REVIEW: Phylogenetic Systematics of Microorganisms Inhabiting Thermal Environments
A. V. Lebedinsky*, N. A. Chernyh, and E. A. Bonch-Osmolovskaya
Winogradsky Institute of Microbiology, Russian Academy of Sciences, pr. 60-letiya Oktyabrya 7, korp. 2, 117312 Moscow, Russia; fax: (499) 135-6530; E-mail: a.lebedinsky@mail.ru* To whom correspondence should be addressed.
Received August 20, 2007
Thermal habitats harbor specialized communities of thermophilic microorganisms, primarily prokaryotes. This review considers modern systematics of prokaryotes and the place of thermophilic archaea and bacteria in it. Among the existing hierarchical classifications of prokaryotes, the bulk of attention is given to the one accepted in the current second edition of “Bergey's Manual of Systematic Bacteriology”, which is primarily based on 16S rRNA phylogeny and phenotypic properties of the organisms. Analysis of the genomics data shows that they on the whole agree with the 16S rRNA-based system, although revealing the significance of the evolutionary role of lateral transfer, duplication, and loss of genes. According to the classification elaborated in the current edition of “Bergey's Manual”, the prokaryotes currently culturable under laboratory conditions are distributed among 26 phyla, two of which belong to the domain Archaea and 24 to the domain Bacteria. Six phyla contain exclusively thermophiles, and eleven phyla contain thermophiles along with mesophiles, thermophiles being usually separated phylogenetically and representing high-level taxa (classes, orders). In light of the data on the topology of the 16S rRNA-based phylogenetic tree and some other data, this review discusses the probable hyperthermophilic nature of the universal common ancestor.
KEY WORDS: genosystematics, phylogenetic systematics, hierarchical classification, prokaryotes, archaea, bacteria, thermophilesDOI: 10.1134/S0006297907120048
Abbreviations: 16S rDNA) 16S rRNA gene or its fragment; Topt) optimal growth temperature; Tmax) maximum growth temperature.
Habitats where temperature considerably exceeds those typical of most of
the Earth's ecosystems and favorable for the majority of living
organisms are called thermal habitats, or thermal environments. These
include diverse hot springs and soils of volcanic and rift zones and
subterranean hot hydrosphere, as well as diverse anthropogenic
biocenoses, where elevated temperature is due to natural factors
(compost, coal piles) or is maintained artificially (bioreactors,
systems of hot water supply, etc.). Thermal habitats harbor specialized
communities of thermophilic microorganisms, primarily prokaryotes. A
few thermophilic eukaryotes are also known, e.g. the red alga
Cyanidium caldarium (Tmax = 56°C, pH 2-3)
[1], the amoeba Echinamoeba thermarum
(Topt = 50°C) [2], and
microscopic fungi (Topt = 45-50°C) [3]. In this review, we confined ourselves to
consideration of the modern systematics of thermophilic prokaryotes.
Bacteria capable of growing at temperatures suppressing the development of the majority of living organisms have been known for more than a century. However, they have usually been isolated from anthropogenic thermal habitats where temperature rises only from time to time, and the upper temperature limit of their growth did not exceed 70°C, whereas the optimum was at 50-60°C [4]. The taxonomic position of thermophilic microorganisms appeared trivial at that time; as a rule, these were thermophilic species belonging to already known genera that mainly contained mesophiles (Bacillus, Desulfotomaculum, Clostridium, Methanobacterium). The investigations of the microbial communities of hot springs of Yellowstone National Park, carried out by T. D. Brock and his students in the 1970s, revealed unique microorganisms [5]. Subsequent intense studies of terrestrial and marine near-shore volcanic habitats, undertaken in the 1980s-1990s by W. Zillig, K. Stetter, and other researchers, yielded breathtaking results [6]. Organisms were discovered whose growth temperature optimum was above 80°C, and the maximum was close to 100°C. Such organisms were termed hyperthermophiles. Great metabolic diversity of thermophilic prokaryotes was revealed: they included aerobes and anaerobes, organotrophs and lithoautotrophs, and were able to utilize virtually the entire spectrum of possible electron donors and acceptors [6]. Organisms showing best growth at extreme values of two parameters, temperature and pH (pH 0.5 or 10), were described [7, 8]. Investigation of high-temperature deep-sea hydrothermal vents, where, at depths of more than 1000 m, the water occurs in the liquid state at temperatures much higher that 100°C, was a new stage in the research on thermophiles [9]. As a result of these studies, the known upper limit of the existence of living organisms shifted first to 115 [10] and then to 121°C [11].
The phylogenetic diversity of thermophilic prokaryotes was not inferior to their phenotypic diversity. The modern systematics of prokaryotic organisms was elaborated in the same period as the main discoveries in the field of thermophile biodiversity were made; moreover, these two processes exerted considerable mutual influence.
CURRENT STATE OF THE ART IN SYSTEMATICS OF PROKARYOTES
Over the whole history of microbiology, systematics of prokaryotic organisms has been a most complicated task, primarily because of the restricted number of recognized phenotypic properties and the uncertainty of their taxonomic weight. Up to the last quarter of the XXth century, the main criteria for subdivision of prokaryotes into large groups was presence or absence of the cell wall outer membrane (or Gram staining as an indirect indication) and certain morphological (spore formation, budding, mycelium formation) and metabolic features (methanogenesis, dissimilatory sulfate reduction, photosynthesis). However, attempts to elaborate for prokaryotes hierarchical classification systems that would include higher taxa failed to gain stable recognition. In each of the successive editions of “Bergey's Manual of Determinative Bacteriology” (the most authoritative manual on bacterial systematics), the list, placement, and subordination of higher taxa changed considerably, until, in the eighth edition [12], the feeling of the insuperability of the difficulties of hierarchical classification of prokaryotes resulted in abandoning the strictly hierarchical principle in favor of a pragmatic fragmentarily hierarchical approach. In spite of the first successes of phylogenetic systematics of higher taxa in the 1970s, the force of inertia and the load of the still unsolved taxonomic problems were so great that in the ninth edition of “Bergey's Manual of Determinative Bacteriology” [13] the hierarchical principle was totally abandoned. The situation in the first edition of “Bergey's Manual of Systematic Bacteriology” [14] was not much better: the classification suggested there was fragmentarily hierarchical.
In the mid-1970s, the classification of lower taxa was much more stable than that of higher taxa. Here, phenotypic argumentation was supported by the first methods of DNA comparison: comparison in terms of G + C content and DNA-DNA hybridization. As early as in 1957, advancing E. Chargaff's works, A. N. Belozersky and his team demonstrated variation of the G + C content among the DNAs of bacterial taxa and came to a conclusion that the similarity of nucleotide compositions is a necessary but not sufficient condition of common taxonomic affiliation [15]. Now, determination of the G + C content of DNA is a prerequisite of a taxonomic description. DNA-DNA hybridization is an important method of delineating prokaryotic species [16]. A DNA-DNA hybridization level between strains of 70% and higher is currently the main genosystematic criterion of species delineation [17, 18]; however, it cannot be excluded that the concept of the prokaryotic species will be reconsidered in response to data provided by genomics. According to J. Johnson's calculations [19, 20], a DNA-DNA hybridization level of 70% corresponds to 96% homology of nucleotide sequences. Analysis of genomics data yields a similar value (94%) [21]. At the same time, it has been shown (first for Escherichia coli strains) that, in the genomes of strains of the same species that fulfill the above condition, as much as one fourth of the genome genes of one strain may have no orthologs in the genome of the other; such facts provide grounds for the species pangenome concept, which implies that the core of genes common to all strains of a species may be rather small and may provide the basis for a novel definition of a prokaryotic species [22, 23].
