Identification of a Highly Conserved Pro-Gly Doublet in Non-animal Small Heat Shock Proteins and Characterization of Its Structural and Functional Roles in Mycobacterium tuberculosis Hsp16.3
Xinmiao Fu1,2,3 and Zengyi Chang1,2*
1State Key Laboratory of Protein Engineering and Plant Genetic Engineering, Peking University, Beijing 100871, China; fax: 86-10-6275-1526; E-mail: changzy@pku.edu.cn2College of Life Science, Peking University, Beijing 100871, China
3Department of Biological Sciences and Biotechnology, Tsinghua University, Beijing 100084, China
* To whom correspondence should be addressed.
Received March 23, 2005; Revision received May 25, 2005
Small heat shock proteins (sHSPs) are highly divergent in primary sequences, with short conserved motifs found in various subfamilies. Here a Pro-Gly doublet was found to be conserved in most non-animal sHSPs by sequence analysis of a total of 344 unique sHSPs (covering the subfamilies: bacterial class A, bacterial class B, archae, fungi, plant, and animal) placed in data banks. In contrast, the residues corresponding to this Pro-Gly doublet in most of animal sHSPs are often charged. Site-directed mutagenesis studies of Mycobacterium tuberculosis Hsp16.3 replacing the Gly (at position 59) residue by Cys or Trp demonstrate that this Gly is likely involved in subunit interactions, which is consistent with that in Methanococcus jannaschii Hsp16.5 and wheat Hsp16.9. Our data suggest that this Pro-Gly doublet in Hsp16.3 is not directly involved in binding of denatured substrate proteins, whereas the corresponding charged residues in bovine alpha-crystallin were instead proposed before to be involved in substrate binding. These observations indicate that the highly conserved Pro-Gly doublet is critical to discriminate between non-animal and animal sHSPs.
KEY WORDS: chaperone, small heat shock protein, evolution, cysteine modification, tryptophan fluorescence, Hsp16.3DOI: 10.1134/S0006297906130141
Abbreviations: DTT) dithiothreitol; DTNB) 5,5´-dithiobis(2-nitrobenzoic acid); sHSPs) small heat shock proteins; L57C, G59C, and D61C) the Hsp16.3 mutant proteins with Leu57, Gly59, and Asp61, respectively, substituted by cysteine; G59A and G59W) the Hsp16.3 mutant proteins with Gly59 substituted by alanine and tryptophan, respectively.
Small heat shock proteins (sHSPs), as one subclass of molecular
chaperones, have been found in almost all organisms [1]. Studies performed in vitro have
demonstrated that sHSPs are able to exhibit chaperone-like activities
and even to enhance the refolding of denatured proteins with help from
other chaperone systems, such as Hsp70/Hsp40 [2-4]. In primary sequence comparison, sHSPs are
characterized by having a relatively conserved alpha-crystallin
domain [5-7]. Nevertheless,
sHSPs have diverged much more during evolution than the large HSPs like
Hsp90, Hsp70, and Hsp60 [1, 5,
7].
Conserved sequence motifs have been revealed in the whole family or various subfamilies of sHSPs. Such motifs conserved in the whole family include the IXI/V motif in the C-terminal extension [8] and consensus I region represented by the sequence of Pro-(X14)-Gly-Val-Leu in the C-termini of the alpha-crystallin domain [7, 9-11], those conserved in plant sHSPs include the consensus II region represented by the sequence of Pro-(X14)-X-Val/Leu/Ile-Val/Leu in the N-termini of the alpha-crystallin domain [9, 10], and those conserved in animal sHSPs include the sequence of SRLFDQFFG in the N-terminal region [5, 11]. Biochemical studies have implicated important structural and functional roles of these motifs for sHSPs [8, 12-14].
