Melatonin Prevents Oxidative Stress and Inhibits Reactive Gliosis Induced by Hyperhomocysteinemia in Rats
G. Baydas1*, M. Ozer1, A. Yasar1, S. T. Koz1, and M. Tuzcu2
1Department of Physiology, Faculty of Medicine, Firat University, Elazig 23119, Turkey; fax: + 90-424-233-3770; + 90-424-237-9138; E-mail: baydas@hotmail.com; gbaydas@firat.edu.tr; ayasar@firat.edu.tr; kozsema@hotmail.com2Department of Biology, Faculty of Science, Firat University, Elazig, Turkey; E-mail: mtuzcu@firat.edu.tr
* To whom correspondence should be addressed.
Received January 4, 2005; Revision received March 22, 2005
Homocysteine (Hcy), an independent risk factor for atherosclerosis, undergoes auto-oxidation and generates reactive oxygen species, which are thought to be main cause of Hcy neurotoxicity. However, the mechanisms leading to neurodegenerative disorders are poorly understood because studies that have investigated the potential neurotoxicity of hyperhomocysteinemia in vivo are scarce. The purpose of this study was to test whether daily administration of methionine, which induces hyperhomocysteinemia, causes glial hyperactivity, and also to investigate the protective effects of melatonin on the brain tissue against oxidative stress of Hcy in rats. There was a significant development of oxidative stress as indicated by an increase in malondialdehyde + 4-hydroxyalkenals in hippocampus and cortex of hyperhomocysteinemic rats, whereas significant reduction was found in the activity of glutathione peroxidase (GSH-Px). Co-treatment with melatonin inhibited the elevation of lipid peroxidation and significantly increased GSH-Px activity in the brain regions studied. Western blot analysis revealed an increase in glial fibrillary acidic protein (GFAP) contents both in hippocampus and frontal cortex (p < 0.001) of hyperhomocysteinemic rats compared to the controls. Administration of melatonin significantly decreased GFAP contents in hippocampus and cortex (p < 0.05). S100B contents increased only in frontal cortex in hyperhomocysteinemic rats compared to the control (p < 0.01) and was inhibited by melatonin treatment (p < 0.01). The present findings show that Hcy can sensitize glial cells, a mechanism which might contribute to the pathogenesis of neurodegenerative disorders, and further suggest that melatonin can be involved in protecting against the toxicity of Hcy by inhibiting free radical generation and stabilizing glial cell activity.
KEY WORDS: melatonin, glial fibrillary acidic protein, S100B protein, lipid peroxidation, glutathione peroxidaseDOI: 10.1134/S0006297906130153
Abbreviations: GFAP) glial fibrillary acidic protein; GSH-Px) glutathione peroxidase; Hcy) homocysteine; LPO) lipid peroxidation; Mel) melatonin.
Homocysteine (Hcy), a thiol-containing amino acid derived from the
metabolism of methionine, is an independent risk factor of
cardiovascular disease [1]. Hcy is an excitatory
amino acid and markedly enhances the vulnerability of neuronal cells to
excitotoxic and oxidative injury in vitro and in vivo [2]. However, the mechanisms underlying the
neurological damage characteristic of chronic hyperhomocysteinemia are
still poorly understood. It can be neurotoxic by promoting thrombosis
and inducing the formation of free oxygen radicals that could cause
brain damage [3]. Furthermore, it can activate the
N-methyl-D-aspartate (NMDA) glutamate receptors to induce its
neurotoxicity [4]. It has been suggested that Hcy
may promote the production of hydroxyl radicals, which initiates lipid
peroxidation, through Hcy autooxidation and thiolactone formation [5, 6]. Moreover, Hcy has the
ability to inhibit the expression of antioxidant enzymes such as
glutathione peroxidase (GSH-Px), which might potentiate the toxic
effects of reactive oxygen species [7].
