REVIEW: Nitrate Reductases: Structure, Functions, and Effect of Stress Factors
E. V. Morozkina* and R. A. Zvyagilskaya
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: Chicelena@yandex.ru* To whom correspondence should be addressed.
Received July 14, 2007
Structural and functional peculiarities of four types of nitrate reductases are considered: assimilatory nitrate reductase of eukaryotes, as well as cytoplasmic assimilatory, membrane-bound respiratory, and periplasmic dissimilatory bacterial nitrate reductases. Arguments are presented showing that eukaryotic organisms are capable of nitrate dissimilation. Data concerning new classes of extremophil nitrate reductases, whose active center does not contain molybdocofactor, are summarized.
KEY WORDS: nitrate reductase, nitrification, denitrification, molybdenum, molybdenum cofactor, molybdopterin-guanine dinucleotides, stressDOI: 10.1134/S0006297907100124
Abbreviations: DMSO) dimethyl sulfoxide; Euk-NR) eukaryotic assimilatory NRase; MGD) molybdopterin-guanine dinucleotide; Mo) molybdenum; Mo-co) molybdenum cofactor; Nap) periplasmic dissimilatory NRase; Nar) membrane-bound respiratory NRase; Nas) cytoplasmic assimilatory NRase; NRase) nitrate reductase; TMAO) trimethylamine N-oxide.
Nitrogen is a constituent of main macromolecules--proteins and nucleic
acids--and is one of most vitally important elements on Earth. It is
incorporated into compounds in different degrees of oxidation (from
N5+ to N3-), the interconversions of which define
the global biochemical cycle of nitrogen. Inorganic nitrogen is
converted to the biologically useful form during nitrogen fixation and
assimilation that provide for nitrogen incorporation into the carbon
backbone of organic compounds. Processes of nitrification (oxidative
conversion of ammonium to nitrate) and denitrification (successive
nitrate reduction to nitrite, nitrogen oxides (NO and N2O))
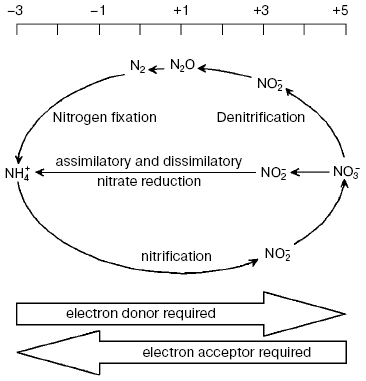
are involved in nitrogen removal from the environment (Fig. 1).
Nitrate reduction, the most important stage of nitrogen turnover in nature, has several functions: i) utilization of NO3- as a source of nitrogen (nitrate assimilation); ii) production of metabolic energy during NO3- utilization as terminal acceptor of electrons (nitrate respiration), and iii) dissipation of excess of reducing energy to maintain oxidation-reduction balance (nitrate dissimilation).Fig. 1. Biogeochemical cycle of nitrogen turnover.
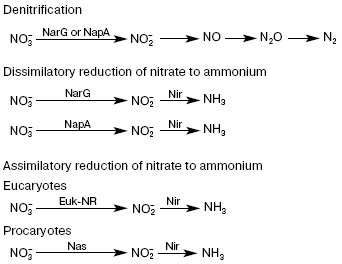
The first, most important stage in the chain of nitrate reduction to nitrites is catalyzed by nitrate reductase (NRase). There are four types of NRases--eukaryotic assimilatory NRase (Euk-NR) [1, 2] and three different bacterial enzymes, i.e. cytoplasmic assimilatory (Nas), membrane-bound respiratory (Nar), and periplasmic dissimilatory (Nap) NRases [3-6] (Fig. 2).
As a rule, all studied NRases share a common property--the presence of molybdenum (Mo) and molybdenum cofactor (Mo-co) in the enzyme active center. Bacterial NRases contain molybdopterin-guanine dinucleotide (MGD), whereas mononucleotide is present in the active center of eukaryotic NRases.Fig. 2. Reactions of nitrogen reduction in pro- and eukaryotes.
In bacterial MGD-binding subunit, molybdenum can be coordinated by four thiolate ligands located in two MGD halves (Fig. 3a). Molybdenum is additionally coordinated by -S, -O, or -Se bonds of cysteine, serine, or selenocysteine residues of the polypeptide chain, as well as by accessible oxo (-O) and hydroxo (-OH) groups or by water (Fig. 3b) in different molybdenum enzymes [7-10].
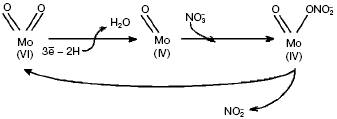
Reactions catalyzed by dimethylsulfoxide (DMSO) and trimethylamine N-oxide (TMAO) reductases follow the oxo-transferase mechanism, in which oxo-group on the oxidized ion Mo(VI) loses OH-/H2O upon reduction to Mo(IV). Nitrate is able to bind Mo in the reduced state and undergo reduction to nitrite, which regenerates the oxo-group on release of Mo(VI) (Fig. 4).Fig. 3. Structure of molybdopterin-guanine-dinucleotide cofactor (MGD cofactor) (a) and active center of NRase (b): I, eukaryotic (Euk-Nar); II, periplasmic (Nap); III, membrane-bound (Nar).
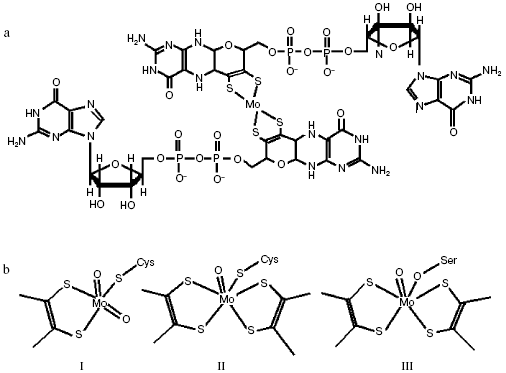
Structures of some catalytic subunits of such MGD family members as formate reductase [8], DMSO reductase [10], TMAO reductase [9], and Nap from D. desulfuromonas [7] have been revealed. However, the number of Mo-bound oxo-groups still attracts attention of researchers. Studying periplasmic NRase (Nap) in P. pantotrophus by EXAFS (Extended X-ray Absorption Fine Structure) pointed to the existence of di-oxo-condition in Mo(VI) [11], whereas the Nap crystalline structure in D. desulfuricans suggests mono-oxo condition in Mo(VI). It should be noted that the amino acid sequence of Nap from D. desulfuricans is similar to that of assimilatory enzymes. There are a number of serine residues that increase the probability of Mo-O-Ser ligands in Nar compared with formation of Mo-S-Cys ligands found for Nap [7] and predicted for Nas. Structural and molecular-genetic peculiarities of different type NRases will be considered below.Fig. 4. Scheme of catalytic cycle of nitrate reduction by Nap of P. denitrificans.