The Pro-Gly doublet, located at the N-terminal end of the alpha-crystallin domain, was previously revealed to be conserved in plant and bacterial sHSPs [11, 15]. Here the sequence analysis of a total of 344 unique sHSP sequences demonstrates that this Pro-Gly doublet is actually highly conserved in almost all non-animal sHSPs. Site-directed mutagenesis studies through Mycobacterium tuberculosis Hsp16.3 demonstrate that the Pro-Gly doublet is very likely involved in subunit interaction but is not directly involved in binding of denatured substrate proteins. These observations will enrich our insights into the evolution of sHSPs from the perspective of protein structure, function, and action mechanism.
MATERIALS AND METHODS
Materials. Dithiothreitol (DTT), 5,5´-dithiobis(2-nitrobenzoic acid) (DTNB), and insulin were all obtained from Sigma (USA). MutantBest Kit was purchased from TaKaRa Biotechnology (Dalian, China). DEAE-Sepharose FastFlow and Q-Sepharose High Performance for chromatography were obtained from Amersham Pharmacia Biotech (Sweden). All other chemical reagents were of analytical purity.
Sequence analysis. All the available amino acid sequences of sHSPs were downloaded from the mirror website of SWISSPROT and TREMBL databases in China (available at http://cn.expasy.org). The fragments and the duplicated sequences were discarded manually and the remaining 344 unique proteins, named by either SWISS_PROT or TREMBL accession number, were grouped as bacteria class A and B [16], archaea, fungi, plant, animal, and other non-animal eukaryotes. The 344 amino acid sequences were aligned using Clustalx (version 1.7).
Plasmid construction and protein purification. The single-site mutants of Hsp16.3 (L57C, G59C, D61C, G59A, G59W) were constructed according to methods described before [17]. The wild type, L57C and D61C mutant Hsp16.3 proteins were purified according to procedures previously described [17, 18]. For the purification of the G59C mutant Hsp16.3 protein, the supernatant of cell lysates was loaded onto a DEAE-Sepharose FastFlow column pre-equilibrated with 50 mM imidazole-HCl (pH 6.5) and was then eluted with a salt gradient of 0.15-0.35 M NaCl. The fractions containing G59C were pooled, dialyzed against 20 mM Tris-HCl (pH 8.5), and loaded onto a Q-Sepharose high performance column before elution with a salt gradient of 0.15-0.4 M NaCl. The G59W and G59A mutant proteins were purified similarly, with both being eluted with a salt gradient of 0.12-0.25 M NaCl from the DEAE column, and the G59W was eluted with a 0.15-0.3 M and G59A with a 0.15-0.4 M salt gradient from the Q-Sepharose column. The purified proteins were dialyzed in deionized water, lyophilized, and stored at -20°C before further analysis. Protein concentrations were determined using the Bio-Rad Protein Assay.
DTNB modification. The exposure status of Cys59 in the G59C mutant Hsp16.3 protein was detected by using DTNB modification as described previously [19]. The optical density of the mixture of protein (1 mg/ml), urea at various concentrations, and DTNB (0.2 mM), dissolved in 50 mM sodium phosphate buffer (pH 7.0), was recorded on a UV-8500 spectrophotometer (Shanghai Techcomp, China) at 412 nm.
Tryptophan intrinsic fluorescence assay. The intrinsic fluorescence of tryptophan in the G59W mutant Hsp16.3 protein was scanned between 300-400 nm while excited at 279 nm on a Hitachi F-4500 fluorescence spectrophotometer, which was connected to a water bath (Boyikang Inc., China). To monitor the urea-induced exposure of tryptophan, the G59W mutant protein (0.2 mg/ml) unfolded by various concentrations of urea from 0.2 to 7 M for 15 h was subjected to fluorescence assay. To monitor the heat-induced exposure of tryptophan, the G59W protein (0.1 mg/ml) was incubated (ranging from 25 to 73°C) at each temperature for 5 min before fluorescence assay was performed.
Chemical cross-linking by glutaraldehyde. Glutaraldehyde (0.5%) and Hsp16.3 proteins (0.2 mg/ml) were reacted at 25°C in 50 mM sodium phosphate buffer (pH 7.0) for 1 or 10 min. The reactions were stopped by quenching with 1 M Tris-HCl for 10 min. The cross-linked samples were analyzed by SDS-PAGE (12%).
