One-Step Purification of Mammalian Deoxyribonucleases I and Differences among Pancreas, Parotid, and Pancreas-Parotid (Mixed) Types Based on Species- and Organ-Specific N-Linked Glycosylation
J. Fujihara, Y. Hieda, Y. Xue, N. Nakagami, K. Takayama, K. Kataoka, and H. Takeshita*
Department of Legal Medicine, Shimane University School of Medicine, 89-1 Enya, Izumo, Shimane 693-8501, Japan; fax: +81-853-20-2155; E-mail: htakeshi@med.shimane-u.ac.jp* To whom correspondence should be addressed.
Received June 16, 2005; Revision received July 14, 2005
Mammalian deoxyribonucleases I (DNase I) are classified into three types, namely, pancreas, parotid, and pancreas-parotid (mixed), based on differences in their tissue concentrations. In this study, DNase I purification by concanavalin A-wheat germ agglutinin mixture-agarose column from rat (parotid type), rabbit (mixed type), and pig (pancreas type) is described. This method permits a relatively easy one-step purification of DNase I from rat and rabbit parotid glands, the rat submaxillary gland, and porcine pancreas. To elucidate differences among the three types, these DNases I were subjected to enzymatic deglycosylation either by peptide N-glycosidase F (PNGase F) or endoglycosidase H (Endo H). Following deglycosylation, digests were separated on DNA-casting polyacrylamide gel electrophoresis. PNGase F produced a single lower mobility product in all samples. Endo H produced a double band in rat and rabbit parotid glands and porcine pancreas, and a single band in the rabbit pancreas corresponding with the PNGase F product. DNase I activity of the porcine pancreas was completely extinguished by deglycosylation, while that of the parotid glands and rabbit pancreas was unaffected. Our results suggest that the distinct properties of DNase I exhibited by the three types may be attributed to differences in the extent of post-translational N-linked glycosylation of the enzyme.
KEY WORDS: Con A, DNase I, glycosylation, pancreas, parotid gland, submaxillary gland, WGADOI: 10.1134/S0006297906130116
Abbreviations: DNase) deoxyribonuclease; Con A) concanavalin A; WGA) wheat germ agglutinin; LCA) Lens culinaris agglutinin; RCA120) Ricinus communis (castor bean) agglutinin; PNGase F) peptide N-glycosidase F; Endo H) endoglycosidase H; PAGE) polyacrylamide gel electrophoresis; SRED) single radial enzyme diffusion; RNase) ribonuclease.
Deoxyribonuclease I (DNase I, EC 3.1.21.1) plays a major role in
alimentary canal digestion, and it is secreted by exocrine glands such
as the pancreas and parotid gland in mammals [1-3]. However, the presence of the enzyme in mammalian
tissues other than digestive organs suggests other function(s) in
vivo [4, 5]. Recent studies
suggest that DNase I may be an essential factor for the prevention of
systemic lupus erythematosus [6-8].
A quick, effective, and scalable procedure for the purification of mammalian DNase I would be of use in studying the biological properties of this protein. However, hitherto, purification of DNase I has entailed numerous time-consuming steps. A prior study reported on the purification of rat parotid DNase I using four-step chromatography, and the product contained contaminations that might have been eliminated with the use of affinity chromatography [9]. Another study on purification of rat DNase I from urine demonstrated an effective but lengthy six-step procedure with two affinity chromatography steps [10]. As mammalian DNase I is a glycoprotein [11-13], examples of the use of lectin-agarose columns to purify this protein have been well documented [14, 15]. Types of carbohydrate moieties of DNase I have been studied only in bovine tissue [16], with no information on carbohydrate moieties for other mammalian DNases I. Our prior study demonstrated that mammalian DNases I could be classified into three types, i.e. pancreas, parotid, and pancreas-parotid (mixed), based on the tissue with highest activity, and that pancreatic-type DNase I is more sensitive to low pH than are the other types [3]. However, to date, differences in the chemical structure of DNases I among these three groups remain unclear.