ASSIMILATORY BACTERIAL NITRATE REDUCTASE (Nas)
Two classes of assimilatory NRases have been found in bacteria: ferredoxin- or flavodoxin-dependent Nas and NADH-dependent Nas, the classification of which is based on the cofactor structure (Fig. 5).
Ferredoxin- or flavodoxin-dependent bacterial and cyanobacterial NRases contain in their active center Mo-co and [Fe-S] clusters and are devoid of FAD or cytochromes [12, 13]. NRases from Azotobacter vinelandii and Plectonema boryanum consist of a single 105 kD subunit incorporating a [4Fe-4S] center and molybdenum (1 atom/molecule) and use flavodoxin as a physiological donor of electrons [14]. Analysis of amino acid sequence of these enzymes has shown the presence of cysteine residues at the N-terminus, which evidently binds the [4Fe-4S] center. Ferredoxin-dependent NRases have been found in A. chroococcum, Clostridium perfringens, and Ectothiorhodospira shaposhnikovii [1].Fig. 5. Organization of genes encoding nitrate assimilation in bacteria Klebsiella oxytoca and Synechococcus PCC7942.
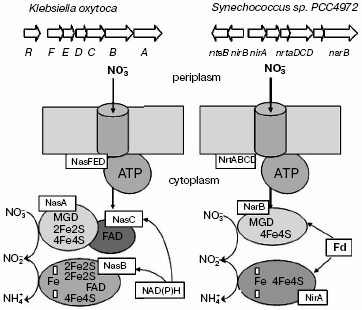
NADH-dependent NRases of Klebsiella oxytoca (pneumoniae) and Rhodobacter capsulatus are heterodimers consisting of the diaphorase FAD-containing subunit of 45 kD and a catalytic subunit of 95 kD with molybdenum cofactor and N-terminal [4Fe-4S] center [15]. Besides, NRase of Klebsiella oxytoca (pneumoniae) contains an additional [2Fe-2S] center bound to a C-terminal cluster of cysteine residues, which resembles by its amino acid sequence the NifU protein, the nifU gene product, involved in formation of [Fe-S] clusters in the nitrogenase active center [16].
This region is absent from the ferredoxin-dependent NRase, whereas in the NADH-dependent NRases it may play the role of the ferredoxin-similar domain carrying out electron transfer. The NADH-dependent NRase of Bacillus subtilis does not contain the NifU-like domain in the catalytic subunit, but contains two tandem NifU-like regions in the diaphorase subunit, which may be involved in [4Fe-4S] or [3Fe-4S] binding [17].
Regulatory and structural genes responsible for nitrate and nitrite uptake (transfer) and reduction are usually located in the same operon. The nasFEDCBA operon of K. oxytoca M5al encodes proteins responsible for nitrate and nitrite assimilation [18, 19], and in this case genes nasFED encode a periplasmic carrier that provides for nitrate and nitrite transfer [19], the NADH-dependent NRase is encoded by genes nasC (diaphorase subunit) and nasA (catalytic subunit), whereas gene nasB is responsible for assimilatory nitrite reductase [20].
The operon encoding enzymes responsible for nitrate assimilation is also well-studied and characterized in A. vinelandii. Structural genes encoding flavodoxin-dependent NRase of A. vinelandii (nasB) are transcribed together with gene nasA responsible for synthesis of nitrite reductase [20]. In most other studied bacteria, genes nirA, nrtABCD, and narB, responsible, respectively, for NRase synthesis, nitrate transport, and nitrite reductase synthesis, make up a single operon [21, 22].
Nitrate transport into a cell involves carriers of the NarK family and ATP-dependent transporters of the ABC family (ATP-binding cassette transporters) [18, 23].
Bacillus subtilis is able to grow using nitrate as a unique source of nitrogen. Assimilatory nitrate and nitrite reductases are encoded by the locus containing genes nasABCDEF. The nasA gene mutant was not able to grow on nitrate, but could grow on nitrite, thus indicating that the NasA protein belongs to the family of the nitrogen oxo-anion transporters (NarK) and is responsible for nitrate transport. NasA homologs were also found in A. aeolicus [24] and Ps. aeruginosa [25].
In Klebsiella and Synechococcus species the ATP-dependent assimilatory uptake of nitrates with involvement of ABC transporters is functioning. The transport systems are encoded by nasFED genes in K. oxytoca [16, 19, 26] and by genes nrtABCD in Synechococcus sp. PCC7942 [22]. Genes nasD, nrtD, and nrtC encode cytoplasmic protein subunits incorporating ATP-binding sites of ABC transporters. Genes nasE and nrtB encode an integral membrane subunit responsible for formation of a channel for nitrate (and nitrite) transport through the cytoplasmic membrane.
The level of nas gene transcription in enterobacteria is under double control, namely, it is the repression by ammonium via the general system of nitrogen regulation (Ntr) and specific induction by nitrate or nitrite [18, 26]. In the photosynthesizing bacterium R. capsulatus assimilatory NRase is induced by nitrate and repressed by low C/N ratio [27]. Positive regulation by nitrate requires nitrate reduction to nitrite, evidently the real activator of the nitrate assimilation gene transcription [28, 29].
BACTERIAL RESPIRATORY MEMBRANE-BOUND NITRATE REDUCTASE
(Nar)
The main membrane-bound NRase (NarGHI) is widespread among enterobacteria and is well studied in E. coli [30]. The enzyme consists of three subunits (alpha, beta, and gamma) and several cofactors and participates in creation of transmembrane proton gradient, which is coupled with nitrate reduction. It has been shown that subunits alpha and beta are localized in the cytoplasm, whereas subunit gamma is localized in the membrane and performs the attachment of subunits alpha and beta to the cytoplasmic side of the internal membrane (Fig. 6).
The 19-28-kD subunit gamma encoded by the narI gene is a hydrophobic protein that forms five transmembrane alpha-helical domains with N-terminus located in the periplasm and C-terminus located in the cytoplasm and containing two b-type hemes [31, 32]. The high-potential heme is adjacent to the cytoplasmic side of the membrane, whereas the low-potential heme is localized in the periplasm. Subunit gamma obtains electrons from quinone and transfers them using b-hemes onto subunit beta via appropriate chains of histidine ligands [33]. Quinone pool oxidation at the membrane periplasmic side forms a transmembrane electrochemical gradient used for ATP formation with involvement of ATP synthase [34].Fig. 6. Organization of genes encoding for proteins of dissimilatory membrane-bound bacterial nitrate reductase.