Further development of molecular methods in the 1970s provided the basis to begin elaboration of a widely recognized hierarchical systematics of prokaryotes. As early as in 1958, Belozersky and Spirin noted [24] that the G + C content in RNA is relatively constant, and this finding provided an impetus to studies that showed the evolutionary conservation of the rRNA genes by the rRNA-DNA hybridization method [25, 26], which was later successfully used in microbiology to establish the structure of some large genera [27, 28].
C. Woese's approach to investigation of phylogenetic relations by comparing 16(18)S rRNA sequences [29] yielded very soon a profound discovery of the evolutionary group of archaebacteria, which turned to be remote from both eubacteria and eucarya [30, 31], and provided the basis to start elaboration of a fundamentally new hierarchical system of living organisms. Instead of the commonly accepted among biologists subdivision of living organisms into the five kingdoms Monera, Protista, Fungi, Animalia, and Plantae [32, 33], Woese proposed subdivision into three higher taxa (first, he called them primary kingdoms [30, 31, 34] and then [35] domains Archaea, Bacteria, and Eucarya). For the domains Archaea and Bacteria, Woese elaborated their inner hierarchical structure [34-36]. Despite the numerous surprises uncorked by the 16S rRNA-based systematics, its results usually could be given phenotypic justification, albeit often an a posteriori one, and in most cases they did not contradict the results of comparative analysis of other genes [34, 35, 37] and data of DNA-DNA hybridization. The interspecies hybridization level (70-100%) corresponded to no less than 97% homology of 16S rRNA sequences [38] (later it was shown that, if there are no sequencing errors, this value is not less than 98.5% [39]). However, it should be mentioned that Woese never strived for elaboration of a strictly defined taxonomic system; for example, his phyla and kingdoms coexisted with groups characterized as probably deserving a phylum (kingdom) status. We will not consider Woese's system in detail but will dwell upon stricter hierarchical taxonomic systems of prokaryotes.
It should be mentioned that there is no official classification of prokaryotes. The “Code of Nomenclature of Prokaryotes” (formerly, “Code of Nomenclature of Bacteria”) [40] only regulates the rules of taxon description. Currently, in addition to Woese's classification, there exist several hierarchical classifications of prokaryotes that are more or less widely used or at least well known. Most of them aim to be phylogenetic, are primarily based on the results of comparative analysis of 16S rRNA sequences, and are fairly similar in the taxonomic conclusions reached. In this review, we will mainly use the hierarchical classification of prokaryotes elaborated in the second edition of “Bergey's Manual of Systematic Bacteriology” [41, 42]. Since this classification takes into account only cultivated organisms, we will also occasionally use the taxonomic systems of NCBI (http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html) and P. Hugenholtz et al. [43] (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi), which contain information on both cultivated and uncultivated organisms but do not contain phenotypic descriptions and are nontransparent with respect to the grounds for outlining higher taxa and assigning to them organisms or phylotypes. It should be said that, in this review, we will only briefly touch upon the diversity of uncultured prokaryotes represented solely by environmental 16S rDNA clones (even if these clones originate from thermal environments); uncultured prokaryotes represent many tens of deep phylogenetic lineages [43-45].
The basis for the currently issued second edition of “Bergey's Manual of Systematic Bacteriology” [41] has been laid since the early 1990s in the course of work that was initiated at the of Bergey's Manual Trust Editorial Office (http://www.bergeys.org/index.html) and proceeded in cooperation with the Ribosomal Database Project (http://rdp.cme.msu.edu/) [46] and the ARB project headed by W. Ludwig (http://www.arb-home.de/) [47]. The approach developed by G. Garrity, which consists in the application of principle component analysis to comparative analysis of 16S rRNA sequences [42, 48], was used as an additional instrument to outline taxonomic groups. It was decided to adopt the subdivision of all living organisms into three domains: Archaea, Bacteria, Eucarya; to refrain from using the kingdom rank in order to avoid conflicts with other nomenclature codes; and to adopt the species-genus-family-order-class-phylum-domain hierarchical scale. Evaluating the results arriving from genomics, the editors of “Bergey's Manual” and other specialists drawn in reached a consensus opinion [42, 49] that comparison of 16S rRNA sequences remains the most reliable instrument to reveal the phylogeny of the core of cellular genes inherited vertically in the course of evolution and that the phylogeny of this core provides reasonable grounds for present-day microbiological classification. Bergey's Manual Trust organized issuing of the “Taxonomic Outline of the Prokaryotes” (http://141.150.157.80/bergeysoutline/main.htm), where a hierarchical system of prokaryotes to be published in “Bergey's Manual” was outlined.
Recently, the “Taxonomic Outline” resumed being updated, although now at another site (http://www.taxonomicoutline.org). It is no longer sponsored by Bergey's Manual Trust and does not declare the goal to promote the elaboration of “Bergey's Manual”. The distinctions between the taxonomic systems of “Bergey's Manual” and the new “Taxonomic Outline” have not become considerable yet.
Worth mentioning is the appearance of a Classifications section at J. P. Euzeby's site List of Prokaryotic names with Standing in Nomenclature (http://www.bacterio.cict.fr/), popular with microbiologists. In this section, Euzeby reproduces (with minor modifications) the hierarchical classification proposed by Garrity et al. in the latest version of the “Taxonomic Outline” (http://www.taxonomicoutline.org) and claims that this classification is the one most widely accepted by the community of microbiologists. Formerly, the site by Euzeby, who is a member of the International Committee on Systematics of Prokaryotes, only contained alphabetic lists of validly published taxa (taxa described in papers published in the International Journal of Systematic and Evolutionary Microbiology (IJSEM) or included in Validation Lists published ibid). For the issue discussed it is important to mention that valid publication of synonymous names (including synonymous names of higher taxa) is admissible. By allowing publication of later homotypic synonyms without abolishing earlier synonyms, the Committee thus refrains from supporting a particular hierarchical system and provides the community of microbiologists the opportunity to form itself its opinion about the adequacy of this or that hierarchical system. Given this traditional stand of the Committee, Euzeby's decision to support a particular hierarchical system is remarkable.
Importantly, the results from the comparison of completely sequenced prokaryotic genomes by methods that proved most adequate for phylogenetic inference (analysis of concatenated alignments of ribosomal proteins, analysis of identity distributions for orthologs, computation of parsimonious evolutionary scenarios) on the whole support the 16S rRNA-based system, although reveal the important evolutionary role of horizontal gene transfer and gene duplication and loss [50-52].
Thus, currently, for the discussion of the problems of prokaryote systematics and phylogeny it is expedient to use the hierarchical system elaborated in the second edition of “Bergey's Manual of Systematic Bacteriology” as the consensus basis for further development. However, we feel it necessary to mention the widely known, although not commonly accepted, alternative classifications: that validly published in IJSEM by Cavalier-Smith [53] and the phylogenetic system elaborated by R. Gupta proceeding primarily from amino acid insertions-deletions in proteins [54, 55].
