each) in sodium acetate buffer, pH 5.2. At 50 mM NaCl concentration, a single peak was obtained which was designated as peak I whereas 100 mM NaCl gave two peaks designated as peak II and peak III. Elution with still higher salt concentrations resulted in separation of a very negligible amount of protein and therefore not used for bioassay and protein quantification. Of these, peak I fraction exhibited maximum antiviral activity, as confirmed through bioassays (table). The CM-Sepharose unadsorbed fraction did not show any antiviral activity. These results are in line with those of antiviral proteins from Phytolacca americana, Mirabilis jalapa, and Bougainvillea xbuttiana [20-22]. On the contrary, a report on Chenopodium amarantricolor has shown that the inhibitor was adsorbed on the DEAE-Sephadex matrix but not on CM-Sephadex [23]. This might be due to non-basic nature of the particular protein. The fractions corresponding to peak I were concentrated in a lyophilizer and subjected to gel-filtration chromatography using Superose-12 resin. No further fractionation or purification was achieved as the fraction again separated into a single peak designated as peak IV.
Purification of antiviral protein from leaf extract of Celosia
cristata
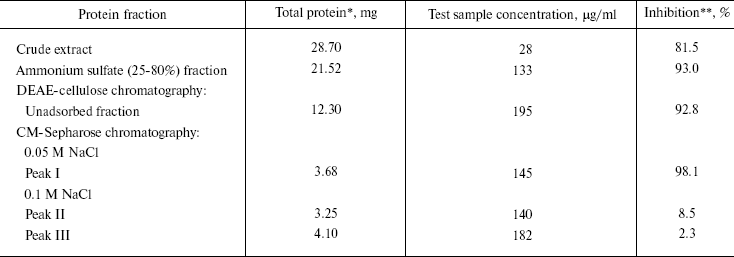
*From 40 g of C. cristata dried leaves.
**Three to four well-developed leaves of four test plants of
Cyamopsis tetragonoloba, each for all sets, were tested with 1
ml of test sample. After 1 h, leaves were washed with distilled
water and blotted dry. The leaves were dusted with carborundum and
inoculated with SRV.
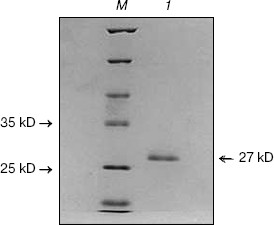
Analysis of this peak on 12% SDS-PAGE showed its correspondence to 27 kD. This Celosia antiviral protein has been designated as CCP-27 (Fig. 1). CCP-27 exhibited ~98% inhibition against SRV in its test host Cyamopsis tetragonoloba at a concentration as low as 145 µg/ml (table and Fig. 2, see color insert, p. 3).
Fig. 1. SDS-PAGE of purified CCP-27. CCP-27 was purified from the dry leaves of the post-flowering stage of Celosia cristata by ion-exchange and gel-filtration chromatography; 10 µg of purified sample was run on 12% SDS-polyacrylamide gel. Lanes: 1) purified CCP-27; M) molecular weight marker.
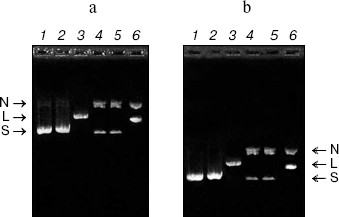
DNase activity. CCP-27 exhibited strong DNase activity. When pBlueScript SK+ DNA having all the three forms, i.e. supercoiled, nicked, and linear (Fig. 3a), was incubated with various amounts of CCP-27, it was found that the extent of supercoiling was obviously altered. The supercoiled form of the pBlueScript SK+ DNA when incubated with 2 and 4 µg of CCP-27, first nicked to give a nicked circular form, which moved significantly slower than the supercoiled DNA through the agarose gel. When the CCP-27 concentration was increased to 6 µg, the linear form of DNA emerged which migrated faster than the nicked circular form but slower than the supercoiled DNA. From this finding, it can be suggested that AVP/RIP-associated DNase activity was conformation specific, i.e. RIPs only cleave supercoiled DNA in a two-step process, first nicking a single strand before linearizing the DNA by second strand cleavage. The results are in line with the earlier findings [8-13]. In order to make sure that band shift was not due to the binding of CCP-27 onto pBlueScript SK+ DNA, attempts were made to separate the two species before electrophoresis. This was achieved by digestion with proteinase K, after incubation of DNA with CCP-27. The DNA was then extracted with phenol-chloroform (1 : 1) and precipitated with ethanol. Subsequent electrophoretic analysis confirmed the same changes in banding patterns (Fig. 3b).Fig. 2. Antiviral bioassay of CCP-27. Purified CCP-27 was subjected to antiviral bioassay by a local lesion test using Cyamopsis tetragonoloba as test host plant and sunnhemp rosette virus (SRV) for infection. Treatments were done as described in “Materials and Methods” and percentage of lesion inhibition was calculated by the formula: % Inhibition = (C - T)/C × 100%. a) Control (buffer and SRV treated); b) CCP-27 (160 µg/ml) and SRV treated.