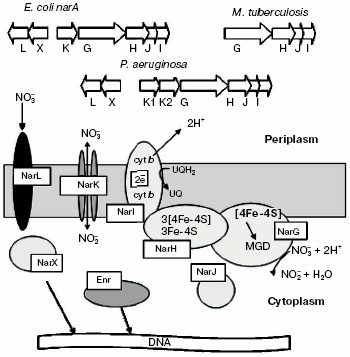
Subunit beta (43-63 kD) encoded by narH gene is a globular protein incorporating [Fe-S] clusters. One [3Fe-4S] and three [4Fe-4S] clusters with different oxidation-reduction potentials were detected by EPR in the alphabeta complex isolated from E. coli. The high potential centers [3Fe-4S] and [4Fe-4S] have the oxidation-reduction potential of +60 and +80 mV, respectively, whereas two low-potential centers [4Fe-4S] have oxidation-reduction potentials of -200 and -400 mV, respectively [35]. It has been supposed recently that only high-potential clusters are able, for thermodynamic reasons, to transfer electrons, whereas the role of the other two clusters is still unknown.
Subunit alpha (104-150 kD) coded by the narG gene contains the [4Fe-4S] cluster and molybdenum cofactor, just on which the nitrate reduction takes place [36]. Molybdenum can exist as Mo(VI), Mo(V), or Mo(IV), and at a low and high pH levels there is pH-dependent equilibrium between forms. X-Ray absorption spectroscopy has detected only a single Mo=O group in NarGHI protein of E. coli. These results agree well with data obtained during studies of crystalline structure of DMSO reductase of Rhodobacter sphaeroides [37]. Structural analysis of the molybdenum center of NarGHI suggests that the enzyme can be a member of the same family of mononuclear molybdenum enzymes like DMSO reductase [38]. Comparison of amino acid sequence of NarGHI with available databases using the BLOCK algorithm [39] has revealed a number of conserved regions characteristic of all prokaryotic molybdopterin-containing oxidoreductases. Three out of four conserved cysteine residues, found in molybdopterin-guanidine-dinucleotide-binding proteins, are located at the N-terminus of the NarGHI alpha subunit.
Another membrane-bound NRase was found in E. coli (and in this organism only) [30]. It was shown by denaturing electrophoresis in polyacrylamide gel that NarZYV consists of two subunits with molecular mass of 150 and 60 kD encoded by narZ and narY genes and contains Mo-co. Subunit gamma of NarZYV encoded by narV was lost during purification, but its existence was proved by spectral methods. The [Fe-S] clusters of NarZYV resemble by their characteristics NarGHI, except the potential of the alpha-subunit high-potential centers. Both enzymes catalyze nitrate and chlorate reduction and are inhibited by azide.
Despite common properties, NarGHI and NarZYV are characterized by different electrophoretic mobility (Rf = 0.23 and 0.38 for NarGHI and NarZYV, respectively). NarZYV is present in E. coli cells in minimal amounts independently of the oxygen partial pressure.
Nitrate reduction is known to be controlled by oxygen at several levels: at the level of gene expression and at the level of nitrogen oxo-anion transport. In E. coli, P. fluorescens, and P. stutzeri the narGHJI operon expression is induced by anaerobiosis and by the presence of nitrate or nitrite [40-42]. The regulatory transcription factor Fnr (regulator of fumarate- and NRases) isolated from E. coli is an anaerobic activator of narGHJI and in structure is a dimer containing [Fe-S] clusters. The protein is inactivated by molecular oxygen as a result of disintegration of [4Fe-4S] clusters and is converted to the monomer form. The Fnr protein monomer can be divided into three functional domains: N-terminal sensor region incorporating the [4Fe-4S] cluster (residues 1-50), central allosteric domain contacting RNA polymerase, and C-terminal regulatory domain incorporating the H-T-H region [43]. In P. aeruginosa and some other Pseudomonas species the Anr protein (anaerobic regulation of anaerobic deiminase and NRase) was identified as a structural and functional homolog of Fnr [44, 45]. This regulator is necessary for denitrification cascade induction during growth on nitrogen oxides [44-46].
Despite significant interest of researchers in the mechanism of nitrate uptake by denitrifying bacteria, this problem is still poorly studied. In a number of articles different mechanisms of nitrate penetration were proposed such as passive uniport, ATP-dependent uniport, transmembrane potential-dependent NO3-/H+ symport, and NO3-/NO2- antiport [47].
BACTERIAL DISSIMILATORY PERIPLASMIC NITRATE REDUCTASE
(Nap)
It is supposed that nitrate reduction in the periplasm plays the key role in aerobic denitrification in Pseudomonas sp. [48], Paracoccus pantotrophus [49, 50], and Rb. sphaeroides f. sp. denitrificans [51, 52], in adaptation to growth under anaerobic conditions in Ralstonia eutropha [53], in the maintenance of the oxidation-reduction equilibrium upon nitrate usage as electron acceptor for reducing energy removal in Rb. capsulatus, Rb. sphaeroides, and Thiosphaera pantotropha [54, 55], or for doubling the nitrate trace amounts in the case of E. coli growing on a nitrate-deficient medium [56-58]. The nap genes were also identified in Ps. putida [59], Ps. aeruginosa [60], Ps. stutzeri, Shewanella putrefaciens, H. influenzae, and quite recently in some pathogenic bacteria like Salmonella typhimurium, Vibrio cholerae, Yersinia pestis, and Campylobacter jejuni. Evidently, as soon as new information concerning genomes of still not studied bacteria will become available, we can expect a significant increase in the number of species incorporating nap genes.
NRases localized in the periplasm are heterodimers consisting of a catalytic 90-kD subunit, incorporating molybdenum cofactor and one [4Fe-4S] center, and of a 13-19-kD subunit incorporating two-heme cytochrome c [61-63] (Fig. 7). They are not involved in formation of transmembrane potential.
Periplasmic NRase (NapABC enzyme) is encoded by the napFDAGHBC operon, in which napA is responsible for synthesis of the catalytic subunit, and napB is the two-heme cytochrome c involved in electron transport onto molybdopterin [64]. Both subunits are synthesized as a precursor with N-terminal signal sequence that defines their transfer through cytoplasmic membrane into the periplasmic space. The NapC, membrane-bound tetra-heme protein of cytochrome c-type, belongs to the NapC/NirT family of periplasmic multi-heme proteins of cytochrome c-type, which provide for the main, cytochrome bc1-independent pathway of electron transfer between quinone of cytoplasmic membrane and water-soluble periplasmic oxidoreductases. The NapC/NirT protein, a homolog of NrfH protein, which transfers electrons between quinone and cytochrome c-nitrite reductase, was identified in Wolinella succinogenes [65]. The cytoplasmic protein NapD is important for NapA maturation [57]. Five different nap genes in different combinations with napDABC were detected in well-studied nap operons. They include napF, napG, and napH encoding the [Fe-S] oxidation-reduction proteins, napE encoding an integral membrane protein whose function is unknown, and napK found only in Rb. sphaeroides [51].Fig. 7. Organization of genes encoding proteins of periplasmic bacterial nitrate reductase.