SPECIFIC FEATURES OF THE PHYLOGENETIC SYSTEMATICS OF THERMOPHILIC
PROKARYOTES
A specific feature of thermophiles, hindering their phylogenetic analysis, is an elevated G + C content of their 16S rRNAs, which determines thermostability (Fig. 1; see also [56, 57]). The increased G + C content results in a lower rate of the fixation of mutations in the 16S rDNAs of thermophiles compared with mesophiles, since the scope for neutral variability is the narrower the more asymmetrical are the contributions of individual nucleotides to the composition of the 16S rRNA molecule; therefore, it is narrower in case of the necessity to maintain the G + C content at a level of 65 mol% (a typical thermophile) than in case of the necessity to maintain the level of 55 mol% (a typical mesophile). On the other hand, at the onset of divergence of thermophilic and mesophilic lineages, fast accumulation of adaptive distinctions in 16S rRNAs should occur. These facts hinder elucidation of the genuine topology of the evolutionary tree, resulting in the attraction of thermophilic lineages to each other and to the root. The importance of this problem for the systematics of prokaryotes was enhanced by the fact that, in the phylogenetic tree of prokaryotes constructed by Woese in the 1980s, the deepest branches were formed by hyperthermophiles (both in the archaeal and bacterial parts of the tree), suggesting a hypothesis about a (hyper)thermophilic nature of the common ancestor [58]. In the works of Woese et al. devoted to the refinement of the phylogenetic position of the thermophilic bacteria of the genus Thermus [59] and of the hyperthermophilic archaea of the genera Archaeoglobus [60] and Methanopyrus [61], it was shown that, as distinct from the G + C content, the content of purines (and, thus, of pyrimidines) in 16S rRNA is virtually constant and does not depend on the temperature optimum of growth. It was suggested that only those distinctions that arise as a result of transversions should be taken into account for the establishment of the phylogenetic position of thermophiles. Trees constructed with the use of different algorithms were compared, and analysis of sequence signatures (specific nucleotides occurring in highly conserved positions) was conducted. Some details of these analysis and their results are discussed below in sections devoted to the above-mentioned microorganisms. The works of Woese et al. on the phylogeny of Thermus, Methanopyrus, and Archaeoglobus highlighted the problem and directed approaches to its solution.
Nowadays, the methods used to determine the positions of thermophiles in the phylogenetic system of prokaryotes include, in addition to analysis of transversions in 16S rDNA, methods used in the phylogenetic systematics of prokaryotes on the whole. First of all, these are various advanced methods of comparative analysis of 16S rRNA (it should be noted that the procedures for the multiple alignment of these molecules have been improved considerably [43, 62]). Attention is paid to the exclusion of variable positions from the analysis and application of various methods of sample variation and various algorithms of tree construction [49]. Among the tree construction algorithms, most popular currently are the maximum parsimony method (e.g. the ARB parsimony tool, which is an ARB-integrated software [47]) and the maximum likelihood method [63, 64]. Analysis of 16S rRNA sequence signatures [34, 61, 65, 66] has shifted behind the scenes, but it is in fact employed during the application of the methods of maximum parsimony and, the more so, maximum likelihood, especially if the positions are weighted by their degree of conservation. Such weighting can be conducted both in the course of a particular analysis or using the entire bulk of available data [67]. Agreement of the results of different analysis where precautions against thermophile-specific artifacts had been taken is considered a criterion of the reliability of the phylogenetic inference. Analysis of alternative phylogenetic markers [49, 68] and comparison of complete genomes by various methods [50, 51, 69, 70] have still been applied to a few thermophilic taxa, primarily higher ones.Fig. 1. Relationship between the optimum growth temperature and the G + C content of the 16S rRNAs of archaea and bacteria. Data for the type strains of all archaeal and bacterial families described in Vol. 1 of the 2nd edition of “Bergey's Manual of Systematic Bacteriology” [41] were used. The 16S rRNA gene sequences were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/).
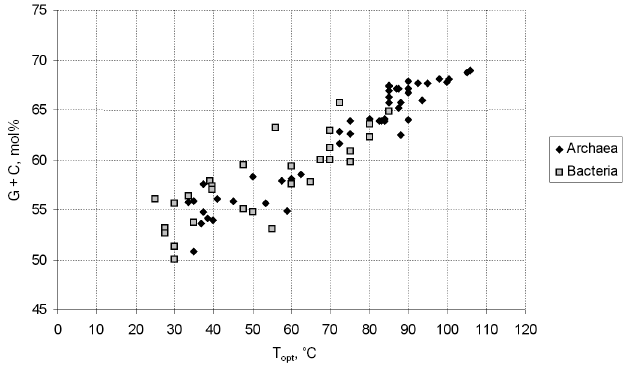
According to the current edition of “Bergey's Manual”, the prokaryotes that are cultured under laboratory conditions are distributed among 26 phyla, two of which belong to the domain Archaea, and 24 to the domain Bacteria. Six phyla contain exclusively thermophiles, and eleven phyla contain thermophiles along with mesophiles, thermophiles being usually separated phylogenetically and representing high-level taxa (classes, orders). Only in nine phyla have no thermophiles yet been found.
PHYLOGENETIC SYSTEMATICS OF THERMOPHILIC ARCHAEA
Hyperthermophilic archaea should undoubtedly be in the center of the description of both phylogenetic and phenotypic diversity of thermophilic prokaryotes. The discovery of hyperthermophiles was directly connected with the interest in archaea aroused by the emergence of Woese's phylogenetic system.
The conceptions of the phylogenetic structure of the domain Archaea were elaborated under the strong influence of Woese's method of signature analysis. In 1991, Woese formulated a definition of the domains Archaea, Bacteria, and Eucarya in terms of their 16(18)S rRNA sequence signatures [65]. Subsequently, signature analysis was widely employed in the analysis of the phylogenetic positions of cultivated archaea (e.g. Methanopyrus [61]) and of archaeal phylogenetic lineages (including thermophilic ones) represented solely by environmental rDNA clones [71-73]. As the result of analysis of the new lineages, the list of signatures characteristic of the archaeal phyla has undergone changes; however, some of the signatures proposed by Woese remain valid. This is one of the reasons to believe that the doubts raised by comparative genome analysis performed by E. Koonin et al. [50, 74] as to the monophyly of Euryarchaeota in relation to Crenarchaeota will prove unjustified; for example, they were not supported by the analysis of ribosomal proteins and translation factors performed by Brochier et al. [75].
The only representatives of the present-day phylum Crenarchaeota (Fig. 2, A1 in “Bergey's Manual”) isolated before the establishment of Woese's phylogenetic system were the moderately thermophilic organisms belonging to the genus Sulfolobus, which are aerobic lithoautotrophs oxidizing sulfur compounds [76]. In the 1980s-1990s, many new crenarchaeotal genera were isolated, which were hyperthermophilic and for the most part anaerobic [6]. Currently, cultivated thermophilic Crenarchaeota are assigned to a sole class Thermoprotei, which includes four orders. The orders Thermoproteales and Desulfurococcales contain anaerobic hyperthermophiles, both lithoautotrophic and organotrophic. Elemental sulfur plays an important role in the metabolism of most of them as an electron acceptor in respiration or facilitated fermentation [77]. The order Sulfolobales mainly contains aerobic thermophilic organisms that oxidize sulfur as the energy substrate. The acidophilic anaerobic hyperthermophiles isolated from Kamchatka hot springs and assigned to the new genus Acidilobus [78] should most probably be described as representing a new order, Acidilobales, on the basis of their phenotypic properties, low level of 16S rRNA homology (<90%) with representatives of other Crenarchaeota orders, and presence of characteristic 16S rRNA sequence signatures.