Our objectives in the present study were (i) to develop an expeditious purification procedure for DNase I from mammalian high-activity tissue using lectin affinity chromatography, and (ii) to examine the differences in the N-linked glycosylation pattern among the three DNase I types and the significance of carbohydrate moieties in enzymatic activity.
MATERIALS AND METHODS
Materials. The parotid and submaxillary glands from a Wister rat, parotid gland and pancreas from a Japanese white rabbit, and a porcine pancreas were harvested. The animals were acquired, maintained, and used in accordance with Guidelines for the Care and Use of Laboratory Animals (NIH, USA; revised 1985). Concanavalin A (Con A)-, wheat germ agglutinin (WGA)-, Lens culinaris agglutinin (LCA)-, and castor bean agglutinin (RCA120)-agaroses were obtained from Seikagaku Kogyo (Tokyo, Japan). Recombinant peptide N-glycosidase F (PNGase F) was purchased from Prozyme, Inc. (USA) and endoglycosidase H (Endo H) was obtained from New England Biolabs (USA). All other chemicals were of reagent grade and commercially available.
Affinity of mammalian DNases I for lectin columns. All procedures were carried out at 4?°C. Each enzyme (about 2.5*10-3 units) was dialyzed against 25 mM Tris-HCl buffer (pH 7.5) containing 0.15 M NaCl. The dialyzed materials were applied to separate lectin-agarose columns, each containing 1 ml of Con A, LCA, RCA120, or WGA. All columns were equilibrated with the above-mentioned buffer before use. Unbound fractions were collected after washing with 8 ml of buffer, and the adsorbed enzymes were eluted with 5 ml 0.2 M methyl-alpha-D-mannopyranoside (Con A column), 0.1 M D-lactose (LCA column), 0.2 M methyl-alpha-D-glucopyranoside (RCA120 column), and 0.2 M N-acetyl-D-glucosamine (WGA column).
One-step purification of mammalian DNases I. All procedures were carried out at 4?°C. The samples (ca. 0.1 g) were minced and homogenized in 1-2 ml 25 mM Tris-HCl buffer (pH 7.5) containing 1 M NaCl and 1 mM phenylmethylsulfonyl fluoride (buffer I). After centrifugation (10,000g, 20 min), the supernatants (crude extracts) were directly applied to a Con A- and WGA-agarose combined column (1 × 1.5 cm) pre-equilibrated with buffer I. The column was then washed with 7 ml of buffer I to collect the unbound fractions. Bound fractions were eluted with 4 ml of buffer I containing 0.2 M N-acetyl-D-glucosamine after first washing with 4 ml buffer I containing 0.2 M methyl-alpha-D-mannopyranoside. Aliquots of the eluted fractions were kept for further analysis.
Enzymatic deglycosylation analysis and DNA-casting PAGE followed by activity staining. DNase I from the salivary glands and pancreas were subjected to deglycosylation either by PNGase F or Endo H without denaturation. Each sample was incubated at 37°C for 10 h in a reaction buffer containing either PNGase F or Endo H. To examine the difference in electrophoretic mobility before and after deglycosylation, digests were separated on DNA-casting polyacrylamide gel electrophoresis (PAGE) (12.5%) followed by activity staining as previously described [17]. Briefly, 1.0% w/v salmon testicular DNA was added to the separating gel and poured into a mold (10 × 10 × 0.1 cm) to 3.0 cm from the top. A stacking gel was prepared in the same manner as for normal PAGE. The samples were prepared by mixing each enzyme with half its volume of sample buffer (0.2 M Tris-HCl, pH 6.8, 40% w/v sucrose and 0.01% w/v bromophenol blue), and then 10 µl of each sample was loaded onto the gel and electrophoresed at 150 V. After electrophoresis, the gel was removed, immersed in 100 mM Tris-HCl, pH 7.5, containing 20 mM MgCl2 and 2 mM CaCl2, incubated at 37°C for 1 h, transferred to fresh sample buffer containing 0.001% w/v ethidium bromide, incubated at 37°C until the dark bands resulting from degradation of DNA by DNase I were observed under ultraviolet light (312 nm).