Recently Maler et al. [8] have shown that Hcy induces cell death of astrocytes in vitro. Astrocytes are known to play an important role in survival of neurons in the brain and they have been implicated in the regulation of ionic environments, which are required for proper physiological function of neurons [9]. Both chemical and mechanical insults to the brain stimulate astrocyte proliferation with hypertrophy and the formation of glial filaments [10]. This phenomenon is called reactive gliosis. It is characterized by an over expression of glial fibrillary acidic protein (GFAP). GFAP, the major protein of the intermediate filaments of astroglial cells, is the most commonly used marker to examine the distribution of astrocytes and the hypertrophy of astrocytes in response to neural degeneration or injury [11, 12]. It has also been suggested that GFAP is essential for the formation of stable astrocytic processes, and this may be critical for morphogenesis of the central nervous system [13].
S100B is also an astroglial protein that has growth factor properties and several functions including interaction with different cytoskeletal proteins to induce cytoskeletal stabilization. S100B is an acidic Ca2+ binding protein that exerts paracrine trophic effects on several neuronal populations [14, 15]. S100B elevates neuronal cytoplasmic free calcium levels, stimulates neurite outgrowth, and promotes neuronal survival. In addition to the over expression of GFAP, astrocytes express high levels of S-100B protein in response to neuronal damage [14]. It can be neurotrophic and antiapoptotic in low concentration but can be neurotoxic in high concentration. Assay of S100B protein and neuron specific enolase can provide qualitative information about extent of brain injury and are sensitive marker of brain damage after stroke and cerebral hypoxia [16].
Recently we found that melatonin has the ability to prevent reactive gliosis both in brain [17] and retina [18], possibly by inhibiting oxidative stress. Numerous studies have demonstrated that melatonin has antioxidant properties that can scavenge oxygen and nitrogen derived radical species [19, 20]. In the present study, we sought to extend the current information on antioxidant effects of melatonin as well as to determine neuronal and glial markers in different brain areas of rats with hyperhomocysteinemia.
MATERIALS AND METHODS
Animals. Male Wistar albino rats weighing 180-220 g were housed in a room at constant temperature of 22 ± 2°C, with 12-h light/dark cycles. They were housed four per cage with free access to food and water. The rats were randomly divided into three groups at the start of the experiment. The first group of rats was assigned as control (n = 10), second group (Hcy group; n = 15) received daily methionine (1 g/kg body weight) dissolved in drinking water [21], and third group (Hcy + Mel group; n = 15) received daily same doses of methionine plus 10 mg/kg melatonin (Mel) (intraperitoneally) for a period of eight weeks.
Sample collection. All rats were fasted overnight and then sacrificed by decapitation. Blood samples were collected and plasma was separated and then stored at -20°C until analysis. Total Hcy levels were determined in plasma with an enzyme immunoassay kit (Axis-Shield AS, Norway). The brain was removed and the hippocampus and frontal cortex were dissected for the biochemical studies. Samples were kept at -70°C until the measurements were performed. The brain areas were taken as follows. The cortex was dissected after the brain was split in the mid-sagittal plane. Following removal of the frontal cortex, the hippocampus was dissected from the undersurface of the corpus callosum. All protocols described were reviewed and approved by the Local Institutional Committee for the Ethical Use of Animals.
Immunoblotting. Tissue samples were homogenized in 10 mM Tris-HCl (pH 7.4), 0.1 mM NaCl, 0.2 mM phenylmethylsulfonyl fluoride, 5 mM EDTA, 2 mM beta-mercaptoethanol, and 1% Triton X-100 containing proteinase inhibitors and centrifuged at 40,000g for 60 min at 4°C. The supernatants containing the tissue extract were collected and their protein concentration measured using a protein assay kit (Sigma, USA).
Samples and molecular weight standards were analyzed by SDS-polyacrylamide gradient gel electrophoresis. Separated proteins were electroblotted onto nitrocellulose membrane (Schleicher & Schuell Inc., USA). Membranes were blocked by incubation with 1% bovine serum albumin. The blots were then incubated with primary antibodies (Santa Cruz Biotechnology, Inc., USA) at 1 : 2000 (anti-GFAP) and 1 : 1000 (anti-S100B) dilutions overnight at 4°C. The membranes were washed extensively in TBS-Tween (25 mM Tris-HCl, 0.2 mM NaCl, 0.1% Tween 20) and then incubated for 1 h at room temperature with a secondary antibody, a goat anti-rabbit Ig peroxidase conjugate (Sigma, UK). Specific binding was detected using diaminobenzidine and H2O2 as substrates. The relative amount of immunoreactive bands on Western blots was quantified in arbitrary units by scanning blots using a computerized software program (LabWorks 4.0; UVP, Inc., Cambridge, UK).