Euryarchaeota, the second (A2) phylum of the domain Archaea, includes, according to “Bergey's Manual”, eight classes. Three of these classes contain exclusively hyperthermophiles (Thermococci, Archaeoglobi, Methanopyri), all of which were isolated over the last decades. As distinct from other archaeal genera, the genus Thermococcus contains many (more than 20) species, which are phenotypically quite homogeneous (anaerobic organotrophic fermenters) but strongly vary in the DNA G + C content (from 33 to 58 mol%) and exhibit a low level of DNA-DNA hybridization [79].Fig. 2. Phylogenetic tree of archaea constructed proceeding from 16S rRNA gene sequences of the type strains of all archaeal orders described in the 2nd edition of “Bergey's Manual of Systematic Bacteriology” [41], as well as some additional sequences. Thermophiles are set in boldface. All of the sequences were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/). The tree was constructed with the use of the neighbor-joining method after calculating the distance matrix with taking into account only distinctions due to transversions. Numerals at the branching points show the reliability of the branching order; only bootstrap values higher that 50% are shown. The software used was MAFFT v.6 (http://align.bmr.kyushu-u.ac.jp/mafft/online/server/) and TREECONW v.1.3b (http://bioinformatics.psb.ugent.be/software/details/TREECON).
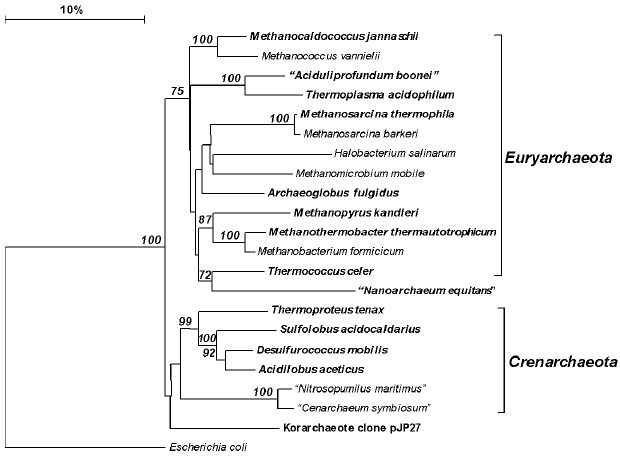
The class Methanopyri is represented by a single species, the hyperthermophilic methanogenic Methanopyrus kandleri, which inhabits deep-sea hydrothermal vents and has a growth temperature optimum of 105°C [80].
The class Archaeoglobi contains a few genera and species of sulfate- and iron-reducers, which belong to physiological groups considered to be highly important by evolutionary biogeochemistry.
The phylum Euryarchaeota also contains many organisms that were successfully cultivated long before the emergence of the 16S rRNA-based phylogenetic system. These are methanogens (the classes Methanobacteria, Methanococci, Methanomicrobia) and halophiles, which grow under conditions of high salinity (class Halobacteria). Among halophiles, no thermophiles have yet been found; among methanogens, thermophiles are fairly numerous. The class Methanomicrobia includes taxonomically dispersed thermophilic species; within the classes Methanobacteria and Methanococci, thermophilic species were recently united into specially established genera [81, 82].
The class Thermoplasmata contains moderately thermophilic acidophilic organisms known since the 1970s, as well as the mesophilic acidophilic family Ferriplasmataceae [83].
The phylogenetic relationships among the euryarchaeotal classes are of considerable interest, since knowledge of the branching order allows the ancientness of methanogenesis and sulfur-dependent metabolism to be judged. The analyses of Woese and coworkers allowed them to arrive to a conclusion that Archaeoglobus does not represent a deep phylogenetic lineage but clusters with Methanomicrobiales [60], whereas Methanopyrus branches near the root of the euryarchaeotal lineage [61]. At present, the discussions about the ancientness of the origin of Archaeoglobus and Methanopyrus is continued and involves data obtained by various methods of genome comparison; the conclusions made seem to be far from final [69, 75, 84, 85].
An important consequence of the elaboration of Woese's phylogenetic system was the opportunity to analyze clone libraries of 16S rDNAs obtained from natural samples and thus to investigate the phylogenetic composition of natural microbial populations. This opportunity produced a breakthrough in microbial ecology; a lot of prokaryotes discovered in this way proved to be unknown in laboratory cultures [45, 86]. Thus, abundant nonthermophilic Crenarchaeota were discovered in marine and fresh waters and sediments, as well as in soils and invertebrate intestines [87]. By now, only one such organism has been isolated as a pure culture [88]; one more organism, which is a mesophilic sponge symbiont, has been rather well studied but has not been isolated in a pure culture [89]. Although thermophilic and mesophilic Crenarchaeota are deeply diverged phylogenetic lineages, this phylum can no more be considered “purely thermophilic” (as it is presented in the current edition of “Bergey's Manual” [42]).
Analysis of 16S rDNA clone libraries obtained from terrestrial and marine hot springs also revealed many new phylogenetic lineages of thermophiles. In the Obsidian Pool hot spring (75-90°C) in Yellowstone National Park, many new archaea were found. Most of them represented new lineages of Crenarchaeota [71]. However, some of the 16S rDNAs retrieved could not be assigned to the known archaeal phyla based on the homology levels and sequence signatures; these phylotypes were termed Korarchaeota. With the use of specific PCR primers, Korarchaeota were then discovered in hot springs in various geographical locations [90]. Completion of the genome sequencing of one of the korarchaeotal components of an enrichment culture was recently announced at the DOE Joint Genome Institute (http://www.jgi.doe.gov) and GOLD Genomes OnLine Database (http://www.genomesonline.org/) web sites. As far as thermophilic Euryarchaeota are concerned, the diversity of their environmental 16S rDNAs proved to be particularly great in deep-sea hydrothermal vents [72]. Recently, the DHVE2 phylogenetic lineage described based on environmental 16S rDNA clones acquired its first cultivated representative, “Aciduliprofundum” [91], which turned out to be an anaerobic thermoacidophilic organotroph.
The difficulties of cultivating new microorganisms led to the situation where many of them, although shown to be phylogenetically distinct by molecular methods, cannot be validly described and/or included in “Bergey's Manual” because they have not been isolated in pure cultures. Thus, archaea of the genus “Nanoarchaeum” [92], whose cell size is about 500 nm, are known to occur only in cultures of the hyperthermophilic archaea of the genus Ignicoccus. The low level of 16S rRNA sequence homology with most closely related taxa allowed a proposal to be made that “Nanoarchaeum” should be considered a representative of a new phylum, Nanoarchaeota. The obligate dependence on the host correlates with the very small size (490 kb) of the “Nanoarchaeum equitans” genome where many genes coding for anabolic enzymes were shown to be lacking [93]. Different methods of comparative genome analysis yield different conclusions about the ancientness of this phylogenetic lineage, which may prove to be the deepest archaeal branch [93, 94] or a lineage within Euryarchaeota [52] or even a fast-evolving (due to the transition to parasitism) euryarchaeotal lineage related to Thermococcales [95, 96].
