Isolation of Active Yeast Telomerase Protein Est3p and Investigation of Its Dimerization in vitro
Yu. S. Sharanov1,2*, E. M. Smekalova1, M. I. Zvereva1, and O. A. Dontsova1
1Chemical Faculty, Lomonosov Moscow State University, 119992 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-3181; E-mail: sharanov@genebee.msu.ru
* To whom correspondence should be addressed.
Received March 1, 2007; Revision received April 8, 2007
In this study we proposed a method for isolation of Est3p modified with various affinity tags, which is applicable for structural and functional studies, and investigated homo- and heterodimer formation with various recombinant forms of Est3p.
KEY WORDS: yeast telomerase, protein Est3p, dimerizationDOI: 10.1134/S0006297907070036
Abbreviations: GST) glutathione-S-transferase; IPTG) isopropyl-beta-D-thiogalactopyranoside; Ni-NTA-agarose) Ni-nitriloacetate agarose.
Telomeres are special DNA-protein complexes located at ends of linear
eukaryotic chromosomes. They maintain stability of chromosomes by
protecting them against degradation and fusion; telomeres are also
involved in processes of cell aging (senescence); they play an
important role in formation of nuclear architecture and regulation of
transcription of neighboring genes [1]. Telomeric
DNA consists of repeated nucleotide sequences and terminates with a
3´-end single-stranded overhang.
Each cell division results in under-replication and telomere shortening. At a certain critical length of telomeres, cells cannot divide and enter senescence and finally die. However, in many cells with unlimited division potential (e.g. single-celled organisms [2, 3] and germinal and cancer cells [4-6]) there is a mechanism responsible for telomere length maintenance. It is based on synthesis of telomeric DNA by a special enzyme called telomerase [7]. Telomerase is a ribonucleoprotein complex, which consists of an RNA molecule, reverse transcriptase, and some other protein components [8].
The single-celled eukaryotic yeast Saccharomyces cerevisiae is a convenient model for telomerase studies. Telomerase complex of this yeast consists of RNA TLC1 [9], which contains a template for synthesis of telomeric repeats, and at least three protein components called Est1p, Est2p, and Est3p [10]. In the complex with TLC1 reverse transcriptase, Est2p can elongate a telomere-like DNA oligonucleotide primer in vitro [5, 11]. Est1p is responsible for attachment of telomerase to the telomere [12-15] and also for enzyme activation in S-phase of the cell cycle [16]. It is suggested that Est3p, a specific component of the S. cerevisiae telomerase complex [17], is required for maintenance of telomere length, because deletion of the gene encoding this protein causes a lethal phenotype of progressive telomere shortening [11]. However, the structure and mechanism of Est3p functioning in the telomerase complex remain basically unknown. This may be partially explained in the absence of a reliable preparative method for isolation of Est3p.
In S. cerevisiae cells, Est3p exists as a truncated and a full-sized protein. The full-sized form is synthesized by means of an unusual mechanism of open reading frame shift, and only this form is required for telomerase functioning in vivo [18]. Overexpression of Est3p in S. cerevisiae may compensate the effects of mutations in the N-terminal regions of Est2; this suggests putative interaction of these two proteins in the telomerase complex [19]. We have recently found that Est3p can bind DNA and RNA and stimulates dissociation of RNA/DNA heteroduplexes [20]. Earlier it was also demonstrated that yeast telomerase contains at least two active sites and may function as a dimer or an oligomer [21]. Moreover, in the case of human telomerase, such oligomerization is functionally important, and it may occur due to RNA-RNA and RNA-protein interactions [22, 23]. When this study was nearly finished, it had been reported that Est3p is possibly involved in dimerization of yeast telomerase [24]. Using gel-filtration, the authors detected formation of Est3p complex and its molecular mass was closed to that of Est3p dimer.
In this paper, we describe a method for isolation of active Est3p suitable for structural and functional analysis and summarize results of studies of its dimerization ability in vitro.
MATERIALS AND METHODS
Materials. The enzymes T4 DNA polymerase, T4 DNA ligase, Taq DNA polymerase, and restriction endonucleases (SphI, SmaI, NheI, HindIII, NcoI, EagI) were produced by MBI Fermentas (Lithuania). Plasmid expression vectors pGEX-4T-1, pQE32, and pET33b+ were produced by Amersham Pharmacia Biotech (USA), Qiagen (Germany), and Novagen (USA), respectively. For transformation we have used E. coli cells of the following strains: JM109, M15[pREP4], and BL21(DE3). Isopropyl-beta-D-thiogalactopyranoside (IPTG) was obtained from Research Organics Inc. (USA).