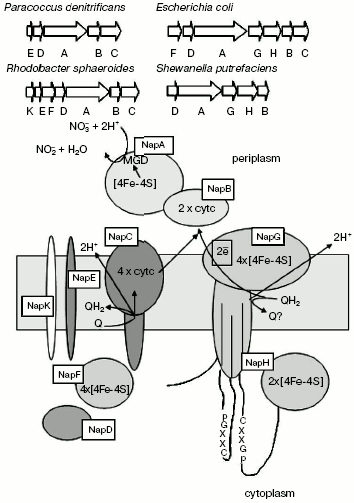
Bacterial periplasmic NRase exhibits various physiological functions and is expressed under different conditions, although usually the NRase activity is revealed under aerobic conditions, and the enzyme synthesis is not repressed by ammonium [66, 67]. In E. coli maximal expression of NRase is observed at low nitrate concentrations under anaerobic conditions, in this case both Fnr and NarP proteins are required [68-70]. On the other hand, the Nap system of Ralstonia eutropha is maximally expressed under aerobic conditions in the stationary growth stage and is not induced by nitrate [53]. In P. pantotrophus maximal nap expression was found in cells grown under aerobic conditions on such reduced carbon sources as butyrate or caproate, but no expression was detected in the case of growth on more oxidized substrates like succinate or malate [71]. In the bacterium R. sphaeroides DSM158 the Nap activity was stimulated, the enzyme synthesis took place both under aerobic and anaerobic conditions, but in the latter case it was more efficient [52, 68].
The periplasmic localization of the enzyme raises important questions concerning NRase transport. The cytochrome c-containing enzyme subunit (NapB) incorporates a typical N-terminal signal sequence that is necessary for Sec (secretion)-dependent transport [72, 73]. However, the catalytic NapA subunit can be assembled in the cytoplasm and exported into the periplasm in a folded conformation via a Sec-independent pathway [72]. It is supposed [73-75] that alternative transport systems may involve unusually long and containing double arginine site N-terminal signal sequences of NapA (and other periplasmic molybdoenzymes), like it was supposed for the pdf (proton driving force)-dependent pathways of thylakoid import. The MGD cofactor is known to be synthesized in the cytoplasm with involvement of five different loci (moa, mob, mod, moe, and mog), but there are still no data concerning the cofactor export in bacteria. Besides, the size of cofactor and its complete internal environment in the protein [7] suggest that the MGD cofactor may be assembled in the cytoplasm before its transport. It has been shown that the cofactor incorporation into apoprotein is a prerequisite for the trimethylamine-N-oxidoreductase transfer in E. coli via Sec-independent pathways [76]. Recently genes have been identified in E. coli that are necessary for Sec-independent export of periplasmic protein that contains the MGD cofactor [77, 78].
ASSIMILATORY EUKARYOTIC NITRATE REDUCTASE (Euk-NRase)
The assimilatory NAD(P)H:nitrate reductase belongs to the NRase family of the class of molybdenum-containing enzymes, which are widespread in plants, fungi, yeasts, and algae [2, 79-81]. It is a water-soluble enzyme consisting of a polypeptide with molecular mass of approximately 100 kD, bound to the molybdenum-molybdopterin molecule (Mo-MPT), heme iron, and FAD. It is known that dimerization of the enzyme is necessary for its activity, and thus the native enzyme is a homodimer having a tendency to further dimerization to a homotetramer (dimer of dimer). The enzyme has two active centers carrying out the internal electron transfer from FAD onto the heme iron and then to Mo-MPT. On the first active center electrons are transferred from FAD via NADH (or NADPH) onto the enzyme; in the second active center two electrons are transferred from reduced Mo(IV) to nitrate and reduce the latter to nitrite and hydroxide. Eukaryotic NRase exhibits its activity with such electron acceptors as ferricyanide and cytochrome c by obtaining electrons directly from FAD and heme iron, respectively. The enzyme has only a limited similarity to prokaryotic NRase that also contains Mo and pterin cofactor. Bacterial cofactor (MGD) differs from eukaryotic MPT by an additional nucleotide. In bacteria, the Mo atom coordinates two pterins. All bacterial NRases contain the [4Fe-4S] oxidation-reduction centers as electron carriers from the electron donor to the enzyme. However, there is still no known eukaryotic NRase that contains [Fe-S] centers as components of the electron transfer chain.
Studying genes encoding NRase of the yeast methylotroph Hansenula polymorpha has shown that they are located in the same locus with genes Yna1 and Yna2 responsible for transcription [82]. A similar gene structure was also observed in different organisms, such as the fungus Aspergillus [83, 84] or algae Chlamydomonas reinhardtii [85, 86], but not in the fungus Neurospora crassa [87].
Despite the fact that many aspects of nitrate transport (NT) in eukaryotes are still not clear, it is assumed that a nitrate carrier really exists. Nitrate transport is carried out in symport with protons, the driving force of which is the transmembrane potential generated at the expense of ATP hydrolysis by H+-ATPase [88]. Individual components of the NT system are regulated in a complex way including induction by nitrate and repression following a feedback mechanism (regulation by nitrate reduction products) [89, 90].
On the basis of expression condition and affinity to nitrate all carriers can be classified as constitutive or inducible with a high or low affinity to the substrate. Studying primary amino acid sequence of nitrate carriers NRT1 and NRT2 in algae and fungi has shown that they belong to the superfamily of peptide transporters (MFS, Major Facilitator Superfamily) [91]. NRT1 belongs to the subgroup of nitrate/nitrite transporters, but effectively transporting nitrate, histidine, and nitrite [92], whereas NRT2 belongs to the subgroup of proton-dependent oligopeptide transporters, transporting peptides, amino acids, nitrate, chlorate, and nitrite [88].