Tissue lipid peroxidation (LPO) as malondialdehyde (MDA) + 4-hydroxyalkenals was determined using a LPO-586 kit (Oxis, Int. Inc. OR, USA); the method is based on a reaction of N-methyl-2-phenylindole with MDA and 4-hydroxyalkenals at 45°C. One molecule of MDA or 4-hydroxyalkenals reacts with two molecules of N-methyl-2-phenylindole to yield a stable chromophore with maximal absorbance at 568 nm. GSH-Px activity was determined according to the method of Lawrence and Burk [22]. The reaction mixture consisted of 50 mM Tris-HCl buffer (pH 7.0), 1 mM glutathione, 1 unit glutathione reductase, and 1.5 mM cumene hydroperoxide (Sigma). The oxidation of NADPH was monitored at 340 nm and 37°C. The unit of enzymatic activity was expressed in nanomoles of NADPH oxidized per min.
RESULTS
Plasma Hcy concentrations of the rats given methionine were significantly higher than those of the control rats (p < 0.001) whereas melatonin administration reduced its level significantly (table; p < 0.05). Analysis of variance demonstrated that the levels of LPO were significantly higher in hippocampus and frontal cortex of Hcy rats compared with controls (p < 0.01) and were reduced by melatonin treatment in both brain parts (p < 0.05). The activity of GSH-Px in hippocampus and frontal cortex of Hcy rats was significantly lower compared with control rats (p < 0.01 and p < 0.05, respectively). Melatonin treatment resulted in a significant alteration in the brain levels of this enzyme towards its control value (p < 0.01).
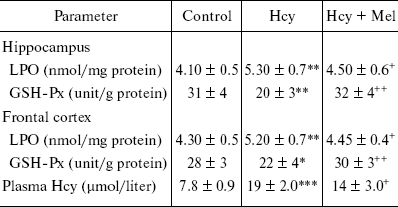
*p < 0.05, **p < 0.01, ***p < 0.001
vs. control values; +p < 0.05, ++p
< 0.01 vs. Hcy group.
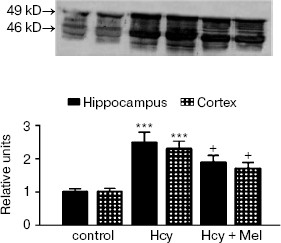
Western blot analysis revealed an increase in GFAP content in rats treated with methionine compared to the controls (p < 0.001; Fig. 1). Co-administration of melatonin with methionine significantly reduced GFAP contents both in hippocampus and cortex (p < 0.05). Similarly, Hcy caused significant elevation in S100B protein levels in frontal cortex compared to the control values (p < 0.01). Administration of melatonin along with methionine significantly inhibited this increase in S100B protein contents (p < 0.01; Fig. 2).
Fig. 1. GFAP levels in rat hippocampus and frontal cortex of control, Hcy, and (Hcy + Mel) groups. Representative portion of Western blots probed with anti-GFAP antibodies. In addition to the normal 49 kD GFAP band, several bands were seen at roughly 44-48 kD molecular weight (15 µg of total protein was applied per lane; computerized densitometry was performed and mean ± SD for all groups are shown in lower panel). Significant elevation in GFAP content was observed in hippocampus and cortex of Hcy group compared to the control values (p < 0.001). Daily administration of melatonin reduced GFAP levels (+p < 0.05). Hcy, homocysteine; Mel, melatonin.