To examine the deglycosylation effect on enzymatic activity, aliquots were removed at several time-points (1.5, 3, 6, 12, and 24 h) following PNGase F or Endo H treatments. Control reactions were performed by incubating samples in reaction buffer without the endoglycosidases. Remaining DNase I activity was determined by comparisons with the controls.
Analytical methods. Enzymatic activity of DNase I was determined using the test tube method [18] and single radial enzyme diffusion (SRED) method as previously described [2]. Activities of DNase II and RNases were also assayed by the SRED method [19, 20]. Protein concentrations were determined with a protein assay kit (Bio-Rad, USA) using bovine serum albumin as a standard. The enzymological properties of each rat, rabbit, and porcine enzyme and the inhibitory effects of specific antibodies on their activities were examined as described previously [10, 21, 22]. SDS-PAGE was performed on a 12.5% polyacrylamide gel [23]; separated proteins were detected with Coomassie brilliant blue R250 (Bio-Rad) or silver staining, or by western blotting with specific antisera.
RESULTS AND DISCUSSION
Lectin column affinity. As mentioned, mammalian DNases I can be classified into pancreas, parotid, and pancreas-parotid (mixed) types based on tissue activity [3]. It can be presumed that chemical structures such as carbohydrate moiety of the DNases I may differ among these three types. We therefore evaluated the affinities of enzymes of parotid glands of the rat (parotid type) and rabbit (mixed type) and of the rabbit and pig pancreas (pancreas type) to four different lectins. The rat submaxillary gland was also analyzed because high DNases I activity (comparable to the parotid gland) was observed in preliminary experiments (Table 1). The rat and rabbit parotid glands and rat submaxillary gland exhibited strong affinities for both Con A and WGA. The porcine pancreas showed a strong affinity for Con A, with a lower affinity for WGA than that of the salivary glands. In the rabbit pancreas, Con A affinity was relatively high, with WGA affinity two orders of magnitude lower than that of the salivary glands. No or very low affinities of the enzymes were observed with all LCA and RCA samples.
Table 1. Binding of mammalian DNase I
derived from salivary gland and pancreas to lectin-affinity columns

Note: Values are mean ± S.D. of triplicate determinations.
These results indicate that salivary enzymes from parotid gland and mixed types possess more complex carbohydrate moieties than do those from the pancreas. The differences between the salivary glands and pancreas may be due to organ-specific post-translational modifications, as have been observed with human secretory-type RNases [24]. It can be postulated that salivary DNase I may undergo more complex glycosylation, which results in greater stability like pH resistance.
One-step protein purification. Considering the strong affinity of DNase I for Con A and WGA, DNase I-purification using a combined Con A-WGA agarose column were performed. Typical purification results for each DNase I are summarized in Table 2. From approximately 0.1 g of each tissue sample, about 30 µg of purified DNase I with a specific activity ranging from 2500 to 40,000 units/mg was obtained. Although the rabbit pancreas tissue has high DNase I activity [3, 21], we were unable to purify the pancreatic enzyme due to its relatively lower lectin binding affinity. All detectable purified enzyme activity was inhibited by 1 mM EDTA and 1 mM EGTA and showed an optimum pH of approximately 7.0 and required divalent cations, 5 mM Mn2+, Mg2+ or Co2+ in the presence of Ca2+. Furthermore, this activity was completely abolished by specific antibodies against each DNase I. The character of each enzyme, such as optimal pH, metal dependency, or actin inhibition was quite similar to those of purified DNase I from other tissue types from the same three kinds of animal [10, 21] (data not shown).