Recently it has been found that eukaryotic cells are able to carry out denitrification [93]. In particular, some species of yeasts and fungi are able to perform the dissimilatory reduction of nitrate to N2O [94-96]. The fungus Cylindrocarpon tonkinense is able to denitrify NO3- despite the presence of ammonium ions in the medium, the presence of sugars being the key condition for switching on denitrification. In this case there is significantly increased level of enzymes involved in NO3- metabolism such as assimilatory NADPH- (or NADH)-dependent NO3- reductase, dissimilatory nitrite reductase, and NO reductase, but not ubiquinone-dependent dissimilatory NRase [96]. Thus, the existence in C. tonkinense of a new type of denitrification was shown, including assimilatory NRase at the first step.
Zhou et al. have shown that the fungus Fusarium oxysporum MT-811 was able to reduce nitrate to ammonium under anaerobic conditions [97]. This process was able to maintain growth of the fungus even under completely anaerobic conditions [97]. Cells grown under microaerophilic conditions contained a noticeable number of intact mitochondria [98], which agrees well with data on mitochondrial localization of the dissimilatory denitrification process [99]. It was assumed previously that soil fungi were obligate aerobes, but new data allow one to consider them as facultative aerobes (or facultative anaerobes).
Kurakov et al. have shown that dissimilatory NRase is synthesized de novo under anaerobic conditions in the fungus F. oxysporum 11dn1. The enzyme significantly differs by its properties (specific activity, molecular mass, temperature optimum) from assimilatory enzyme [100].
Japanese scientists have identified a relationship between activity of NRase found in mitochondrial fraction of the fungus F. oxysporum MT-811 and ATP synthesis and also solubilized and partially purified the enzyme [99, 101]. Intact mitochondria of F. oxysporum exhibited NRase activity with physiological respiratory substrates (malate + pyruvate, succinate, formate) and with synthetic electron donors (reduced methyl viologen, MVH). Detergent treatment of mitochondrial fraction resulted in a loss of NRase activity in the presence of respiratory substrates, but had no effect on the MVH-dependent enzyme activity. These results confirm the supposition concerning the NRase relationship with the respiratory chain. Besides, fungal NRase exhibited similarity by spectral characteristics, molecular mass, and cofactor to dissimilatory NRase of E. coli and other denitrifying bacteria containing molybdopterin, cytochrome b, and Fe-S proteins.
ALTERATIONS OF NITRATE REDUCTASE PROPERTIES AND STRUCTURE CAUSED
BY STRESS FACTORS
At this stage of investigations properties and structure of NRases of organisms (microorganisms, plants) grown under stress conditions are still poorly studied. Nevertheless, this information is extremely important both for revealing general mechanisms of adaptation to stress and for applied investigations with due regard to the role of NRase in nitrogen turnover in nature.
It is known that many hyperthermophilic (Pyrobaculum aerophilum [102], Halobacterium denitrificans [103]) and thermophilic (Thermus thermophilus [104], Caldithrix abyssi [105], Thermovibrio ammonificans [106]) bacteria are able to utilize nitrate as electron acceptor under anaerobic conditions. The process of the extremophile denitrification is studied insufficiently, but NRase of some archaebacteria (P. aerophilum [107], Haloarcula marismortui [108]) and certain Haloferax species [109] has been studied.
Nitrate reductase of P. aerophilum is structurally similar to the enzyme of Haloferax volcanii consisting of three subunits of 100, 60, and 31 kD [110] and is a membrane-bound enzyme, whereas NRases of Haloferax denitrificans, Haloferax mediterranei, and Haloarcula marismortui are localized in the periplasm [108, 109]. There may be an additional protein that binds NRase to the cytoplasmic membrane, NRase of H. marismortui, evidently, differs from the rest enzymes because it is a homotetramer consisting of 63 kD subunits. All NRases of halophilic archaebacteria, except H. denitrificans, require an increased salt concentration for revealing activity and structural stabilization, but the increase in salt concentration resulted in a shift of temperature optimum of the H. mediterranei enzyme activity towards higher temperatures.
Nitrate reductase of hyperthermophilic archaebacterium P. aerophilum was isolated from cells grown under anaerobic conditions in the presence of nitrate and low amounts of tungsten and molybdenum. The isolated enzyme has been characterized [107, 110]. The enzyme contained molybdenum (0.8 mol/mol protein) in its active center and was structurally similar to NRases of mesophilic organisms [30], but differed from them by unusually high specific activity (326 U/mg), measured at 75°C with benzyl viologen as an electron donor; the activity measured at 95°C was even higher (526 U/mg). Like other dissimilatory NRases of mesophilic organisms, the enzyme of P. aerophilum was also able to utilize chlorate as a substrate at a rate exceeding that of nitrate utilization. The NRase affinity to nitrate and chlorate 4-60 times higher than this is described for dissimilatory NRases of mesophilic organisms for which the Km value is 2-3 mM [30].
It should be noted that anaerobic growth of this bacterium on the nitrate-containing medium required the presence of 0.7-1.5 µM tungstate, which could not be replaced by molybdate [107]. In most prokaryotes and eukaryotes, tungsten is an antagonist of molybdenum and easily replaces the latter within inactive analogs of molybdenum-containing enzymes [111]. Owing to this, tungsten is often used as a tool for studying structure and properties of molybdenum enzymes [112]. The addition of 100 µM molybdate to culture medium completely inhibited bacterial growth. At low tungstate concentrations (0.3 µM and lower) nitrite accumulation in the growth medium was observed, which was prevented by tungsten addition in higher concentrations. The dependence of P. aerophilum growth on tungsten suggests the incorporation of tungsten-containing enzymes into metabolic pathways, but the key enzyme of nitrate dissimilation NRase contained molybdenum in its active center [110].
A similar tungsten effect was also observed in the salt-tolerant yeast Rhodotorula glutinis on a medium containing molybdenum and tungsten in equimolecular amounts (1 mM). Tungsten stimulated Mo-co synthesis and following increase in NRase activity, but tungsten was not found in NRase; its activity was revealed only at the level of Mo-co expression [113, 114].
During the past decades, a number of tungsten-containing enzymes have been isolated from cells of thermophilic archaebacteria. Among them there are formate dehydrogenase, aldehyde:ferredoxin oxidoreductase, formaldehyde:ferredoxin oxidoreductase, etc. containing tungsten in their active center and not able to replace it with molybdenum. At the same time a number of enzymes were found whose catalytic activity can be exhibited both with molybdenum and tungsten (formyl methanofuran dehydrogenase, trimethylamine-N-oxide reductase, etc.). Molybdopterin, able to coordinate both molybdenum and tungsten, is the active center of these enzymes [112].
Very recently two NRases, free of molybdenum and molybdenum cofactor, have been isolated from the cells of vanadate-reducing bacterium Pseudomonas isachenhovii growing under high content of vanadium (0.5 g/liter) [115]. The periplasmic NRase was a 55-kD monomer, containing vanadium, whereas the membrane-bound NRase was a heterodimer of 130 and 67 kD subunits and no metal was found in the active center [115].