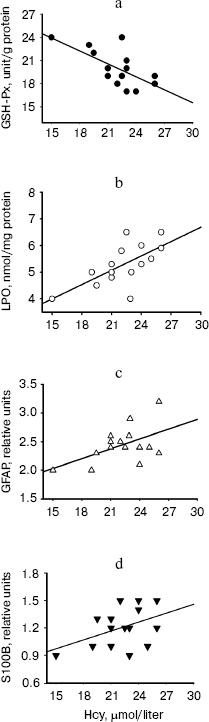
Correlations of the levels of plasma Hcy with the values of hippocampal LPO, GFAP, S100B, and GSH-Px activity in the Hcy group are shown in Fig. 3. A significant negative correlation existed between the levels of Hcy and the activity of GSH-Px (r = -0.631, p = 0.01), whereas significant positive correlations existed between the levels of Hcy and LPO (r = 0.670, p = 0.006) and between the levels of Hcy and GFAP (r = 0.050, p = 0.04). There was a positive but insignificant correlation between Hcy levels and S100B contents in hippocampus of hyperhomocysteinemic rats (r = 0.440, p = 0.09).Fig. 2. Expression of S100B protein in hippocampus and frontal cortex from control, Hcy, and (Hcy + Mel) groups. (A total of 10 µg protein was applied per lane; the results of densitometric analysis were presented as mean ± SD in the lower panel.) Hyperhomocysteinemia significantly elevated the S100B protein levels in cortex (**p < 0.01) while no significant effect was found in hippocampus (p > 0.05) compared to the control group. Treatment with melatonin significantly prevents the increase in S100B content induced by hyperhomocysteinemia (++p < 0.01). Hcy, homocysteine; Mel, melatonin.
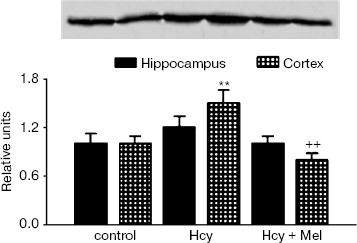
Fig. 3. Analysis of the correlation between plasma Hcy levels and activity of GSH-Px (a), levels of LPO (b), contents of GFAP (c), and levels of S100B (d) in hippocampus of Hcy rats. Regression analysis revealed that there was a significant negative correlations between plasma Hcy levels and GSH-Px activity (r = -0.631, p = 0.01) whereas significant positive correlations existed between the levels of Hcy and LPO (r = 0.670, p = 0.006) and between the levels of Hcy and GFAP (r = 0.550, p = 0.04) in Hcy rats. The correlation between Hcy levels and S100B contents (d) of hippocampus was not significant (r = 0.440, p = 0.09).
DISCUSSION
In the present work, we suggest that chronic methionine supplementation significantly increases the plasma Hcy levels and melatonin at least partly prevents this elevation. In our previous studies we first suggested that pinealectomy confers an increase in Hcy level and melatonin administration normalizes its concentration [23]. In addition to its protective effects against oxidative stress, melatonin at least in pharmacological doses may protect tissues by reducing plasma Hcy levels [24].
Hcy is a neurotoxic amino acid that accumulates in several neurological disorders [25]. The pathogenic mechanism of Hcy on the nervous system is not fully understood. Hcy may produce its effects by increasing smooth muscle proliferation, decreasing endothelial DNA synthesis, inhibiting anticoagulant factors, increasing platelet aggregation, and impairing endothelial-dependent arterial vasodilatation [26]. Furthermore, it has been shown that neurotoxic effects of Hcy are involved in NMDA stimulation, nitric oxide synthase activation, and associated free radical formation [27]. Hcy is able to generate reactive toxic oxygen species such as superoxide, hydroxyl radicals, and hydrogen peroxide [28, 29] and to initiate lipid peroxidation. Consistent with these reports, in the present study we show that Hcy stimulated lipid peroxidation. This was supported by the previous studies, which report that Hcy can increase malondialdehyde + 4-hydroxyalkenals levels both in vitro [30] and in vivo [31] and administration of melatonin reduces this lipid peroxidation product. Hcy is readily oxidized as a consequence of auto-oxidation and during oxidation of the sulfhydryl groups oxygen derived free radicals are formed, which promotes lipid peroxidation [28, 29]. We also show here that Hcy significantly reduced GSH-Px activity in the brain regions. Hcy can increase oxidative stress by inhibiting expression and function of antioxidant enzymes such as superoxide dismutase and GSH-Px [7, 32]. Thus, in addition to generating reactive oxygen and reactive nitrogen species during auto-oxidation, Hcy impairs crucial enzymes responsible for the inactivation or reduction of these reactive species. In agreement with previous studies [30, 31], herein we found that melatonin significantly reversed the oxidative stress seen in Hcy rats and normalized their antioxidant enzyme activity in different areas of the brain.