Table 2. Purification of mammalian DNase I
from salivary gland and pancreas

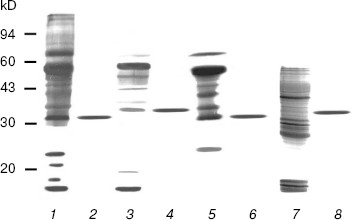
SDS-PAGE analyses of the purified enzymes revealed only one band for each enzyme (Fig. 1). The DNase I molecular masses of the rat and rabbit parotid glands, the rat submaxillary gland, and the porcine pancreatic DNase I under reducing conditions were approximately 32, 35, 32, and 34.5 kD, respectively, all in accord with previous studies [10, 21, 22]. Gel filtrations on Superdex 75 revealed each of the three types of DNase I had only one protein peak and the molecular mass of each enzyme was consistent with those estimated by SDS-PAGE (data not shown). These peaks coincided with DNase I activity. No DNase II, secretory RNase, or non-secretory RNase activities were detected in any of the purified DNases I.
This is the first report on the purification of DNase I from rabbit parotid gland and from mammalian submaxillary glands. In the present experiment, the one-step purification for DNase I was achieved solely in high-activity mammalian tissues. This procedure allowed DNases I to be easily and reproducibly isolated and purified to electrophoretic homogeneity, not with gradient elution but with stepwise elution.Fig. 1. SDS-PAGE patterns of purified mammalian pancreatic and salivary gland DNase I by silver staining. Samples were treated with 2% w/v SDS containing 25 mM dithiothreitol at 100°C for 3 min, electrophoresed in 12.5% T gel, and stained as described in “Materials and Methods”. One microgram of each protein was loaded in each lane. Lanes: 1) crude extract of rat parotid gland; 2) purified rat parotid gland DNase I; 3) crude extract of rabbit parotid gland; 4) purified rabbit parotid gland DNase I; 5) crude extract of rat submaxillary gland; 6) purified rat submaxillary gland DNase I; 7) crude extract of porcine pancreas; 8) purified porcine pancreatic DNase I.
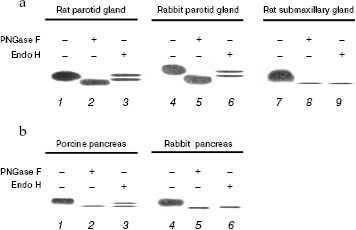
Enzymatic deglycosylation of mammalian DNase I. Based on our results on DNase I affinity for lectin columns, we investigated the differences in N-linked glycosylation patterns among the three types of DNase I, either by PNGase F or Endo H digestion. PNGase F cleaves any type of N-linked sugar, whereas Endo H cleaves high mannose and certain types of hybrid sugars. DNases I eluted by combined Con A-WGA agarose column were subjected to deglycosylation analysis. After 10 h digestion at 37°C, the samples were subjected to DNA-casting PAGE followed by activity staining (Fig. 2). In all samples, PNGase F digestion produced a lower mobility product when compared to the native enzyme, indicating that these mammalian DNases I are modified by N-linked glycosylation. On the other hand, the pattern of Endo H digestion differed among the samples. In the rat and rabbit parotid glands (Fig. 2a) and porcine pancreas (Fig. 2b), Endo H produced a double band: one had the same mobility as the native sample, and the other a slightly larger product than that of PNGase F digestion. This implies that Endo H is unable to completely digest parotid gland and porcine pancreas DNase I, and that the N-acetylglucosamine moieties remain on the asparagines residues of the DNase I following endo H digestion. On the other hand, the rat submaxillary gland (Fig. 2a) and rabbit pancreas (Fig. 2b) showed a single band following endo H digestion corresponding with PNGase F product. These results suggest that (i) DNases I from the rat and rabbit parotid gland and porcine pancreas are composed of both N-linked high-mannose/hybrid and complex-type carbohydrate moieties, and that (ii) DNases I from the rat submaxillary gland and rabbit pancreas possess N-linked high-mannose/hybrid type moieties fully sensitive to endo H digestion.