The reductase complex isolated from the dissimilatory iron-reducing bacterium Geobacter metallireducens did not contain molybdenum and molybdenum cofactor in the active center, but contained cytochrome c and was able to reduce both nitrate and nitrite [116].
In our laboratory a highly active NRase of 130-140 kD was isolated from cells of a halophilic denitrifying bacterium of the Halomonas genus, strain AGJ 1-3. The enzyme was purified to homogeneity, and it contained no molybdenum or molybdo-cofactor in the active center [117]. The enzyme was characterized by thermal stability and relative insensitivity to NaCl in high concentrations, had temperature optimum at 70-80°C, and exhibited polyreductase activity by reducing nitrates and other oxo-anions.
The above-mentioned data from the literature allow us to conclude unambiguously that stress factors in culture medium have a substantial effect on structural and functional characteristics of NRases. Attempts to systematize leading mechanisms of the pro- and eukaryotic organisms' “responses” to the environmental stress conditions were not successful, probably due to not enough experimental data. However, analysis of data from the literature indicates that, as a rule, alterations take place at the structural level of the active center, which results in a shift of the enzyme activity optimum to the region of higher temperatures, an increase in thermal stability, and changes in substrate specificity towards polyfunctionality; changes also happen at the level of synthesis of specific molecules that prevent or decrease the effect of stress factors on the enzyme.
REFERENCES
1.Campbell, W. H. (1999) Annu. Rev. Plant Physiol.
Plant Mol. Biol., 50, 277-303.
2.Campbell, W. H. (2001) Cell. Mol. Life Sci.,
58, 194-204.
3.Moreno-Vivian, C., Cabello, P.,
Martínez-Luque, M., Blasco, R., and Castillo, F. (1999) J.
Bacteriol., 181, 6573-6584.
4.Richardson, D. J. (2000) Microbiology,
146, 551-571.
5.Richardson, D. J., Berks, B. C., Russell, D. A.,
Spiro, S., and Taylor, C. J. (2001) Cell. Mol. Life Sci.,
58, 165-178.
6.Stolz, J. F., and Basu, P. (2002)
ChemBioChem., 3, 198-206.
7.Dias, J. M., Than, M. E., Humm, A., Huber, R.,
Bourenkov, G. P., Bartunik, H. D., Bursakov, S., Calvete, J., Caldeira,
J., Carneiro, C., Moura, J. J. G., Moura, I., and Romao, M. J. (1999)
Structure Fold Des., 7, 65-79.
8.Boyington, J. C., Gladyshev, V. N., Khangulov, S.
V., Stadtman, T. C., and Sun, P. D. (1997) Science, 275,
1305-1308.
9.Czjzek, M., Dos Santos, J. P., Pommier, J.,
Giordano, G., Mejean, V., and Haser, R. (1998) J. Mol. Biol.,
284, 435-447.
10.McAlpine, A. S., McEwan, A. G., and Bailey, S.
(1998) J. Mol. Biol., 275, 613-623.
11.Butler, C. S., Charnock, J. M., Bennett, B.,
Sears, H. J., Reilly, A. J., Ferguson, S. J., Garner, C. D., Lowe, D.
J., Thomson, A. J., Berks, B. C., and Richardson, D. J. (1999)
Biochemistry, 38, 9000-9012.
12.Rubio, L. M., Flores, E., and Herrero, A. (1999)
FEBS Lett., 462, 358-362.
13.Rubio, L. M., Flores, E., and Herrero, A. (2002)
Photosynth. Res., 72, 13-26.
14.Mikami, B., and Ida, S. (1984) Biochim. Biophys.
Acta, 791, 294-304.
15.Blasco, R., Castillo, F., and Martinez-Luque, M. (1997)
FEBS Lett., 414, 45-49.
16.Lin, J. T., and Stewart, V. (1998) Adv. Microb.
Physiol., 38, 1-30.
17.Ogawa, K. I., Akagawa, E., Yamane, K., Sun, Z. W.,
LaCelle, M., Zuber, P., and Nakano, M. M. (1995) J. Bacteriol.,
177, 1409-1413.
18.Lin, J. T., and Stewart, V. (1996) J. Mol. Biol.,
256, 423-435.
19.Wu, Q., and Stewart, V. (1998) J. Bacteriol.,
180, 1311-1322.
20.Ramos, F., Blanco, G., Gutierrez, J. C., Luque, F., and
Tortolero, M. (1993) Mol. Microbiol., 8, 1145-1153.
21.Frias, J. E., Flores, E., and Herrero, A. (1997) J.
Bacteriol., 179, 477-486.
22.Omata, T. (1995) Plant Cell Physiol., 36,
207-213.
23.Moir, J. W., and Wood, N. J. (2001) Cell. Mol. Life
Sci., 58, 215-224.
24.Deckert, G., Warren, P. V., Gaasterland, T., Young, W.
G., Lenox, A. L., Graham, D. E., et al. (1998) Nature, 392,
353-358.
25.Kerschen, E. J., Irani, V. R., Hassett, D. J., and Rowe,
J. J. (2001) J. Bacteriol., 183, 2125-2131.
26.Wu, S. Q., Chai, W., Lin, J. T., and Stewart, V. (1999)
J. Bacteriol., 181, 7274-7284.
27.Castillo, F., Dobao, M. M., Reyes, F., Blasco, R.,
Roldan, M. D., Gavira, M., Caballero, F. J., Moreno-Vivian, C., and
Martinez-Luque, M. (1996) Curr. Microbiol., 33, 341-346.
28.Herrero, A., Muro-Pastor, A. M., and Flores, E. (2001)
J. Bacteriol., 183, 411-425.
29.Maeda, S., and Omata, T. (2004) J. Bacteriol.,
186, 2107-2114.
30.Zumft, W. G. (1997) Microbiol. Mol. Biol. Rev.,
61, 533-616.
31.Berks, B. C., Ferguson, S. J., Moir, J. W., and
Richardson, D. J. (1995) Biochim. Biophys. Acta, 1232, 97-173.
32.Philippot, L., and Hojberg, O. (1999) Biochim.
Biophys. Acta, 1446, 1-23.
33.Magalon, A., Rothery, R. A., Giordano, G., Blasco, F.,
and Weiner, J. H. (1997) J. Bacteriol., 179, 5037-5045.
34.Jormakka, M., Byrne, B., and Iwata, S. (2003) FEBS
Lett., 545, 25-30.
35.Jepson, B. J., Anderson, L. J., Rubio, L.M., Taylor, C.