The major findings of this study are that Hcy stimulates overproduction of glial markers (GFAP and S100B), and treatment with melatonin prevents Hcy-induced augmented production of GFAP and S100B. These data suggest that Hcy induces glial reactivity, a universal cellular reaction to central nervous system damage. During reactive gliosis, glial cells secrete neurotoxic substances, such as excitatory amino acids, pro-inflammatory cytokines or free radicals, which kill neurons and have been proposed to be the major causes of neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease [33]. Reactive glial cells and the factors they produce actively contribute to disease progression and pathology [34]. Increases in S100B levels induce an elevation in intra-neuronal free calcium concentrations, which are neurotoxic. Thereby Hcy-induced reactive gliosis may alleviate oxidant injury to neuronal components and cell death. The mechanism by which Hcy induces glial reactivity is not clear. However, Hcy-induced oxidative reaction accompanied by the generation of reactive oxygen species, such as hydrogen peroxide or superoxide anion, and reduction in the activity or expression level of GSH-Px might induce reactive gliosis. Glial cells are situated between vessels and neurons to protect neurons from insults. Thus, glial cells are the targets and mediators of many insults to the nervous system. These results support a model in which cortical astrocytes have been found to be affected by Hcy cytotoxicity in a time- and dose-dependent manner [8].
In the present study we found that melatonin prevents reactive gliosis induced by Hcy. To our knowledge this is the first report to show that melatonin inhibits Hcy-induced reactive gliosis. Recently we have reported that increase in GFAP levels was found to be related with elevated oxidative stress [17, 35], and administration of melatonin significantly reduced the reactive gliosis both in brain and in retina [17, 18]. Furthermore, we also found that functional pinealectomy caused an elevation in total and degraded levels of GFAP in different brain parts and these elevations were reversed by the melatonin treatment [36].
Our experimental data show that Hcy can sensitize glial cells the mechanism which might contribute to the pathogenesis of neurodegenerative disorders and further suggest that melatonin can be involved in protecting against the toxicity of Hcy by inhibiting of reactive oxygen species generation and stabilizing glial cell activity.
This work was supported by the Firat University Research Foundation (FUBAP, project No. 1056).
REFERENCES
1.Finkelstein, J. D. (2000) Semin. Thromb.
Hemost., 26, 219-225.
2.Ho, P. I., Ortiz, D., Rogers, E., and Shea, T. B.
(2002) J. Neurosci. Res., 70, 694-702.
3.Hankey, G. J., and Eikelboom, J. W. (1999)
Lancet, 354, 407-413.
4.Lipton, S. A., Kim, W. K., Choi, Y. B., Kumar, S.,
D'Emilia, D. M., Rayudu, P. V., Arnelle, D. R., and Stamler, J. S.
(1997) Proc. Natl. Acad. Sci. USA, 94, 5923-5928.
5.Stamler, J. S., Osborne, J. A., Jaraki, O.,
Rabbani, L. E., Mullins, M., Singel, D., and Loscalzo, J. (1993) J.
Clin. Invest., 91, 308-318.
6.Heinecke, J. W. (1988) in Oxyradicals in
Molecular Biology and Pathology (Cerruti, P. A., Fridovich, I., and
McCord, J. M., eds.) Alan R. Liss, New York, pp. 443-457.
7.Upchurch, G. R., Jr., Welch, G. N., Fabian, A. J.,
Freedman, J. E., Johnson, J. L., Keaney, J. F., Jr., and Loscalzo, J.
(1997) J. Biol. Chem., 272, 17012-17017.
8.Maler, J. M., Seifert, W., Huther, G., Wiltfang,
J., Ruther, E., Kornhuber, J., and Bleich, S. (2003) Neurosci.
Lett., 347, 85-88.
9.Moonen, G., Rogister, B., Leprince, P., Rigo, J.
M., Delree, P., Lefebvre, P. P., and Schoenen, J. (1990) Progr.
Brain Res., 86, 63-73.
10.Reier, P. J., and Houle, J. D. (1988) Adv.
Neurol., 47, 87-138.
11.O'Callaghan, J. P., Jensen, K. F., and Miller, D.