PHYLOGENETIC SYSTEMATICS OF THERMOPHILIC BACTERIA
In the domain Bacteria, the phyla Aquificae, Thermotogae, Thermodesulfobacteria, Thermomicrobia, and Dictyoglomi contain thermophiles only. Thermophilic taxa also occur in the phyla Deinococcus-Thermus, Nitrospirae, Deferribacteres, Chloroflexi, Cyanobacteria, Proteobacteria, Firmicutes, Actinobacteria, and Spirochaetes (Fig. 3).
While most thermophilic archaea are hyperthermophiles, i.e. have a growth temperature optimum above 80°C and upper limit at about 100°C, thermophilic bacteria are usually so-called extreme thermophiles (Topt = 70°C, Tmax = 80°C) or moderate thermophiles (Topt = 50-60°C, Tmax = 70°C). Hyperthermophilic bacteria occur only in the most deeply branching phyla: Aquificae (B1), Thermotogae (B2), and Thermodesulfobacteria (B3).Fig. 3. Phylogenetic tree showing the evolutionary relationships of the bacterial phyla. The tree is reproduced with minor modifications from W. Ludwig and H. P. Klenk's introductory paper to the 2nd edition of “Bergey's Manual of Systematic Bacteriology” [49]. The tree was constructed based on the analysis of 16S rRNA genes with the use of the maximum parsimony method (ARB parsimony tool). The arrow indicates the position of the root. Framed are phyla that include thermophiles; phyla containing thermophiles exclusively are marked with gray background. Of the 24, 20 bacterial phyla are shown. Of the phyla that include thermophiles, Thermodesulfobacteria and Thermomicrobia are not presented; of the mesophilic phyla, Chrysiogenetes and Gemmatimonadetes are not shown.
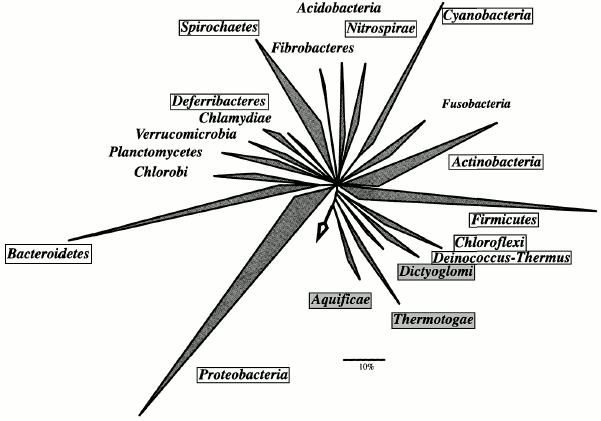
The phylum Aquificae (B1; a sole class Aquificae and a sole order Aquificales) comprises numerous genera of extremely thermophilic and hyperthermophilic prokaryotes with the lithoautotrophic type of metabolism, which use the energy of molecular hydrogen and/or sulfur compounds for growth under aerobic or anaerobic conditions. The distinct phylogenetic position of this group is supported by specific 16S rRNA sequence signatures [66].
The phylum Thermotogae (B2; a sole class Thermotogae and a sole order Thermotogales) comprises thermophilic organotrophs varying in their growth temperature characteristics from hyperthermophiles (Thermotoga) to organisms that hardly fit the thermophily criteria (Geotoga with its growth maximum at 45°C and lower temperature limit of 30°C). While Aquificales are typical inhabitants of terrestrial and marine hydrothermal vents, Thermotogales, in addition to these environments, are widespread in subterranean ecosystems, where, at depths of greater than 2000 m, the temperature reaches 80°C and more [97]. Thermotogales also occur in thermophilic bioreactors [98]. Most Thermotogales genera occur in various types of thermal habitats; an exception is Marinitoga [99, 100], which is endemic to deep-sea hydrothermal vents.
The phylogenetic tree constructed proceeding from concatenated alignment of ribosomal proteins allowed the existence of a common ancestor of Thermotogales and Aquificales to be assumed [50], although the authors themselves present this conclusion with certain caution.
One more phylum represented solely by thermophiles was named Thermodesulfobacteria (B3) after its first isolated representative, the extremely thermophilic sulfate-reducing bacterium Thermodesulfobacterium commune [101, 102]. This phylum also includes Geothermobacterium ferrireducens [103], the organism that has the highest growth characteristics in the domain Bacteria. This anaerobic bacterium, which uses a single energy source (molecular hydrogen) and a single electron acceptor (ferric oxide), shows best growth at 90°C and has an upper temperature limit of 110°C. Both of the above-mentioned bacteria were isolated from the hot springs of Yellowstone National Park, which are also the habitat of one more thermophilic sulfate-reducing bacterium, Thermodesulfovibrio yellowstonii, first thought to cluster phylogenetically with Thermodesulfobacterium [102], but assigned to the phylum Nitrospirae (B8) in the current edition of “Bergey's Manual”.
Thermophilic bacteria of the genus Thermus were discovered by T. Brock in the course of his investigations of the hot springs of Yellowstone National Park [104]. They are aerobic organotrophs widespread in thermal environments. These bacteria became widely known beyond microbiology as producers of a thermostable DNA polymerase used in PCR. Investigations of deep-sea hydrothermal vents resulted in the isolation of new Thermaceae genera whose representatives are distinguished by their ability to grow lithoheterotrophically with molecular hydrogen and by microaerophily [105, 106]. The family Thermaceae belongs to phylum B4, named Deinococcus-Thermus; this phylum includes, along with the thermophilic Thermaceae, the widely known extremophilic genus Deinococcus, which is highly resistant to radiation and desiccation. Woese [36] considers the discovery of phylogenetic proximity of Thermus and Deinococcus to be one of the greatest successes of his method. Interestingly, the first indications of this proximity were given by methods that have a relatively low resolving power: 5S rRNA analysis [107] and comparison of oligonucleotide catalogs of 16S rRNA [108]. Further investigation of the phylogenetic position of Thermus with the use of sequenced 16S rRNA genes promoted the development of the methods of phylogenetic systematics as applied to thermophiles. The phylogenetic trees constructed by the neighbor-joining method widely used at that time clustered Thermus with other deeply branching thermophilic lineages but not with Deinococcus [109]. Woese suggested that this clustering might be artificial and related to the high G + C content of the 16S rRNA of thermophiles [59]. Restriction of the analysis to the distinctions determined by transversions only or to low-variability positions [59] or the inclusion in the analysis of the sequence of Meiothermus, whose temperature characteristics are more moderate than those of Thermus [110], or assessment (in the same work [110]) of the evolutionary remoteness of lineage divergence with the use of the phenomenon of slower evolutionary accumulation of transversions as compared to transitions [60, 111] convincingly demonstrated the specific relatedness of Thermus and Deinococcus. Simultaneously, a number of approaches to phylogenetic systematics of thermophiles were elaborated.