Chaperone-like activity assay. Chaperone-like activities of the Hsp16.3 wild type and mutant proteins (0.3 mg/ml, wild type, G59A, or G59W) were assayed by measuring their capacity to suppress the DTT-induced (20 mM) aggregation of insulin (0.3 mg/ml) at different temperatures (from 25 to 65°C). The measurements were performed with the UV-8500 spectrophotometer as described previously [20, 21].
RESULTS
Sequence alignment reveals a Pro-Gly doublet highly conserved in non-animal sHSPs. The sequence alignment of 344 sHSPs unanimously identified a highly conserved doublet Pro-Gly in the non-animal sHSPs (data presented in Table 1, Fig. 1a, and supplementary data in Fig. 2). The 344 unique sHSP sequences were obtained from the SWISSPROT and TREMBL databases, covering the seven subfamilies of sHSPs, i.e., bacterial class A (32 sequences), bacterial class B (64), archaea (35), fungi (10), plant (115), animal (79), and other eukaryotes (9) (see supplementary data in Table 2). The corresponding positions of the Pro-Gly doublet are, instead, often occupied by charged residues in animal sHSPs (Table 1 and Fig. 2). One exception among the non-animal sHSPs is that most of the members of the bacteria class A sHSPs have an Ala at the position of the highly conserved Pro (Fig. 2), somehow suggesting a distinct evolutionary history of bacterial class A sHSPs from the rest of the non-animal sHSPs. Additionally, the sequence alignments (Fig. 2) also revealed that the two residues sandwiching the Pro-Gly doublet in almost all the sHSPs are hydrophobic.
Table 1. The conserved Pro-Gly doublet in
non-animal sHSPsa
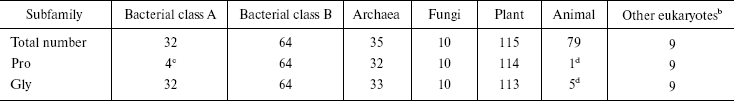
aThe data in this table were obtained from the sequence alignment results displayed in Fig. 2.
bThese eukaryotes contain three kingdoms: Alveolata (seven sequences), Entamoebidae (one sequence), Rhodophyta (one sequence).
cThe corresponding residue is Ala, instead of Pro, in 26 out of 32 members of bacterial class A sHSPs.
dThe residues corresponding to Pro and Gly are charged residues, respectively, in 38 and 51 out of 79 members of animal sHSPs.
Fig. 1. Characterization of the G59C mutant Hsp16.3 protein. a) Sequence alignment of representative sHSPs, covering bacterial class B (M. tuberculosis Hsp16.3), bacterial class A (E. coli IbpA), archaea (M. jannaschii Hsp16.5), plant (pea Hsp18.1 and wheat Hsp16.9), fungi (yeast Hsp26), and animal (the other four sequences). For simplicity, only a part of the residues are shown here. The secondary structure elements were arbitrarily assigned according to the crystal structure of M. jannaschii Hsp16.5 [22]. The consensus II region identified in plant sHSPs [10] is shown here. b) SDS-PAGE analysis results for the G59C, L57C, and D61C mutant Hsp16.3 proteins with DTT added in half of the loaded samples (lanes 2, 4, and 6). Positions of the dimers (lane 3) generated by the spontaneous formation of disulfide bonds in the G59C mutant protein and monomers are indicated by the arrows on the right. c) The crystal structure of Hsp16.5 dimer (cartoon model). The black, dark gray, and light gray balls are Gly62, Asn64, and Leu60 in Hsp16.5, respectively, which correspond to Gly59, Asn61, and Leu57 in Hsp16.3, respectively. d) The time-dependent change in the absorption (at 412 nm) of DTNB (added to a final concentration of 0.2 mM) during its reaction with sulfhydryl groups of the G59C mutant Hsp16.3 protein (1 mg/ml) monitored at room temperature in the presence of different urea concentrations (0, 1, 4, and 8 M urea for the curves 1, 2, 3, and 4, respectively).