B. (1995) Neurochem. Int., 26, 115-124.
12.Baydas, G., Nedzvetskii, V. S., Tuzcu, M., Yasar,
A., and Kirichenko, S. V. (2003) Eur. J. Pharmacol., 462,
67-71.
13.Liedtke, W., Edekmann, W., Bieri, P. L., Chiu, F.
C., Cowan, N. J., Kucherlapati, R., and Raine, C. S. (1996)
Neuron, 17, 607-615.
14.Cerutti, S. M., and Chadi, G. (2000) Cell.
Biol. Int., 24, 35-49.
15.Kligman, D., and Marshak, D. R. (1985) Proc.
Natl. Acad. Sci. USA, 82, 7136-7139.
16.Persson, L., Hardemark, H. G., Gustafsson, J.,
Rundstrom, G., Mendel-Hartvig, I., Esscher, T., and Pahlman, S. (1987)
Stroke, 18, 911-918.
17.Baydas, G., Reiter, R. J., Yasar, A., Tuzcu, M.,
Akdemir, I., and Nedzvetskii, V. S. (2003) Free Rad. Biol. Med.,
35, 797-804.
18.Baydas, G., Tuzcu, M., Yasar, A., and Baydas, B.
(2004) Acta Diabetol., 41, 123-128.
19.Tan, D. X., Chen, L. D., Poeggeler, B.,
Manchester, L. C., and Reiter, R. J. (1993) Endocrine J.,
1, 57-60.
20.Zhang, H., Squadrito, G. L., and Pryor, W. A.
(1998) Biochem. Biophys. Res. Commun., 251, 83-87.
21.Bagi, Z., Cseko, C., Toth, E., and Koller, A.
(2003) Am. J. Physiol. Heart. Circ. Physiol., 285,
2277-2283.
22.Lawrence, R. A., and Burk, R. F. (1976)
Biochem. Biophys. Res. Commun., 71, 952-958.
23.Baydas, G., Gursu, M. F., Cikim, G., and Canatan,
H. (2002) J. Pineal Res., 32, 63-64.
24.Baydas, G., Yilmaz, O., Celik, S., Yasar, A., and
Gursu, M. F. (2002) Arch. Med. Res., 33, 515-519.
25.Kim, J. P., Koh, J. Y., and Choi, D. W. (1987)
Brain Res., 437, 103-110.
26.Reutens, S., and Sachdev, P. (2002) Int. J.
Geriatr. Psychiatry, 17, 859-864.
27.Jara-Prado, A., Ortega-Vazquez, A.,
Martinez-Ruano, L., Rios, C., and Santamaria, A. (2003) Neurotox.
Res., 5, 237-243.
28.Dayal, S., Arning, E., Bottiglieri, T., Boger, R.
H., Sigmund, C. D., Faraci, F. M., and Lentz, S. R. (2004)
Stroke, 35, 1957-1962.
29.Goth, L., and Vitai, M. (2003) Free Rad. Biol.
Med., 35, 882-888.
30.Osuna, C., Reiter, R. J., Garcia, J. J.,
Karbownik, M., Tan, D. X., Calvo, J. R., and Manchester, L. C. (2002)
Pharmacol. Toxicol., 90, 32-37.
31.Baydas, G., Kutlu, S., Naziroglu, M., Canpolat,
S., Sandal, S., Ozcan, M., and Kelestimur, H. (2003) J. Pineal
Res., 34, 36-39.
32.Yamamato, M., Hara, H., and Adachi, T. (2000)
FEBS Lett., 486, 159-162.
33.Bates, K. A., Fonte, J., Robertson, T. A.,
Martins, R. N., and Harvey, A. R. (2002) Neuroscience,
113, 785-796.
34.Banati, R. B., Gehrmann, J., Schubert, P., and
Kreutzberg, G. W. (1993) Glia, 7, 111-118.
35.Baydas, G., Reiter, R. J., Nedzvetskii, V. S.,
Yasar, A., Tuzcu, M., Ozveren, F., and Canatan, H. (2003) Toxicol.
Lett., 137, 169-174.
36.Baydas, G., Reiter, R. J., Nedzvetskii, V. S.,
Nerush, P. A., and Kirichenko, S. V. (2002) J. Pineal Res.,
33, 134-139.