There may be species- and/or organ-specific differences in glycosylation of analogs of DNase I. Two potential N-linked glycosylation sites are known for mammalian enzymes. N18 (N-A-T) and N106 (N-D-T) are well conserved in the rat and porcine enzymes, whereas rabbit enzyme has only one potential site at N18 (N-A-T) [10, 21, 22]. Prior study has shown that the number of sugar chains in bovine DNase I differed by tissue: DNase I isolated from parotid glands and submaxillary glands were shown to have two sugar chains, while pancreatic DNase I has only one [16]. Interestingly, in contrast to the rabbit pancreas, the deglycosylation pattern of porcine pancreas was identical to that of the parotid gland (Fig. 2). In addition, lectin affinity of porcine pancreatic DNase I was similar to that of parotid gland, and not to that of the rabbit pancreas (Table 1). Feasibly, these results suggest that porcine pancreatic DNase I undergoes effective glycosylation causing it to exhibit a similar function to parotid gland DNase I. Moreover, it is noteworthy that differences in glycosylation patterns between the rat parotid and submaxillary glands and the rabbit parotid gland and pancreas have been shown for the first time (Fig. 2a). The functions of DNase I may be different despite the tissues coming from the same animal.Fig. 2. Electrophoretic patterns of purified DNase I from mammalian salivary gland (a) and pancreas (b) followed by digestion with PNGase F or Endo H for 10 h at 37°C. a) Purified DNase I (1, 4, 7), PNGase F digests (2, 5, 8), and Endo H digests (3, 6, 9) were subjected to DNA-casting PAGE (12.5%) followed by activity staining. b) Purified DNase I (1) and partially purified DNase I (4), PNGase F digests (2, 5), and Endo H digests (3, 6) were subjected to DNA-casting PAGE followed by activity staining. The values below each band indicate the activity following 10 h incubation at 37°C with or without endoglycosidase treatment.
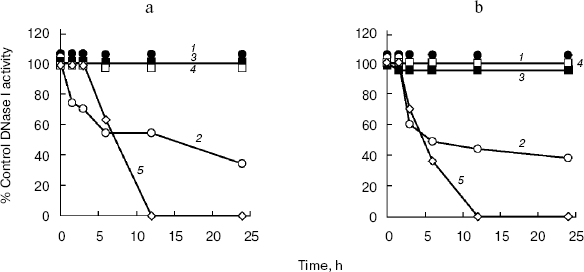
Effect of deglycosylation on DNase I activity. In DNA-casting PAGE analysis, the rat submaxillary gland and porcine pancreas showed a faint band following PNGase F and Endo H treatment (Fig. 2), which may indicate activity loss due to deglycosylation. Therefore, in this experiment, effects of deglycosylation on DNase I activities were investigated (Fig. 3). DNase I activity of the porcine pancreas was completely abolished by 12 h incubation both with PNGase F and Endo H. The rat submaxillary gland showed a 50% decrease at 6 h, and 60% decrease at 24 h from both enzyme treatments. On the other hand, DNase I activities in the rat and rabbit parotid glands and rabbit pancreas were unaffected by enzymatic deglycosylation.
N-Glycosylation plays important roles not only in protein folding but also in function [25-28]. Several studies have shown that enzymatic inactivation is caused by the loss of N-glycosylated moieties. Deglycosylation by PNGase F showed time-dependent decrease in ectonucleoside triphosphate diphosphohydrolase 3 activity [29]. Tsiakas et al. have shown that mutation of the N-glycosylation site in human CLN2 protein, which encodes lysosomal tripeptidyl peptidase-I, results in loss of enzymatic activity [30]. In pigs, the enzymatic activity of thyroid peroxidase was inhibited by PNGase F and Endo H treatments [31]. In the present study, N-glycosylated and/or high mannose sugar has been shown to be essential for full DNase I activity of the rat submaxillary gland, and would be indispensable for porcine DNase I activity.Fig. 3. Effects of PNGase F (a) and Endo H (b) on the DNase I activities of mammalian tissues. Each sample was incubated at 37°C in a reaction buffer containing endoglycosidase. Aliquots were removed at several time-points (1.5, 3, 6, 12, and 24 h) following the addition of endoglycosidases. 1) Rat parotid gland; 2) rat submaxillary gland; 3) rabbit parotid gland; 4) rabbit pancreas; 5) porcine pancreas. Control samples were incubated in reaction buffer without either endoglycosidase. The residual DNase I activities were determined by the SRED method. DNase I activity is given as percentage activity of controls.