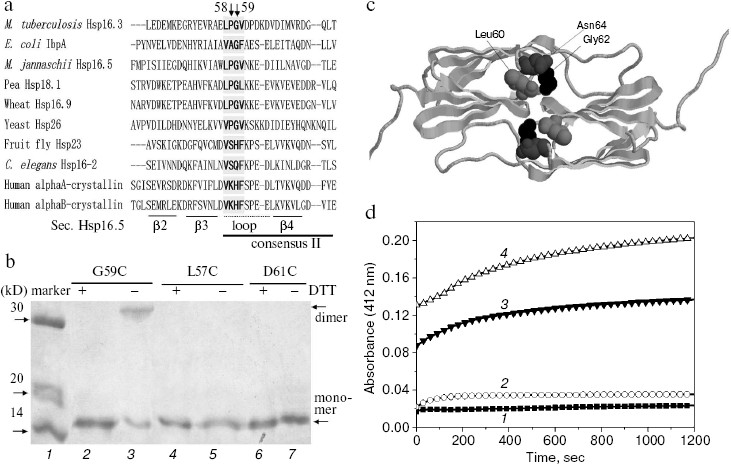
Table 2. Total sHSPs (344 unique sequences)Fig. 2. Sequence alignment of seven sHSPs subfamilies. The full sequences of 344 sHSPs (with names shown in Table 2 including bacterial class A and B, archaea, fungi, plant, animal, and other eukaryote sHSPs) were aligned using Clustalx. For simplicity, only the Pro-Gly doublet and its neighboring residues are shown here.
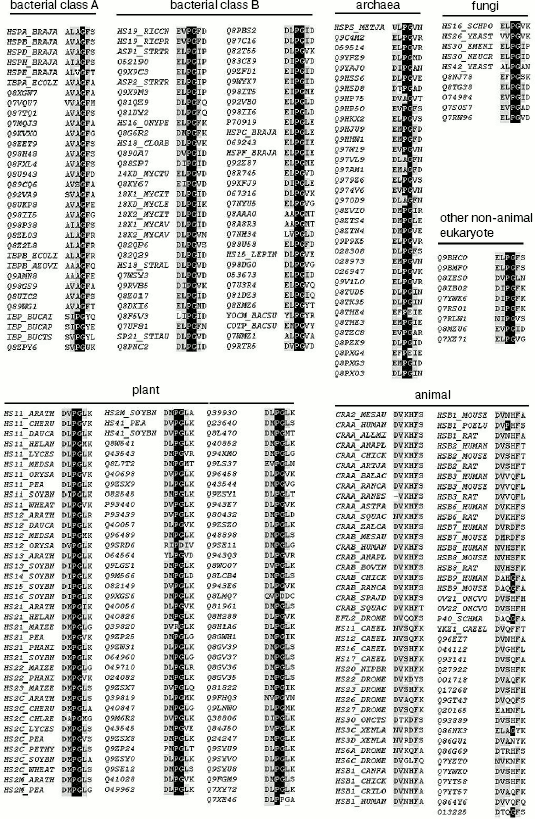

aThe total number of sHSPs in each subfamily is indicated.
bThe name of each sHSP is either TREMBL or SWISS_PROT number.
Inter-subunit disulfide bond is formed spontaneously in the G59C mutant Hsp16.3 protein having a buried Cys59 residue. What are the structural and functional roles of the highly conserved Pro-Gly doublet in non-animal sHSPs? Hints were first sought by examining the spatial positions of this Pro-Gly doublet in the crystal structures of archaea M. jannaschii Hsp16.5 and plant wheat Hsp16.9 [22, 23]. This reveals that they are located in a loop that connects two anti-parallel beta-strands (as indicated in Fig. 1a) and is localized at the interface of subunit-subunit interactions in the oligomers [22] (or implicated in Fig. 1c).