Plasmid construction (preparation of pGEX_EST3, pQE32_EST3, pET33b+_6His-EST3, and pET33b+_EST3-6His constructs). Construction of pGEX_EST3 has been exhaustively described in [20]. The plasmid pGEX_EST3 was used for construction of pQE32_EST3, pET33b+_6His-EST3, and pET33b+_EST3-6His; it served as the source of the EST3 gene with open reading frameshift for expression yielding expression of full-sized Est3p only. In the case of pQE32_EST3 construct for gene amplification in polymerase chain reaction (PCR), we used the following oligonucleotides as forward and reverse primers, respectively: E3FwdSphI, 5´-GGGCATGCCGAAAGTAATTCTGGAGTCTC-3´; E3RevSmaI, 5´-TCGACCCGGGTCATAAATATTTATATAC-3´. The ends of these primers have recognition sites for restriction endonucleases SphI and SmaI (underlined). The PCR product was purified through agarose gel and treated with restriction endonucleases SphI and SmaI and inserted by ligation into expression vector pQE32 pretreated with the same restriction endonucleases.
The constructs pET33b+_6His-EST3 and pET33b+_EST3-6His were obtained by insertion of corresponding DNA fragments into the pET33b+ vector. These fragments were obtained using PCR and the following primers: for pET33b+_6His-EST3, 5´-AAAAAAGCTAGCATGCCGAAAGTAATT-3´ (forward E3FwdNheI) and 5´-GGTCGAAAGCTTTCATAAATA-3´ (reverse E3RevHindIII); for pET33b+_EST3-6His, 5´-CCGAAAGTAATTCTGGA-3´ (forward, E3Fwd) and 5´-TTTCGGCCGTCAATGATGATGATGATGATGTAAATATTTATATACAAATGGGA-3´ (reversed E3Rev6HisEagI). All PCR products were purified through agarose gel. In the case of the pET33b+_6His-EST3 construct, the vector pET33b+ and corresponding PCR product were pretreated with restriction endonucleases NheI and HindIII before ligation. For construction of pET33b+_EST3-6His, the vector pET33b+ was sequentially treated with restriction endonuclease NcoI, T4 DNA polymerase, and restriction endonuclease EagI and then ligated with the PCR product pretreated with restriction endonuclease EagI. In all cases, primary structure of DNA containing EST3 gene was determined by Sanger sequencing [25]. DNA sequencing was carried out using kits ABI PRISM® BigDye™ Terminator v.3.1 followed by subsequent analysis of reaction products using the DNA sequencer ABI PRISM 3100-Avant.
Isolation of Est3p. The recombinant Est3p protein containing glutathione-S-transferase (GST) as the N-terminal affinity tag was purified using the protocol described in [20]. In the case of Est3p protein containing N-terminal six histidine residues (6His), overnight cultures of E. coli M15[pREP4] transformed with pQE32_EST3 plasmid and E. coli BL21(DE3) transformed with the pET33b+_6His-EST3 plasmid overnight in the LB medium [26] with either ampicillin (0.1 mg/ml) or kanamycin (0.05 mg/ml) for pQE32_EST3 and pET33b+_6His-EST3, respectively, were diluted 100-fold with the same medium containing the same antibiotics and grown at 37°C under intensive aeration up to 0.5 OD600. Expression was induced by IPTG (0.05, 0.1, 0.5, and 1 mM). After incubation at 19°C for 12-16 h under stirring, cells were collected by centrifugation at 5000g for 10 min at 4°C, washed with water, resuspended in buffer A containing 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 40 mM NH4Cl, 10 mM MgCl2, pH 7.3, and sonicated using an ultrasound disintegrator. Cell debris was removed by centrifugation at 15,000g for 20 min at 4°C. The resulting supernatant was mixed with Ni-NTA-agarose, and the mixture was incubated at 4°C for 60 min under mild stirring. The affinity sorbent was separated by centrifugation at 10,000g for 5 min with subsequent supernatant decanting. Ni-NTA-agarose with the immobilized protein was washed twice in buffer A supplemented with 50 mM imidazole. Protein was eluted with buffer containing 50 mM NaH2PO4, 300 mM NaCl, pH 7.6, 300 mM imidazole. Protein preparations were analyzed by SDS-PAGE [27].