There is a possibility that post-translational glycosylation is involved in acid stability. Takeshita et al. demonstrated that amphibian DNase I has no glycosylation site and is sensitive to low pH [32]. Previous study found that compared to that of other mammals, porcine DNase I was more sensitive to low pH: acute loss of activity was observed with pH of 5.0, while enzymes of the parotid and mixed (containing parotid) types were stable ranging from pH 3.0 to 6.0 [3]. It would be interesting to investigate the involvement of glycosylation in acid stability of DNase I using the porcine enzyme because it is inactivated both by low-pH condition and by deglycosylation. Recently, the piscine DNase I family has been shown to be remarkably sensitive to low pH despite the presence of a glycosylation site [33]. The extent of glycosylation may influence enzymatic stability.
In summary, we address the first attempt to purify mammalian DNases I using a one-step procedure. Glycosylation analysis showed parotid gland DNase I was efficiently glycosylated to acquire a stable property. The porcine pancreas DNase I showed a glycosylation pattern similar to that of the rat and rabbit parotid glands rather than rabbit pancreas, indicating a similar function to parotid gland DNase I. Porcine pancreatic enzyme was rendered inactive by deglycosylation. To our knowledge, this study is the first report demonstrating species- and organ-specific glycosylation of mammalian DNases I. Further comparative study of the DNase I family is needed to elucidate inter-species or inter-organ differences in the post-translational modification. Additionally, similar studies of other enzymes, which show dual expression in the pancreas and parotid, would contribute to a deeper understanding of the evolutionary aspects of enzymatic stability and activity by glycosylation.
We thank Mrs. Izumi Okui for her excellent secretarial assistance. This work was supported in part by Grants-in-Aid from Japan Society for the Promotion of Science (17790407 to JF and 16689015 to HT), the Sekisui Integrated Research Foundation, the Japan Securities Scholarship Foundation, Japan Foundation of Cardiovascular Research, and Kyowa Medex Research Foundation.
REFERENCES
1.Moore, S. (1981) in The Enzymes (Boyer, P.
D., ed.) Academic Press, New York, pp. 281-296.
2.Nadano, D., Yasuda, T., and Kishi, K. (1993)
Clin. Chem., 39, 448-452.
3.Takeshita, H., Mogi, K., Yasuda, T., Nakajima, T.,
Nakashima, Y., Mori, S., Hoshino, T., and Kishi, K. (2000) Biochem.
Biophys. Res. Commun., 269, 481-484.
4.Polzar, B., Zanotti, S., Stephan, H., Rauch, F.,
Peitsch, M. C., Irmler, M., Tschopp, J., and Mannherz, H. G. (1994)
Eur. J. Cell Biol., 64, 200-210.
5.Mannherz, H. G., Peitsch, M. C., Zanotti, S.,
Paddenberg, R., and Polzar, B. (1995) Curr. Top. Microbiol.
Immunol., 198, 61-174.
6.Chitrabamrung, S., Rubin, R. L., and Tan, E. M.
(1981) Rheumatol. Int., 1, 55-60.
7.Napirei, M., Karsunky, H., Zevnik, B., Stephan, H.,
Mannherz, H. G., and Moroy, T. (2000) Nat. Genet., 25,
177-181.
8.Yasutomo, K., Horiuchi, T., Kagami, S., Tsukamoto,
H., Hashimura, C., Urushihara, M., and Kuroda, Y. (2001) Nat.