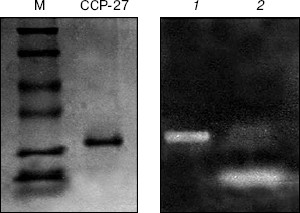
RNase activity. For RNase activity test, as the gel was stained with Toluidine blue, which interacts with RNA to give blue color, the unstained portion indicated the position of RNase activity. Figure 4 (lane 1) shows a single unstained band that was found to co-migrate with purified CCP-27 (2 µg), indicating that CCP-27 does possess a strong RNase activity. The presence of only one band with RNase activity corresponding to the position of CCP-27 suggests that there was no other contaminating nuclease with the purified CCP-27 preparation. Lane 2 (Fig. 4) shows the position of positive control (2 µg RNase A). The ribonucleolytic activity on naked rRNA was also reported in case of alpha- and beta-momorcharin [14]. Masayuki et al. [24] observed 50% inhibition of local lesion formation in cowpea leaves when treated with a 23 kD RNase like glycoprotein (figaren) from Cucumis figarei, 24 h before to 1 h after incubation with cucumber mosaic virus. Figaren also digested double stranded RNase extracted from cucumber mosaic virus infected tobacco tissues. Thus, there may be a positive correlation between antiviral activity and RNase activity.Fig. 3. a) DNase activity test. When incubated with 2 or 4 µg of CCP-27, pBlueScript SK+ DNA supercoiled form (S) first gives rise to nicked form (N) (lanes 4 and 5) and with 6 µg of CCP-27 gives rise to linear form (L) (lane 6). Lanes: 1) plasmid; 2) control (pBlueScript SK+ DNA was incubated without CCP-27); 3) plasmid restricted with EcoRI. b) DNase activity test after proteinase K treatment. Same changes in banding patterns indicate that band shifting was not due to binding of the protein with plasmid DNA.
In conclusion, the present study suggests that CCP-27, the antiviral protein purified from leaves of the post-flowering stage of Celosia cristata, is multifunctional as it possesses both DNase and RNase activity and these activities may have a positive correlation with the inhibition of viral infection.Fig. 4. RNase activity test. CCP-27 was separated using 12% SDS-PAGE with Torula yeast RNA. Following electrophoresis, the gel was stained with Toluidine blue O to show the bands with RNase activity. The unstained portions indicate the presence of RNase activity. Lanes containing the protein marker M and CCP-27 were cut from the gel and stained with Coomassie blue to indicate location on the protein. Lanes: 1) 4 µg CCP-27; 2) 2 µg RNase A (positive control).
The senior author is thankful to the Council of Scientific and Industrial Research, New Delhi for awarding Junior and Senior Research Fellowship during the study.
REFERENCES
1.Lin, Q., Chen, Z. C., Antoniw, J. F., and White, R.
F. (1991) Plant Mol. Biol., 1, 7609-7614.
2.Kataoka, J., Noriyuki, H., Masahiri, F., and
Masashi, M. (1991) J. Biol. Chem., 266, 8426-8430.
3.Legname, G., Bellosta, P., Gromo, G., Modena, D.,
Keen, J. N., Roberts, L. M., and Lord, J. M. (1991) Biochim.
Biophys. Acta, 119, 1090.
4.Kumar, D., Hridya, N., Verma, N. T., and Krishna,
K. T. (1997) Plant Mol. Biol., 33, 745-751.
5.Kwon, Y. S., An, C. S., Liu, J. R., and Peak, K.
(1997) Biosci. Biotech. Biochem., 61, 1613-1614.
6.Guo, Y. J., Demin, J., Man Li, W., Guo, B., Bin,
W., Jin, D. M., Weng, M. L., Guo, B. T., and Wang, B. (1999) Acta
Botan. Sin., 41, 334-336.
7.Van Damme, E. J. M., Hao, Q., Chen, Y., Barre, A.,
Vandenbussche, F., Desmyter, S., Roughe, P., and Peumans, W. J. (2001)
Crit. Rev. Plant Sci., 20, 395-465.
8.Tomas, T. M., Go, H. W., Yeung, and Fong, W. P.
(1992) Life Sci., 51, 1347-1353.
9.Ling, J., Liu, W. Y., and Wang, T. P. (1994)
FEBS Lett., 345, 143-146.
10.Roncuzzi, L., and Gasperi-Campani, A. (1996)
FEBS Lett., 392, 16-20.
11.Nicolas, E., Beggs, J. M., Haltiwanger, B. M.,
and Taraschi, T. F. (1997) FEBS Lett., 406, 162-164.
12.Nicolas, E., Goodyer, I. D., and Taraschi, T. F.
(1997) Biochem. J., 327, 413-417.
13.Wang, P., and Tumer, N. E. (1999) Nucleic
Acids Res., 27, 1900-1905.
14.Mock, J. W., Ng, T. B., Wong, R. N., Yao, Q. Z.,
Yeung, H. W., and Fong, W. P. (1996) Life Sci., 59,
1853-1859.
15.Balasubrahmanyam, A., Baranwal, V. K., Lodha, M.
L., Varma, A., and Kapoor, H. C. (2000) Plant Sci., 154,
13-21.
16.Lowry, O. H., Rosenbrough, N. J., Farr, A. L.,
and Randall, R. J. (1951) J. Biol. Chem., 193,
265-275.
17.Laemmli, U. K. (1970) Nature, 227,
680-685.
18.Ahn, S. C., Back, B. S., Oh, T., Song, C. S., and
Chatterjee, B. (2000) Biotechniques, 29, 466-468.
19.Yen, X., and Green, J. (1991) Plant Phy.,
97, 1487-1493.
20.Irvin, J. D. (1975) Arch. Biochem.
Biophys., 169, 522-528.
21.Takanami, Kuwata, S., Ikeda, T., and Kubo, S.
(1990) Ann. Phytopathol. Soc. Jap., 5, 488-494.
22.Narwal, S., Balasubrahmanyam, A., Lodha, M. L.,
and Kapoor, H. C. (2001) Indian J. Biochem. Biophys., 38,
342-347.
23.Taniguchi, T., and Goto, T. (1979) Ann.
Phytopathol. Soc. Jap., 45, 135-141.
24.Masayuki, F., Takeshi, K., Satoshi-T, O., and
Takeshi, O. (2001) J. Gen. Plant Pathol., 67,
152-158.