Est3p protein with C-terminal tag of histidine residues was isolated using a similar method. Escherichia coli cells, strain BL21(DE3), were grown in LB medium with kanamycin (0.05 mg/ml). Expression was induced by 0.05 mM IPTG. Ni-NTA-agarose with the immobilized protein was additionally washed two times with 50 mM NaH2PO4, 1 M NaCl, pH 7.6, 50 mM imidazole and then two times with 50 mM NaH2PO4, 300 mM NaCl, pH 7.6, 80 mM imidazole. After elution, protein preparations were stored in the presence of 30% glycerol.
Concentrations of all proteins were evaluated spectrophotometrically [28].
Tests for dimer formation. A 100-µl aliquot of recombinant Est3p protein (10 µM) with C-terminal tag of six histidine residues (Est3p-6His) was mixed with 1 ml of Est3p with N-terminal GST tag (GST-Est3p) in buffer containing 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3. Three concentrations of GST-Est3p were used to obtain molar ratios Est3p-6His/GST-Est3p of 1 : 0.1, 1 : 1, and 1 : 10, respectively. In control samples solution of Est3p-6His was mixed with 1 ml of protein free buffer or 1 ml of buffer containing 10 µM GST. Resultant mixtures were incubated for 30 min at 30°C, and Est3p-6His was then isolated using the affinity sorbent Ni-NTA-agarose as described above. Protein fractions were analyzed by electrophoresis in 15% polyacrylamide gel [27] containing 0.1% SDS and in 12% polyacrylamide gel containing 0.01% SDS. In the latter case the upper electrode buffer contained 0.01% SDS and lower electrode buffer did not contain SDS.
RESULTS AND DISCUSSION
Est3p with various affinity tags at N- and C-ends. Among approaches that can be employed for study of putative Est3p dimerization, one can use affinity chromatography applicable for a mixture of derivatives of this protein that differ by affinity tags (or lack thereof) at N- and C-ends. Co-purification of one Est3p derivative with another would indicate the existence not only of monomeric forms of this protein. We have chosen a fragment of glutathione-S-transferase (GST) as the affinity tag, which has been placed at the N-terminus of Est3p. Yeast proteins are often poorly soluble during expression of corresponding yeast genes in E. coli. The presence of GST fragment increases solubility of a recombinant protein in extracts of E. coli cells [20]. For preparation of such recombinant protein, we placed EST3 gene into the expression vector pGEX-4T-1 so that GST reading frame coincided with Est3p reading frame; these encoding sequences were separated by the sequence encoding signal peptide recognizable by specific protease.
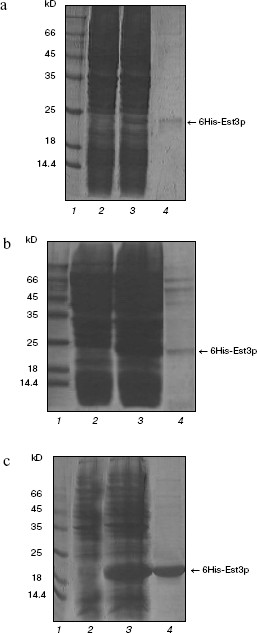
Treatment of resultant recombinant protein with protease yields the protein lacking the affinity tag. Indeed, GST-modified protein was readily soluble and was easily purified by affinity chromatography on glutathione-Sepharose; however, removal of the affinity tag was accompanied by significant loss of Est3p in solution due to precipitation of this protein [20]. Thus, we could not use unmodified Est3p in this study. The Est3p derivative with hexahistidine tag (6His) was used as the second component. For preparation of such derivative, we used the plasmid vectors pQE32 and pET33b+; using these vectors, it is possible to regulate expression level of the recombinant protein in E. coli cells. This could be an important precondition for preparation of the soluble protein. Escherichia coli cells, transformed with pQE32_EST3 or pET33b+_6His-EST3, produced Est3p containing six histidine residues at the N-end. As expected, expression level was higher in the case of pET33b+_6His-EST3 compared with pQE32_EST3. However, in both cases, the major proportion of the recombinant protein was located in inclusion bodies, and after isolation only trace amounts of 6His-Est3p were detected (Fig. 1, a and b).