The Gly59 in M. tuberculosis Hsp16.3 (belonging to bacterial class B subfamily [16]) existing as nonamers and exhibiting chaperone-like activities in vitro [18, 24] was then replaced via site-directed mutagenesis to understand the roles of this Pro-Gly doublet. For this purpose, we took advantage of the fact that the wild type Hsp16.3 protein contains neither cysteine nor tryptophan residues, two of the residues whose side chains can be effectively probed in the protein due to the special chemical reactivity of the former and high fluorescence intensity of the latter [25].
First, Gly59 in the Pro-Gly doublet of Hsp16.3 was replaced by cysteine. Data presented in Fig. 1b clearly demonstrate that an intersubunit disulfide bond was spontaneously formed in the G59C mutant Hsp16.3 protein (lane 3). Such an intersubunit disulfide bond, however, was not detected in the L57C and D61C mutant Hsp16.3 proteins (lanes 4-7 in Fig. 1b). Given that disulfide bonds are only formed between two cysteine residues being within about 6 Å from each other [26], these observations suggest that Gly59 residues on a certain pair of subunits are close to each other in the wild type Hsp16.3 protein. Supporting this speculation is that the distance between the side chains of a pair of Gly62 residues in Hsp16.5 dimer (residue corresponding to Gly59 in Hsp16.3) is slight closer than that between Leu60 residues, and is much closer than that between Asn64 residues (Fig. 1c).
To probe the topological location of Gly59 in the wild type Hsp16.3 oligomers, we examined the exposure status of the cysteine residues in the G59C mutant protein using DTNB, which is able to access and react with those exposed sulfhydryl groups [19]. Data presented in Fig. 1d demonstrate that the Cys59 residue in the G59C mutant protein is buried and not accessible to DTNB under normal conditions, but becomes exposed and thus accessible to DTNB in the presence of urea at a relatively high concentration. Examination of the exposure status of Cys59 upon heating was not successful due to the formation of aggregates of the G59C mutant protein at high temperatures. The cysteine modification curves of 3 and 4 (Fig. 1d) obviously contain two phases (fast and slow). A reasonable explanation for this observation is that the fast phase might represent a fast unfolding and/or dissociation of the mutant protein in the presence of high concentrations of urea, and the slow phase a limited subunit exchange process between the dissociated mutant protein oligomers since the time scale of this phase is close to that of subunit exchange as previously observed [27].
The tryptophan residue in the G59W mutant Hsp16.3 protein is also buried and is located within or near the subunit interfaces. Second, Gly59 in the Hsp16.3 protein was replaced by tryptophan with its topological location in the protein examined by recording its intrinsic fluorescence. The introduction of a tryptophan residue indeed dramatically increases the apparent intrinsic fluorescence of the Hsp16.3 protein (insert in Fig. 3a). The fact that the maximum emission wavelength of fluorescence is around 321 nm (insert in Fig. 3a) suggests that Trp59 is buried in a hydrophobic environment. The exposure of Trp59 in the presence of urea or upon heating is indicated by the red shift of the maximum emission wavelengths (Figs. 3a and 3b).
A very striking observation is that the urea-induced exposure of Trp59 in the G59W mutant protein was surprisingly accompanied with a dramatic increase in the fluorescence intensity (Fig. 3a). The most likely explanation for this is that the urea induces the oligomers to dissociate and this will in turn reduce the self-quenching of the tryptophan residues located close to each other at the subunit interfaces [28]. Nevertheless, the priority of Trp fluorescence red shift to its intensity upon urea increase (Fig. 3a) may imply an unfolding process leading to the exposure of Trp occurring ahead of the subunit dissociation.Fig. 3. Characterization of the G59W mutant Hsp16.3 protein. a) The tryptophan maximum emission wavelength (curve 1) and fluorescence intensity at the spectral maximum (curve 2) of the G59W mutant Hsp16.3 protein (0.2 mg/ml) are plotted against the urea concentration. The inset represents the intrinsic fluorescence spectra of the wild type (curve 1), G59A (curve 2), and G59W (curve 3) mutant Hsp16.3 proteins (all being at 0.1 mg/ml). b) The tryptophan emission maximum wavelength of the G59W mutant Hsp16.3 protein (0.1 mg/ml) is plotted against temperature increase applied to unfold the protein. c) SDS-PAGE analysis results of the wild type (lanes 2-4), G59A (lanes 5-7), and G59W (lanes 8-10) mutants of Hsp16.3 protein after cross-linking by glutaraldehyde for the indicated time intervals. d) The relative chaperone-like activities of the wild type, G59A, and G59W mutants of Hsp16.3 protein (curves 1-3, respectively) in suppressing of the aggregation of insulin B molecules at the indicated temperatures. Each value represents the average of two independent experiments.