Genet., 28, 313-314.
9.Kreuder, V., Dieckhoff, J., Sittig, M., and
Mannherz, H. G. (1984) Eur. J. Biochem., 139,
389-400.
10.Takeshita, H., Yasuda, T., Nadano, D., Iida, R.,
and Kishi, K. (1995) J. Biochem., 118, 932-938.
11.Catley, B. J., Moore, S., and Stein, W. H. (1969)
J. Biol. Chem., 244, 933-936.
12.Price, P. A., Liu, T.-Y., Stein, W. H., and
Moore, S. (1969) J. Biol. Chem., 244, 917-923.
13.Baranovskii, A. G., Buneva, V. N., and Nevinsky,
G. A. (2004) Biochemistry (Moscow), 69, 587-601.
14.Wadano, A., Hobus, P. A., and Liao, T. H. (1979)
Biochemistry, 18, 1424-1429.
15.Kijimoto-Ochiai, S., Katagiri, Y. U., Hatae, T.,
and Okuyama, H. (1989) Biochem. J., 257, 43-49.
16.Nishikawa, A., and Mizuno, S. (2001) Biochem.
J., 355, 245-248.
17.Nakajima, T., Yasuda, T., Nakashima, Y., Hosomi,
O., Takeshita, H., and Kishi, K. (1998) Immunol. Invest.,
27, 145-152.
18 Yasuda, T., Awazu, S., Sato, W., Iida, R., Tanaka, Y., and Kishi, K.
(1990) J. Biochem., 108, 393-398.
19.Nadano, D., Yasuda, T., Sawazaki, K., Takeshita,
H., and Kishi, K. (1993) Analyt. Biochem., 212,
111-116.
20.Yasuda, T., Takeshita, H., Iida, R., Nakajima,
T., Hosomi, O., Nakashima, Y., and Kishi, K. (1998) J. Biol.
Chem., 273, 2610-2616.
21.Yasuda, T., Takeshita, H., Nakajima, T., Hosomi,
O., Nakashima, Y., and Kishi, K. (1997) Biochem. J., 325,
465-473.
22.Mori, S., Yasuda, T., Takeshita, H., Nakajima,
T., Nakazato, E., Mogi, K., Kaneko, Y., and Kishi, K. (2001)
Biochim. Biophys. Acta, 1547, 275-287.
23.Laemmli, U. K. (1970) Nature, 227,
680-685.
24.Yasuda, T., Mizuta, K., Sato, W., and Kishi, K.
(1990) Eur. J. Biochem., 191, 523-529.
25.Wyss, D. F., and Wagner, G. (1996) Curr. Opin.
Biotechnol., 7, 409-416.
26.Imperiali, B., and O'Connor, S. E. (1999)
Curr. Opin. Chem. Biol., 3, 643-649.
27.Parodi, A. J. (2000) Annu. Rev. Biochem.,
69, 69-93.
28.Helenius, A., and Aebi, M. (2001) Science,
291, 2364-2369.
29.Murphy, D. M., and Kirley, T. L. (2003) Arch.
Biochem. Biophys., 413, 107-115.
30.Tsiakas, K., Steinfeld, R., Storch, S., Ezaki,
J., Kominami, E., Kohlschütter, A., Ullrich, K., and Braulke, T.
(2004) Glycobiology, 14, 1C-5C.
31.Long, Y., Franc, J. L., Kaniewski, J., Lanet, J.,
and Giraud, A. (1991) Eur. J. Biochem., 202, 501-505.
32.Takeshita, H., Yasuda, T., Iida, R., Nakajima,
T., Mori, S., Mogi, K., Kaneko, Y., and Kishi, K. (2001) Biochem.
J., 357, 473-480.
33.Yasuda, T., Takeshita, H., Iida, R., Ueki, M.,
Nakajima, T., Kaneko, Y., Mogi, K., Kominato, Y., and Kishi, K. (2004)
Biochim. Biophys. Acta, 1672, 174-183.