The relatedness between Deinococcus and Thermus was then confirmed by analysis of several alternative phylogenetic markers, e.g. protein RecA [112], and by comparison of complete genomes, which, in addition, allowed the adaptive nature of thermophily in Thermus to be assumed [70].
Some of the thermophilic genera turned out to be so distinct phylogenetically that each of them was assigned to a special separate phylum. These are the aerobic genus Thermomicrobium, represented by a sole species, Thermomicrobium roseum [113] and forming the phylum Thermomicrobia (B7), and the anaerobic genus Dictyoglomus (phylum Dictyoglomi, B23), which contains fermentative bacteria isolated from continental hydrothermal vents [114].
The phylum Deferribacteres (B9), in addition to two mesophilic genera, contains the thermophilic genus Deferribacter, which comprises bacteria with various types of anaerobic respiration [115, 116].
A number of thermophilic genera and/or species are present in the voluminous and evolutionary important phyla Cyanobacteria (B10), Actinobacteria (B14), and Bacteroidetes (B20). Thermophilic cyanobacteria develop in terrestrial hot spring in the form of cyanobacterial mats at temperatures of 40-70°C. These mats also contain thermophilic anoxygenic phototrophs of the order Chloroflexales. Together with the mesophilic Herpetosiphonales, they form the phylum Chloroflexi.
The phylum Proteobacteria (B12) comprises a large number of taxa, mainly mesophilic, which are distributed among five classes (alpha, beta, gamma, delta, epsilon). Thermophiles are known in all five classes. These are mainly moderate thermophiles; an exception is Thermothrix thiopara [117]--a bacterium belonging to the class Betaproteobacteria and performing oxidation of sulfide with atmospheric oxygen in sulfide-containing terrestrial hot springs with a temperature of about 70°C. The sulfur-reducing moderately thermophilic bacteria of the genus Desulfurella [118] occupy a phylogenetically distinct order-level position in the class Deltaproteobacteria. Thermophilic representatives of the class Epsilonproteobacteria will be discussed below in the context of the narration about cultivation of “uncultivated” bacteria.
The phylum Firmicutes (B13) is a group of bacteria that are easy to cultivate, and it is therefore voluminous and well studied. Many of the thermophilic bacteria belonging to it have long been known. However, there is currently a tendency toward reclassifying them into separate genera based on the analysis of 16S rRNA. A vivid example is the genus Geobacillus, established to include most thermophilic species of bacilli [119]. It is probable that the fate of thermophilic representatives of Desulfotomaculum will be the same. Firmicutes also include the order Thermoanaerobacteriales, which comprises a number of anaerobic organotrophs widespread in hydrothermal vents [120]. It is quite possible that isolation of phenotypically new representatives of Firmicutes will result in description of new high-level taxa within this well-studied phylogenetic lineage. A good example is the new class Thermolithobacteria, established to include anaerobic thermophilic gram-positive bacteria utilizing molecular hydrogen and carbon monoxide [121].
The Spirochaetes (B17) is a phylum in which the phylogenetic position correlates with unique and uniform morphology: it unites spirochetes, among which various extremophiles (thermophiles and alkaliphiles) occur.
Investigation of bacterial 16S rDNA clone libraries obtained from hydrothermal vents of various types revealed phylogenetic diversity of thermophilic bacteria that compares well with the diversity of thermophilic archaea. In the above-mentioned Obsidian Pool hot spring, 12 new phylum-level lineages of thermophilic bacteria were found [122]. Nowadays, the number of reports on new uncultured lineages greatly exceeds the number of reports about successful attempts of their cultivation. However, eventually, new phylogenetic groups detected by molecular methods acquire their cultured representatives, which in future will allow them to find their place in “Bergey's Manual”. For example, an early diverged bacterial phylogenetic lineage was detected at shallow submarine hydrothermal vents in the Aegean Sea [123]. Several years later, its culturable representative was isolated from deep-sea hydrothermal vents of the Mid-Atlantic Ridge. This moderately thermophilic anaerobic bacterium, named Caldithrix abyssi, is capable of fermentation and hydrogen oxidation with nitrate reduction [124]; it has not yet been assigned to any of the recognized phyla.
For a long time it was considered that the class Epsilonproteobacteria comprises only mesophilic bacteria. However, numerous investigations of environmental 16S rDNA clone libraries indicated wide occurrence of this group in deep-sea hydrothermal vents [66]. The moderately thermophilic bacterium Nautilia lithotrophica [125] was the first cultured representative of this group, which has later been described as the order Nautiliales [126], currently already containing two genera and three species.
One more example of the emergence of thermophiles in high-level taxa is the isolation of a thermophilic representative of the phylum Verrucomicrobia (B22). This phylum comprises difficult-to-cultivate mesophilic bacteria; this lineage is also represented by numerous 16S rDNA clones isolated from mesophilic habitats. Recently, a Verrucomicrobia representative that can grow aerobically on methane at 60°C and pH 2.0 was isolated from a hot spring of Kamchatka [127].
ON THE PROBABLE HYPERTHERMOPHILIC NATURE OF THE UNIVERSAL COMMON
ANCESTOR
Investigations of the last decades showed that thermophiles dominate the early diverged phylogenetic lineages of prokaryotes. This, together with data on the geochemical conditions on ancient Earth [128], prompted the hypothesis about the hyperthermophilic nature of the universal common ancestor [34, 129]. This viewpoint remains predominant [130], although not proven [131]. Alternative hypotheses exist, e.g. one of them suggests that the topology of the phylogenetic tree that we can reconstruct was determined by Earth's heating due to heavy meteorite bombardment with consequent frustration of mesophilic forms of life [132]. One more alternative scenario is that implied by the already mentioned phylogenetic system by Cavalier-Smith [53]; in this scenario, thermophily is a late evolutionary invention, and, moreover, the emergence of archaea is a late evolutionary event driven by adaptation to thermal environments. However, the theory of Cavalier-Smith contradicts a too large bulk of data obtained by analyses of phylogenetic markers and genome comparisons to be argued against here.
In recent years, most of the works devoted to the nature of the universal common ancestor or common ancestors of particular phylogenetic lineages did not touch upon the probable temperature characteristics of the ancestors, probably because it became evident that the resolving power of the existing methods of analysis is insufficient to make final conclusions. Almost no qualitative leaps have yet been revealed between the enzymatic systems or genomes of mesophiles and thermophiles. Among enzymes specific for (hyper)thermophiles, one may name reverse gyrase [96] and several hypothetical enzyme systems predicted based on the analysis of genomes [133, 134]. Distinctions between orthologous proteins of mesophiles and thermophiles are not qualitative leaps and include differences in amino acid compositions, loop lengths, number of salt bridges, strength of hydrophobic interaction, number of disulfide bonds, etc. [135, 136].
However, comparison of the complete genomes of Thermus thermophilus and Deinococcus radiodurans allowed Koonin et al. to suggest that thermophily in Thermus is an acquired secondary trait [70]. An analogous assumption can be made, proceeding from general considerations, about the nature of thermophily in those bacterial branches that occur in mesophilic portions of the phylogenetic tree. Such an evolutionary scenario seems to be more parsimonious, although the problem certainly requires special investigations.
As for the probable temperature characteristics of the bacterial common ancestor, the high ratio of archaeal genes found in the genomes of Thermotoga (24%) [137] and Aquifex (16%) [138, 139] can be interpreted both as a reflection of the thermoadaptation process in these bacteria [137] and as a result of sharing common ecological niches with archaea [139].