We could not overcome these difficulties either by varying concentrations of IPTG (expression inductor) or synthesis of the recombinant protein at 16°C. So we developed a new construct, pET33b+_EST3-6His, which would yield Est3p with the hexahistidine tag located at C-end rather than at N-end. Using the plasmid pET33b+_EST3-6His, the level of EST3 gene expression in E. coli cells (Fig. 1c) and the amount of Est3p in soluble fraction and after isolation by affinity chromatography on Ni-NTA-agarose was significantly higher than in the case of pQE32_EST3p and ET33b+_6His-EST3 (Fig. 1). Using this system, we were able to get Est3p solution with rather high protein concentration (0.5-0.6 mg/ml). It is possible that transition of 6His from N- onto C-end of Est3p improved correct folding of this protein in E. coli and increased its solubility. Using our own earlier developed functional tests in vitro [20], we found that the presence of the hexahistidine tag at C-end did not influence biochemical properties of the resulting protein. Est3p-6His as well as protein lacking affinity tags or protein containing the GST tag at N-end [20] bound DNA- and RNA-oligonucleotides and stimulated dissociation of DNA/RNA heteroduplexes. Thus, we have developed a method for rapid and convenient isolation of rather high concentration of active Est3p containing the hexahistidine tag at the C-end.
Est3p dimerization. For several years, there has been active discussion about a model of telomerase functioning as dimer or even oligomer in the process of telomere elongation [29, 30]. Recently a possibility of Est3p dimerization has also been proposed [24]. Having Est3-6His and GST-Est3p available, we have also investigated putative ability of Est3p to form dimers and oligomers. First of all, we have analyzed the Est3-6His preparation by PAGE under non-denaturing conditions in the presence of 0.01% SDS. Usually such low detergent concentration in gel and buffers for electrophoresis does not cause dissociation of protein complexes into subunits, but reduces duration of the electrophoretic procedure and improves the quality of images after gel staining with Coomassie brilliant blue R-250 due to small change in charge of protein molecules. The Est3-6His preparation applied onto such polyacrylamide gel (Fig. 2a, lane 1) was characterized by the presence of a major band corresponding to Est3p monomer and a band of protein with larger molecular mass. The latter band was not observed when the sample was heated in a buffer with 8 M urea before application onto the gel (Fig. 2a, lane 2). This indicated oligomerization ability of Est3p protein. Intensity of a band corresponding to oligomer was comparable to that corresponding to the monomeric protein; this means that in a given protein preparation about 50% of protein molecules form an oligomer.Fig. 1. Isolation of the hexahistidine tagged derivatives of Est3p protein. Total protein fractions of E. coli cells transformed by plasmids pQE32_EST3 (a), pET33b+_6His-EST3 (b), and pET33b+_EST3-6His (c) and grown in the absence (a-c, lanes 2) and in the presence of expression inducer (a-c, lanes 3) were analyzed by PAGE in 15% polyacrylamide gel in the presence of 0.1% SDS. Lanes: 4) eluates from Ni-NTA-agarose; 1) molecular weight markers.
For independent validation of oligomerization, we mixed two protein preparations, Est3-6His and GST-Est3p, and then isolated the hexahistidine tag-containing protein by affinity chromatography on Ni-NTA-agarose. Analysis of resultant protein fraction by SDS-PAGE (Fig. 2b) revealed the presence of both Est3-6His and GST-Est3p. This implies interaction between these two proteins. In control samples, GST added instead of GST-Est3p was not co-isolated with Est3-6His during affinity chromatography (Fig. 2b, lane 5). The amount of co-isolated GST-Est3p did not increase with the increase of molar ratio Est3p-6His/GST-Est3p from 1 : 1 to 1 : 10 and did not exceed amount of Est3p-6His (Fig. 2b, lanes 3 and 4). This indicates that the oligomeric form is clearly a dimer.Fig. 2. Est3p dimerization in vitro. a) Analysis of Est3p-6His (1) and the same preparation preheated at 95°C in buffer containing 8 M urea (2) by electrophoresis in 12% polyacrylamide gel in the presence of 0.01% SDS. b) Analysis of protein fractions obtained by affinity chromatography of a mixture of proteins Est3p-6His and GST-Est3p (1 : 0.1) (2), Est3p-6His and GST-Est3p (1 : 1) (3), Est3p-6His and GST-Est3p (1 : 10) (4), and Est3p-6His and GST (1 : 10) (5) by electrophoresis in 15% polyacrylamide gel in the presence of 0.1% SDS. Lane 1: affinity chromatography of Est3p-6His on Ni-NTA-agarose in the absence of other proteins.

Using PAGE under non-denaturing conditions, we did not detect heterodimer formation between Est3-6His and GST-Est3p (data not shown). This suggest that interaction between GST-Est3p and Est3-6His is much weaker than in the case of interaction between two molecules of Est3-6His, possibly due to the presence in GST-Est3p of the large affinity tag, GST.