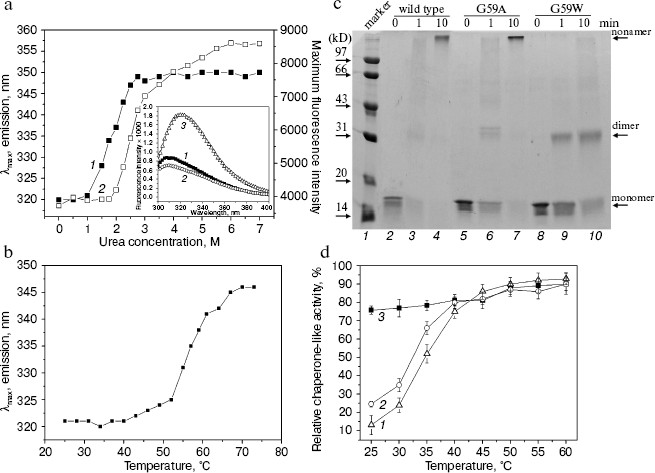
The G59W mutant Hsp16.3 protein exists predominantly as dimers rather than nonamers and exhibits efficient chaperone-like activity even at low temperature. It was subsequently examined whether the replacement of Gly59 in the Hsp16.3 protein would alter its oligomeric structure and chaperone-like activity. Results of chemical cross-linking analyses (Fig. 3c) demonstrate that the G59W mutant Hsp16.3 protein predominantly exists as dimer, whereas the G59A mutant protein (being a control mutant protein) still appears as a nonamer similar to the wild type protein. This observation indicates that Gly59 in Hsp16.3 is indeed located in a position that is important for the assembly of the protein.
Given that the Trp59 residue is buried under native conditions and exposed upon heating (Fig. 3b), it is then asked whether such exposure correlates with any increase in chaperone-like activity, which would implicate the involvement of the Pro-Gly doublet in substrate binding for the wild type Hsp16.3. Data presented in Fig. 3d show that the G59W mutant protein exhibits chaperone-like activity at 25°C as efficiently as it does at 60°C, in contrast to the wild type protein, which exhibited little at 25°C and high activity only at a higher temperature (see also ref. [18]).
Parallel examination on the G59C mutant protein revealed that it exists as polydisperse oligomers rather than as nonamers, is different in secondary structure content, and exhibits lower chaperone-like activity when compared with the wild type protein (data not shown). However, it is not very easy to explain these observations since disulfide bonds are known to be critical for the folding and assembly of proteins in general [29] and cysteine itself may interfere with the chaperone-like activity of chaperones [17].
DISCUSSION
The Pro-Gly doublet is often involved in the formation of beta-turn structure in proteins due to the unique properties of the Pro and Gly residues [30, 31]. Our site-directed mutagenesis studies of Gly59 for Hsp16.3 of M. tuberculosis (a residue in a Pro-Gly doublet highly conserved in non-animal sHSPs) strongly demonstrate its important direct role in the subunit-subunit interactions. This conclusion is also well supported by the analysis on the crystal structures of Hsp16.5 and Hsp16.9 [22, 23] (Fig. 1c).