P. Forterre et al. consider the question about the common ancestor via analysis of the topology of the phylogenetic tree of reverse gyrases [96, 140]. From the topology of the tree constructed initially [140], the authors inferred monophyly of the archaeal part of the tree and several times repeated transfer of reverse gyrase from archaea to bacteria; a logical consequence of these inferences was the adaptive, secondary nature of thermophily in bacteria. The topology of the latest tree [96] rather suggests inheritance of reverse gyrase by archaeal and bacterial hyperthermophiles from the common ancestor; however, Forterre has not abandoned the initial hypothesis.
One more approach to the investigation of the temperature characteristics of the universal common ancestor consists in the reconstruction of the most likely primary structure of its rRNA with subsequent evaluation of the organism's growth temperature optimum from the rRNA G + C content. Opposite conclusions have been reached upon application of different reconstruction methods [141, 142].
A similar approach employs reconstruction of the most likely primary structure of the proteins of the universal common ancestor and evaluation of their temperature characteristics either from the amino acid composition (in case of purely conceptual reconstruction [143]) or by studying the properties of “ancestral” proteins after the expression of cloned genes with required mutations introduced by PCR site-directed mutagenesis [144]. These works led their authors to a conclusion about the hyperthermophilic nature of the universal common ancestor.
The development of microbiology over the last three decades occurred under a strong influence of the phylogenetic systematics of prokaryotes. The choice of the 16S rRNA gene as a universal phylogenetic marker allowed placing all organisms into a common system with a quantitative measure of evolutionary relatedness. This system is not final and is subject to constant changes. The occurrence of debatable issues and, occasionally, voluntarism in certain taxonomic decisions should be noted. However, stable advancement toward ordering microbial taxonomy is obvious, in spite of the voluminous supply of information from genomics and molecular ecology of microbial communities, which, by the way, originated from the analysis of 16S rDNAs isolated from natural samples. One more research field that gained strong impetus from phylogenetic systematics is the study of microbial biodiversity, since the information on the nontrivial phylogenetic position often stimulates an in-depth study of the phenotype.
However, it becomes evident that the selectivity of the cultivation methods may seriously distort our view of the genuine phylogenetic diversity. For example, some of the early diverged prokaryotic lineages revealed as yet only by a molecular method seem to be mesophilic judging from the habitat or from the G + C content of 16S rRNA (e.g. the bacterial lineage OP11 [44, 145]). We thus come back to the questions about the primary or secondary nature of thermophily in prokaryotes belonging to various taxa and about the thermophilic or mesophilic nature of the common ancestor, a question to which no final answer seems to be possible yet.
This work was supported by the Russian Foundation for Basic Research (project No. 05-04-49311) and programs of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology” and “Origin and Evolution of the Biosphere”.
REFERENCES
1.Doemel, W. N., and Brock, T. D. (1970) Arch.
Microbiol., 72, 326-332.
2.Baumgartner, M., Yapi, A., Grobner-Ferreira, R.,
and Stetter, K. O. (2003) Extremophiles, 7, 267-274.
3.Maheshwari, R., Bharadwaj, G., and Bhat, M. K.
(2000) Microbiol. Mol. Biol. Rev., 64, 461-488.
4.Bonch-Osmolovskaya, E. A., Miroshnichenko, M. L.,
Sokolova, T. G., and Slobodkin, A. I. (2004) in Proceedings of
Winogradsky Institute of Microbiology [in Russian], Issue 12,
Nauka, Moscow, pp. 29-40.
5.Brock, T. D. (1978) Thermophilic Microorganisms
and Life at High Temperatures, Springer-Verlag, New York.
6.Stetter, K. O. (1996) FEMS Microbiol. Rev.,
18, 149-158.
7.Schleper, C., Puehler, G., Holz, I., Gambacorta,
A., Janekovic, D., Santarius, U., Klenk, H. P., and Zillig, W. (1995)
J. Bacteriol., 177, 7050-7059.
8.Li, Y., Mandelco, L., and Wiegel, J. (1993)
Int. J. Syst. Bacteriol., 43,
450-460.
9.Jannasch, H. W., and Mottl, M. J. (1985)
Science, 229, 717-725.
10.Blochl, E., Rachel, R., Burggraf, S., Hafenbradl,
D., Jannasch, H. W., and Stetter, K. O. (1997) Extremophiles,
1, 14-21.
11.Kashefi, K., and Lovley, D. R. (2003)
Science, 301, 934.
12.Buchanan, R. E., and Gibbons, N. E. (eds.) (1974)
Bergey's Manual of Determinative Bacteriology, 8th Edn.,
Williams & Wilkins, Baltimore.
13.Holt, J. G., Krieg, N. R., Sneath, P. H. A.,
Staley, J. T., and Williams, T. (eds.) (1994) Bergey's Manual of
Determinative Bacteriology, 9th Edn., Williams & Wilkins,
Baltimore.
14.Holt, J. G., et al. (eds.) (1984-1989)
Bergey's Manual of Systematic Bacteriology, Vols. 1-4, 1st Edn.,
Williams & Wilkins, Baltimore.
15.Spirin, A. S., Belozersky, A. N., Shugaeva, N.
V., and Vanyushin, B. F. (1957) Biokhimiya, 22,
744-753.
16.Rossello-Mora, R. (2005) in Molecular
Identification, Systematics, and Population Structure of
Prokaryotes (Stackebrandt, E., ed.) Springer, Heidelberg, pp.
23-50.
17.Wayne, L. G., Brenner, D. J., Colwell, R. R.,
Grimont, P. A. D., Kandler, O., Krichevsky, M. I., Moore, L. H., Moore,
W. E. C., Murray, R. G. E., Stackebrandt, E., Starr, M. P., and Truper,
H. G. (1987) Int. J. Syst. Bacteriol., 37, 463-464.
18.Stackebrandt, E., Frederiksen, W., Garrity, G.
M., Grimont, P. A. D., Kampfer, P., Maiden, M. C. J., Nesme, X.,
Rossello-Mora, R., Swings, J., Truper, H. G., Vauterin, L., Ward, A.
C., and Whitman, W. B. (2002) Int. J. Syst. Evol. Microbiol.,
52, 1043-1047.
19.Johnson, J. L. (1973) Int. J. Syst.
Bacteriol., 23, 308-315.
20.Stackebrandt, E. (2006) in The Prokaryotes
(Dworkin, M., Falkow, S., Rosenberg, E., Scleifer, K.-H., and
Stakebrandt, E., eds.) Vol. 1, Springer-Verlag, New York, pp.
29-57.
21.Konstantinidis, T. K., and Tiedge, J. M. (2005)
Proc. Natl. Acad. Sci. USA, 102, 2567-2572.
22.Nesbo, C. L., Dlutek, M., and Doolittle, W. F.
(2006) Genetics, 172, 759-769.
23.Doolittle, W. F. (2006) Microbiol. Today,
Nov 06, pp. 148-151.
24.Belozersky, A. N., and Spirin, A. S. (1958)
Nature, 182, 111-112.
25.Doi, R. H., and Igarashi, R. T. (1965) J.
Bacteriol., 90, 384-390.