Thus, in this work we have developed an effective method for single-step isolation of the active component of S. cerevisiae telomerase complex, protein Est3p, and demonstrated its homodimerization ability.
This work was supported by the Russian Foundation for Basic Research (grant No. 05-04-49715), HFSP RGP 0032/005, and also by grants for Leading Scientific Schools (NSh-1877 and NSh RI-112/001/358).
REFERENCES
1.Blackburn, E. H. (1991) Nature, 350,
569-572.
2.Blackburn, E. H. (1992) Annu. Rev. Biochem.,
61, 113-129.
3.Cohn, M., and Blackburn, E. H. (1995)
Science, 269, 396-400.
4.Harley, C. B., Futcher, A. B., and Greider, C. W.
(1990) Nature, 345, 458-460.
5.Counter, C. M., Avilion, A. A., LeFeuvre, C. E.,
Stewart, N. G., Greider, C. W., Harkey, C. B., and Bacchetti, S. (1992)
EMBO J., 11, 1921.
6.Vaziri, H., Dragowska, W., Allsopp, R. C., Thomas,
T. E., Harley, C. B., and Landsdorp, P. M. (1994) Proc. Natl. Acad.
Sci. USA, 91, 9857-9860.
7.Greider, C. W., and Blackburn, E. H. (1985)
Cell, 43, 405-413.
8.Collins, K. (2000) Curr. Opin. Cell Biol.,
12, 378-383.
9.Singer, M. S., and Gottschling, D. E. (1994)
Science, 266, 404-409.
10.Lendvay, T. S., Morris, D. K., Sah, J.,
Balasubramanian, B., and Lundblad, V. (1996) Genetics,
144, 1399-1412.
11.Lingner, J., Cech, T. R., Hughes, T. R., and
Lundblad, V. (1997) Proc. Natl. Acad. Sci. USA, 94,
11190-11195.
12.Evans, S. K., and Lundblad, V. (2002)
Genetics, 162, 1101-1115.
13.Evans, S. K., and Lundblad, V. (1999)
Science, 286, 117-120.
14.Qi, H., and Zakian, V. A. (2000) Genes
Dev., 14, 1777-1788.
15.Pennock, E., Buckley, K., and Lundblad, V. (2001)
Cell, 104, 387-396.
16.Taggart, A. K., Teng, S. C., and Zakian, V. A.
(2002) Science, 297, 1023-1026.
17.Hughes, T. R., Evans, S. R., Weilbaecher, R. G.,
and Lundbland, V. (2000) Curr. Biol., 10, 809-812.
18.Morris, D. K., and Lundbland, V. (1997) Curr.
Biol., 7, 969-976.
19.Friedman, K. L., Heit, J. J., Long, D. M., and
Cech, T. R. (2003) Mol. Biol. Cell., 14, 1-13.
20.Sharanov, Yu. S., Zvereva, M. I., and Dontsova,
O. A. (2006) FEBS Lett., 580, 4683-4690.
21.Prescott, J., and Blackburn, E. H. (1997)
Genes Dev., 11, 2790-2800.
22.Beattie, T. L., Zhou, W., Robinson, M. O., and
Harrington, L. (2001) Mol. Cell Biol., 21, 6151-6160.
23.Moriarty, T. J., Huard, S., Dupuis, S., and
Autexier, C. (2002) Mol. Cell Biol., 22, 1253-1265.
24.Cui-Ping Yang, Yong-Bin Chen, Fei-Long Meng, and
Jin-Qiu Zhou (2006) Nucleic Acids Res., 34, 407-416.
25.Sanger, F., Nicklen, S., and Coulson, A. R.
(1977) Proc. Natl. Acad. Sci. USA, 74, 5463-5467.
26.Sambrook, J., Fritch, E. F., and Maniatis, T.
(1989) in Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor, N. Y.
27.Laemmli, U. K. (1970) Nature, 227,
680-685.
28.Walker, J. M. (1996) in The Protein Protocols
Handbook, Humana Press Inc., New Jersey, pp. 3-6.
29.Wenz, C., Enenkel, B., Amacker, M., Kelleher, C.,
Damm, K., and Lingner, J. (2001) EMBO J., 20,
3526-3534.
30.Wang, L., Dean, S. R., and Shippen, D. E. (2002)
Nucleic Acids Res., 30, 4032-4039.