Our observations, summarized below, however, suggest the Gly59 residue of Hsp16.3 is not directly involved in binding denatured substrate proteins. First, the G59W mutant Hsp16.3 protein does not expose Trp59 at room temperature but exhibits efficient chaperone-like activity (Figs. 3b and 3d), and the exposure of Trp upon heating is not correlated with a change in chaperone-like activity. Second, the wild type Hsp16.3 protein exhibits efficient chaperone-like activity and maintains its nonameric structure in the presence of low concentrations of urea (0.8 M) [27, 32], at which Trp59 in the G59W mutant protein is not exposed (Fig. 3a). Supporting this claim is the previous demonstration that the N-terminal region (2-35 residues) of the wild type Hsp16.3 protein is critical for substrate binding [33]. This claim, however, should not be taken as exclusive, given that it is based on observations of the behavior of the G59W mutant Hsp16.3 protein, where the degree of the exposure of the Trp residue was examined in the absence of denatured substrate proteins, which may induce such an exposure.
The substitution of this Pro-Gly doublet by charged residues in the animal sHSPs may suggest a different role of this region. For instance, one of the substrate-binding sites for bovine alphaA- and alphaB-crystallin was identified as a region containing these charged residues [34, 35]. These charged residues, on the other hand, may also be important for protein oligomerization of animal sHSPs since the region containing these charged residues in alphaB-crystallin was found to be involved in an interaction with alphaA-crystallin [36].
In our preliminary study [37], far-UV CD spectroscopy analysis revealed that the replacement of Gly59 by Trp leads to a significant disturbance of the secondary structure of the Hsp16.3 protein, consistent with its effect on oligomeric structure. More importantly, the behavior of G59W mutant Hsp16.3 protein is very similar to that of another mutant Hsp16.3 protein lacking nine residues from the C-terminus [33] in that both predominantly exist as dimers and exhibit efficient chaperone-like activities even at room temperature. These observations strengthened the view that sHSPs, like Hsp16.3, exhibit chaperone-like activities via oligomeric dissociation [18, 21, 27, 38]. Moreover, the existence of both as dimers is in line with the recent report that Hsp16.3 protein was a dodecamer using dimers as the assembling units [39].
The authors wish to thank members of Dr. Chang's laboratory for their suggestions and discussions (especially Zhang Xuefeng, Jiao Wangwang, and Liu Chong).
This work was funded by the National Key Basic Research Foundation of China (grant No. G1999075607) and the National Natural Science Foundation of China (grant No. 30270289).
REFERENCES
1.Kappe, G., Leunissen, J. A., and de Jong, W. W.
(2000) Progr. Mol. Subcell. Biol., 28, 1-17.
2.Horwitz, J. (1992) Proc. Natl. Acad. Sci.
USA, 89, 10449-10453.
3.Jakob, U., Gaestel, M., Engel, K., and Buchner, J.
(1993) J. Biol. Chem., 268, 1517-1520.
4.Lee, G. J., Roseman, A. M., Saibil, H. R., and
Vierling, E. (1997) EMBO J., 16, 659-671.
5.Plesofsky-Vig, N., Vig, J., and Brambl, R. (1992)
J. Mol. Evol., 35, 537-545.
6.De Jong, W. W., Leunissen, J. A. M., and Voorter,
C. E. M. (1993) Mol. Biol. Evol., 10, 103-126.
7.Vierling, E. (1991) Annu. Rev. Plant Physiol.
Plant Mol. Biol., 42, 579-620.
8.Studer, S., Obrist, M., Lentze, N., and Narberhous
F. (2002) Eur. J. Biochem., 269, 3578-3586.
9.Waters, E. R., Lee, G., and Vierling, E. (1996)
J. Exp. Bot., 47, 325-338.
10.Waters, E. R. (1995) Genetics, 141,
785-795.
11.De Jong, W. W., Caspers, G. J., and Leunissen, J.
A. (1998) Int. J. Biol. Macromol., 22, 151-162.
12.Pasta, S. Y., Bakthisaran, R., Tangirala, R., and
Rao, C. M. (2003) J. Biol. Chem., 278, 51159-51166.
13.Mao, Q., and Chang, Z. (2001) Biochem.
Biophys. Res. Commun., 289, 1257-1261.