J., Butler, C. S., Flores, E., Herrero, A., Butt, J. N., and Richardson, D. J.
(2004) J. Biol. Chem., 279, 32212-32218.
36.Gregory, L. G., Bond, P. L., Richardson, D. J., and
Spiro, S. (2003) Microbiology, 149, 229-237.
37.Schindelin, H., Kisker, C., Hilton, J., Rajagopalan, K.
V., and Rees, D. C. (1996) Science, 14, 1615-1621.
38.Hille, R., Retey, J., Bartlewski-Hof, V., Reichenbechen,
W., and Schink, B. (1999) FEMS Microbiol. Rev., 22, 489-501.
39.Henikoff, S., and Henikoff, J. G. (1991) Nucleic Acids
Res., 19, 6565-6572.
40.Khoroshilova, N., Popescu, C., Munck, E., Beinert, H.,
and Kiley, P. J. (1997) Proc. Natl. Acad. Sci. USA, 94,
6087-6092.
41.Hartig, E., Schiek, U., Vollack, K. U., and Zumft, W. G.
(1999) J. Bacteriol., 181, 3658-3665.
42.Philippot, L., Mirleau, P., Mazurier, S., Siblot, S.,
Hartmann, A., Lemanceau, P., and Germon, J. C. (2001) Biochim. Biophys.
Acta, 1517, 436-440.
43.Bates, D. M., Lazazzera, B. A., and Kiley, P. J. (1995)
J. Bacteriol., 177, 3972-3978.
44.Galimand, M., Gamper, M., Zimmermann, A., and Haas, D.
(1991) J. Bacteriol., 173, 1598-1606.
45.Sawers, R. G. (1991) Mol. Microbiol., 5,
1469-1481.
46.Ye, R. W., Haas, D., Ka, J. O., Krishnapillai, V.,
Zimmermann, A., Baird, C., and Tiedje, J. M. (1995) J. Bacteriol.,
177, 3606-3609.
47.Berks, B. C., Page, M. D., Richardson, D. J., Reilly, A.,
Cavill, A., Outen, F., and Ferguson, S. J. (1995) Mol. Microbiol.,
15, 319-331.
48.Bedzyk, L., Wang, T., and Ye, R. W. (1999) J.
Bacteriol., 181, 2802-2806.
49.Butler, C. S., Charnock, J. M., Bennett, B., Sears, H.
J., Reilly, A. J., Ferguson, S. J., Garner, C. D., Lowe, D. J., Thomson, A. J.,
Berks, B. C., and Richardson, D. J. (1999) Biochemistry, 38,
9000-9012.
50.Ellington, M. J. K., Sawers, G., Sears, H. J., Spiro, S.,
Richardson, D. J., and Ferguson, S. J. (2003) Microbiology, 149,
1533-1540.
51.Reyes, F., Gavira, M., Castillo, F., and Moreno-Vivian,
C. (1998) Biochem. J., 331, 897-904.
52.Gavira, M., Roldan, M. D., Castillo, F., Moreno-Vivian,
C. (2002) J. Bacteriol., 184, 1693-1702.
53.Siddiqui, R. A., Warnecke-Eberz, U., Hengsberger, A.,
Schneider, B., and Friedrich, B. (1993) J. Bacteriol., 175,
5867-5876.
54.Bell, L. C., Richardson, D. J., and Ferguson, S. J.
(1990) FEBS Lett., 265, 85-87.
55.Dobao, M. M., Martinez-Luque, M., Moreno-Vivian, C., and
Castillo, F. (1994) Can. J. Microbiol., 40, 645-650.
56.Brondijk, T. H. C., Fiegen, D., Richardson, D. J., and
Cole, J. A. (2002) Mol. Microbiol., 44, 245-255.
57.Potter, L. C., Millington, P. D., Thomas, G. H., Rothery,
R. A., Giordano, G., and Cole, J. (2000) FEMS Microbiol. Lett.,
185, 51-57.
58.Potter, L., Angove, H., Richardson, D., and Cole, J. A.
(2001) Adv. Microb. Physiol., 45, 51-112.
59.Carter, J. P., Richardson, D. J., and Spiro, S. (1995)
Arch. Microbiol., 163, 159-166.
60.Vollack, K., Xie, J., Hartig, E., Romling, U., and Zumft,
W. G. (1998) Microbiology, 144, 441-448.
61.Berks, B. C., Richardson, D. J., Reilly, A., Willis, A.
C., and Ferguson, S. J. (1995) Biochem. J., 309, 983-992.
62.Philippot, L., Clays-Josserand, A., Lensi, R.,
Trinsoutrot, I., Normand, P., and Potier, P. (1997) Biochim. Biophys.
Acta, 1350, 272-276.
63.Stewart, V., Lu, Y., and Darwin, A. J. (2002) J.
Bacteriol., 184, 1314-1323.
64.Flanagan, D. A., Gregory, L. G., Carter, J. P.,
Karakas-Sen, A., Richardson, D. J., and Spiro, S. (1999) FEMS Microbiol.
Lett., 177, 263-270.
65.Simon, J., Gross, R., Einsle, O., Kroneck, P. M. H.,
Kroger, A., and Klimmek, O. (2000) Mol. Microbiol., 35,
686-696.
66.Kiley, P. J., and Beinert, H. (1998) FEMS Microbiol.
Rev., 22, 341-352.
67.Philippot, L. (2002) Biochim. Biophys. Acta,
1577, 355-376.
68.Darwin, A. J., Ziegelhoffer, E. C., Kiley, P. J., and
Stewart, V. (1998) J. Bacteriol., 180, 4192-4198.
69.Stewart, V., Bledsoe, P. J., and Williams, S. B. (2003)
J. Bacteriol., 185, 5862-5870.
70.Wang, H., Tseng, C.-P., and Gunsalus, R. P. (1999) J.
Bacteriol., 181, 5303-5308.
71.Sears, H. J., Sawers, G., Reilly, A., Berks, B. C.,
Ferguson, S. J., and Richardson, D. J. (2000) Microbiology, 146,
2977-2985.
72.Eser, M., and Ehrmann, M. (2003) Proc. Natl. Acad.
Sci. USA, 100, 13231-13234.
73.Fekkes, P. A., and Driessen, J. M. (1999) Microbiol.
Mol. Biol. Rev., 63, 161-173.
74.Dilks, K., Rose, R. W., Hartmann, E., and Pohlschroder,
M. (2003) J. Bacteriol., 185, 1478-1483.
75.Berks, B. C., Sargent, F., and Palmer, T. (2000) Mol.
Microbiol., 35, 260-274.
76.Santini, C. L., Ize, B., Chanal, A., Muller, M.,
Giordano, G., and Wu, L. F. (1998) EMBO J., 17, 101-112.
77.Sargent, F., Bogsch, E. G., Stanley, N., Wexler, M.,
Robinson, C., Berks, B. C., and Palmer, T. (1998) EMBO J., 17,
3640-3650.
78.Weiner, J. H., Bilous, P. T., Shaw, G. M., Lubitz, S. P.,
Frost, L., Thomas, G. H., Cole, J. A., and Turner, R. J. (1998) Cell,
93, 93-101.
79.Inokuchi, R., Kuma, K., Miyata, T., and Okada, M. (2002)
Physiol. Plant., 116, 1-11.
80.Joseph-Horne, T., Hollomon, D. W., and Wood, P. M. (2001)
Biochim. Biophys. Acta, 1504, 179-195.
81.Siverio, J. M. (2002) FEMS Microbiol. Rev.,
26, 277-284.
82.Avila, J., Gonzalez, C., Brito, N., Machin, F., Perez, M.
D., and Siverio, J. M. (2002) Yeast, 19, 537-544.
83.Amaar, Y. G., and Moore, M. M. (1998) Curr.
Genet., 33, 206-215.
84.Hall, N., and Tomsett, A. B. (2000) Microbiology,
146, 1399-1406.
85.Llamas, A., Igeno, M., Galvan, A., and Fernandez, E.
(2002) Plant J., 30, 261-271.
86.Zhou, J.-J., Fernandez, E., Galvan, A., and Miller, A. J.
(2000) FEBS Lett., 466, 225-227.
87.Okamoto, P. M., and Marzluf, G. A. (1993) Mol. Gen.
Genet., 240, 221-230.
88.Galvan, A., and Fernandez, E. (2001) CMLS, Cell. Mol.
Life Sci., 58, 225-233.
89.Daniel-Vedele, F., Filleur, S., and Caboche, M. (1998)
Curr. Opin. Plant Biol., 1, 235-239.
90.Forde, B. (2000) Biochim. Biophys. Acta,
1465, 219-235.
91.Crawford, N. M., and Glass, A. M. D. (1998) Trends
Plant Sci., 3, 389-395.
92.Pao, S. S., Paulsen, I. T., and Saier, M. H. (1998)
Microbiol. Mol. Biol. Rev., 62, 1-34.
93.Takaya, N. (2002) J. Biosci. Bioeng., 94,
506-510.
94.Shoun, H., Kim, D.-H., Uchiyama, H., and Sugiyama, J.
(1992) FEMS Microbiol. Lett., 94, 277-282.
95.Tsuruta, S., Takaya, N., Zhang, L., Shoun, H., Kimura,
K., Hamamoto, M., and Nakase, T. (1998) FEMS Microbiol. Lett.,
168, 105-110.
96.Watsuji, T., Takaya, N., Nakamura, A., and Shoun, H.
(2003) Biosci. Biotech. Biochem., 67, 1115-1120.
97.Zhou, Z., Takaya, N., Nakamura, A., Yamaguchi, M., Takeo,
K., and Shoun, H. (2002) J. Biol. Chem., 277, 1892-1896.
98.Takaya, N., Kuwazaki, S., Adachi, Y., Suzuki, S.,
Kikuchi, T., Nakamura, H., Shiro, Y., and Shoun, H. (2003) J. Biochem.,
133, 461-465.
99.Kobayashi, M., Matsuo, Y., Takimoto, A., Suzuki, S.,
Maruo, F., and Shoun, H. (1996) J. Biol. Chem., 271,
16263-16267.
100.Kurakov, A.V., Nosikov, A. N., Skrynnikova, E. V., and
L'vov, N. P. (2000) Curr. Microbiol., 41, 114-119.
101.Uchimura, H., Enjoji, H., Seki, T., Taguchi, A.,
Takaya, N., and Shoun, H. (2002) J. Biochem., 131, 579-586.
102.Volkl, P., Huber, R., Drobner, E., Rachel, R.,
Burggraf, S., Trincone, A., et al. (1993) Appl. Environ. Microbiol.,
59, 2918-2926.
103.Tomlinson, G. A., Jahnke, L. L., and Hochstein, L. I.
(1986) Int. J. Syst. Bacteriol., 36, 66-70.
104.Ramirez-Arcos, S., Fernandez-Herrero, L. A., and
Berenguer, J. (1998) Biochim. Biophys. Acta, 1396, 215-227.
105.Miroshnichenko, M. L., Kostrikina, N. A., Chernyh, N.
A., Pimenov, N.V., Tourova, T. P., Antipov, A. N., Spring, S., Stackebrandt,
E., and Bonch-Osmolovskaya, E. A. (2003) Int. J. Syst. Evol. Microbiol.,
53, 323-329.
106.Vetriani, C., Speck, M. D., Ellor, S. V., Lutz, R. A.,
and Starovoytov, V. (2004) Int. J. Syst. Evol. Microbiol., 54,
175-181.
107.Afshar, S., Kim, C., Monbouquette, H. G., and Schroder,
I. (1998) Appl. Environ. Microbiol., 64, 3004-3008.
108.Yoshimatsu, K., Sakurai, T., and Fujiwara, T. (2000)
FEBS Lett., 470, 216-220.
109.Hochstein, L. I., and Lang, F. (1991) Arch. Biochem.
Biophys., 288, 380-385.
110.Afshar, S., Johnson, E., de Vries, S., and Schroder, I.
(2000) J. Bacteriol., 183, 5491-5495.
111.Hille, R. (2002) Trends Biochem. Sci.,
27, 360-367.
112.L'vov, N. P., Nosikov, A. N., and Antipov, A. N. (2002)
Biochemistry (Moscow), 67, 196-200.
113.Morozkina, E. V., Nosikov, A. N., Zvyagilskaya, R. A.,
and L'vov, N. P. (2005) Biochemistry (Moscow), 70, 809-814.
114.Nosikov, A. N., Chichikalo, E. V., Golubeva, L. I.,
Zvyagilskaya, R. A., and L'vov, N. P. (2000) Biochemistry (Moscow),
65, 204-207.
115.Antipov, A. N., Lyalikova, N. N., Khijniak, T. V., and
L'vov, N. P. (1998) FEBS Lett., 441, 257-260.
116.Murillo, M. F., Gugliuzza, T., Senko, J., Basu, P., and
Stolz, J. F. (1999) Arch. Microbiol., 172, 313-320.
117.Antipov, A. N., Morozkina, E. V., Sorokin, D. Yu.,
Golubeva, L. B., Zvyagilskaya, R. A., and L'vov, N. P. (2005) Biochemistry
(Moscow), 70, 799-803.