14.Muchowski, P. J., Wu, G. J. S., Liang, J. J. N.,
Adman, E. T., and Clark, J. I. (1999) J. Mol. Biol., 289,
397-411.
15.Narberhaus, F. (2002) Microbiol. Mol. Biol.
Rev., 66, 64-93.
16.Munchbach, M., Nocker, A., and Narberhaus, F.
(1999) J. Bacteriol., 181, 83-90.
17.Fu, X., Li, W., Mao, Q., and Chang, Z. (2003)
Biochem. Biophys. Res. Commun., 308, 627-635.
18.Gu, L., Abulimiti, A., Li, W., and Chang, Z.
(2002) J. Mol. Biol., 319, 517-526.
19.Ellman, G. L. (1959) Arch. Biochem.
Biophys., 82, 70-77.
20.Sanger, F. (1949) J. Biol. Chem.,
45, 563-574.
21.Haslbeck, M., Walke, S., Stromer, T.,
Ehrnsperger, M. E., White, H., Chen, S. R., Raibil, H., and Buchner, J.
(1999) EMBO J., 18, 6744-6751.
22.Kim, K. K., Kim, R., and Kim, S. H. (1998)
Nature, 394, 595-599.
23.Van Montfort, R. L. M., Basha, E., Fieldrich, K.
L., and Viering, E. (2001) Nature Struct. Biol., 8,
1025-1030.
24.Chang, Z., Primm, T. P., Jakana, J., Lee, I. H.,
Serysheva, I., Chiu, W., Gilbert, H. F., and Quiocho, F. A. (1996)
J. Biol. Chem., 271, 7218-7224.
25.Creighton, T. E. (1993) Proteins Structures
and Molecular Properties, 2nd Edn., W. H. Freeman Company.
26.Perry, L. J., and Wetzel, R. (1984)
Science, 226, 555-557.
27.Fu, X., Liu, C., Liu, Y., Feng, X., Gu, L., Chen
X., and Chang, Z. (2003) Biochem. Biophys. Res. Commun.,
310, 412-420.
28.Renard, D., Lefebvre, J., Griffin, M. C. A., and
Griffin, W. G. (1998) Int. J. Biol. Macromol., 22,
41-49.
29.Gilbert, H. F. (1994) in Mechanisms of Protein
Folding (Pain, R. H., ed.) IRL Press, Oxford, p. 107.
30.Thakur, A. K., and Wetzel, R. (2002) Proc.
Natl. Acad. Sci. USA, 99, 17014-17019.
31.Venkatraman, J., Shankaramma, S. C., and Balaram,
P. (2001) Chem. Rev., 101, 3131-3152.
32.Yang, H., Huang, S., Dai, H., Gong, Y., Zheng,
C., and Chang, Z. (1999) Protein Sci., 8, 174-179.
33.Fu, X., Zhang, H., Zhang, X., Cao, Y., Jiao, W.,
Liu, C., Song, Y., Abulimiti, A., and Chang, Z. (2005) J. Biol.
Chem., 280, 6337-6348.
34.Sharma, K. K., Kumar, G. S., Murphy, A. S., and
Kester, K. (1998) J. Biol. Chem., 273, 15474-15478.
35.Sharma, K. K., Kumar, R. S., Kumar, G. S., and
Quinn, P. T. (2000) J. Biol. Chem., 275, 3767-3771.
36.Sreelakshmi, Y., Santhoshkumar, P.,
Bhattacharyya, J., and Sharma, K. K. (2004) Biochemistry,
43, 15785-15795.
37.Chen, X., Fu, X., Ma, Y., and Chang, Z. (2005)
Biochemistry (Moscow), 70, 913-919.
38.Fu, X., and Chang, Z. (2004) Biochem. Biophys.
Res. Commun., 316, 291-299.
39.Kennaway, C. K., Benesch, J. L., Gohle, U., Wang,
L., Robinson, C. V., Orlova, E. V., Saibi, H. R., and Keep, N. H.
(2005) J. Biol. Chem., 280, 33419-33425.