26.Dubnau, D., Smith, I., Morell, P., and Marmur, J.
(1965) Proc. Natl. Acad. Sci. USA, 54, 491-498.
27.Palleroni, N. J., Kunisawa, R., Contopoulou, R.,
and Doudoroff, M. (1973) Int. J. Syst. Bacteriol., 23,
333.
28.Johnson, J. L., and Francis, B. S. (1975) J.
Gen. Microbiol., 88, 229-244.
29.Woese, C. R., Sogin, M. L., and Sutton, L. A.
(1974) J. Mol. Evol., 3, 293-299.
30.Woese, C. R., and Fox, G. E. (1977) Proc.
Natl. Acad. Sci. USA, 74, 5088-5090.
31.Woese, C. R., Magrum, L. J., and Fox, G. E.
(1978) J. Mol. Evol., 11, 245-252.
32.Whittaker, R. H. (1969) Science,
163, 150-163.
33.Whittaker, R. H., and Margulis, L. (1978)
Biosystems, 10, 3-18.
34.Woese, C. R. (1987) Microbiol. Rev.,
51, 221-271.
35.Woese, C. R., Kandler, O., and Wheelis, M. L.
(1990) Proc. Natl. Acad. Sci. USA, 87, 4576-4579.
36.Woese, C. R. (2006) in The Prokaryotes
(Dworkin, M., Falkow, S., Rosenberg, E., Scleifer, K.-H., and
Stackebrandt, E., eds.) Vol. 1, Springer-Verlag, New York, pp.
3-23.
37.Woese, C. R., Gibson, J., and Fox, G. E. (1980)
Nature, 283, 212-214.
38.Stackebrandt, E., and Goebel, B. M. (1994)
Int. J. Syst. Bacteriol., 44, 846-849.
39.Stackebrandt, E., and Ebers, J. (2006)
Microbiol. Today, 10, 152-155.
40.Lapage, S. P., Sneath, P. H. A., Lessel, E. F.,
Skerman, V. B. D., Seeliger, H. P. R., and Clark, W. A. (eds.) (1992)
International Code of Nomenclature of Bacteria (1990 Revision),
ASM Press; http://www.bacterio.cict.fr/code.html
41.Garrity, G. M., et al. (eds.) (2001, 2005)
Bergey's Manual of Systematic Bacteriology, 2nd Edn.,
Springer-Verlag, New York-Berlin-Heidelberg, Vols. 1, 2.
42.Garrity, G. M., Bell, J. A., and Lilburn, T. G.
(2005) in Bergey's Manual of Systematic Bacteriology, 2nd Edn.
(Garrity, G. M., et al., eds.) Vol. 2, Springer-Verlag, New
York-Berlin-Heidelberg, pp. 159-187.
43.DeSantis, T. Z., Hugenholtz, P., Larsen, N.,
Rojas, M., Brodie, E. L., Keller, K., Huber, T., Dalevi, D., Hu, P.,
and Andersen, G. L. (2006) Appl. Environ. Microbiol., 72,
5069-5072.
44.Hugenholtz, P. (2002) Genome Biol.,
3, reviews 0003.1-0003.8.
45.Robertson, C. E., Harris, J. K., Spear, J. R.,
and Pace, N. R. (2005) Curr. Opin. Microbiol., 8,
638-642.
46.Cole, J. R., Chai, B., Farris, R. J., Wang, Q.,
Kulam, S. A., McGarrell, D. M., Garrity, G. M., and Tiedje, J. M.
(2005) Nucleic Acids Res., 33, Database issue
D294-D296.
47.Ludwig, W., Strunk, O., Westram, R., Richter, L.,
Meier, H., Yadhukumar, I., Buchner, A., Lai, T., Steppi, S., Jobb, G.,
Forster, W., Brettske, I., Gerber, S., Ginhart, A. W., Gross, O.,
Grumann, S., Hermann, S., Jost, R., Konig, A., Liss, T., Lussmann, R.,
May, M., Nonhoff, B., Reichel, B., Strehlow, R., Stamatakis, A.,
Stuckmann, N., Vilbig, A., Lenke, M., Ludwig, T., Bode, A., and
Schleifer, K. H. (2004) Nucleic Acids Res., 32,
1363-1371.
48.Lilburn, T. G., and Garrity, G. M. (2004) Int.
J. Syst. Evol. Microbiol., 54, 7-13.
49.Ludwig, W., and Klenk, H.-P. (2001) in
Bergey's Manual of Systematic Bacteriology, 2nd Edn. (Garrity,
G. M., et al., eds.) Vol. 1, Springer-Verlag, New
York-Berlin-Heidelberg, pp. 49-65.
50.Wolf, Y. I., Rogozin, I. B., Grishin, N. V.,
Tatusov, R. L., and Koonin, E. V. (2001) BMC Evol. Biol.,
1, 8.
51.Mirkin, B. G., Fenner, T. I., Galperin, M. Y.,
and Koonin, E. V. (2003) BMC Evol. Biol., 3, 2.
52.Makarova, K. S., and Koonin, E. V. (2005)
Curr. Opin. Microbiol., 8, 586-594.
53.Cavalier-Smith, T. (2002) Int. J. Syst. Evol.
Microbiol., 52, 7-76.
54.Gupta, R. S. (1998) Microbiol. Mol. Biol.
Rev., 62, 1435-1491.
55.Griffiths, E., and Gupta, R. S. (2004) Int.
Microbiol., 7, 41-52.
56.Galtier, N., and Lobry, J. R. (1997) J. Mol.
Evol., 44, 632-636.
57.Kimura, H., Sugihara, M., Kato, K., and Hanada,
S. (2006) Appl. Environ. Microbiol., 72, 21-27.
58.Achenbach-Richter, L., Gupta, R., Stetter, K. O.,
and Woese, C. R. (1987) Syst. Appl. Microbiol., 9,
34-39.
59.Weisburg, W. G., Giovannoni, S. J., and Woese, C.
R. (1989) Syst. Appl. Microbiol., 11, 128-134.
60.Woese, C. R., Achenbach, L., Rouviere, P., and
Mandelco, L. (1991) Syst. Appl. Microbiol., 14,
364-371.
61.Burggraf, S., Stetter, K. O., Rouviere, P., and
Woese, C. R. (1991) Syst. Appl. Microbiol., 14,
346-351.
62.Cole, J. R., Chai, B., Marsh, T. L., Farris, R.
J., Wang, Q., Kulam, S. A., Chandra, S., McGarrell, D. M., Schmidt, T.
M., Garrity, G. M., and Tiedje, J. M. (2003) Nucleic Acids Res.,
31, 442-443.
63.Olsen, G. J., Matsuda, H., Hagstrom, R., and
Overbeek, R. (1994) Comput. Appl. Biosci., 10, 41-48.
64.Stamatakis, A., Ludwig, T., and Meier, H. (2005)
Bioinformatics, 21, 456-463.
65.Winker, S., and Woese, C. R. (1991) Syst.
Appl. Microbiol., 14, 305-310.
66.Reysenbach, A.-L., Longnecker, K., and Kirshtein,
J. (2000) Appl. Environ. Microbiol., 66, 3798-3806.
67.Wuyts, J., van de Peer, Y., and De Wachter, R.
(2001) Nucleic Acids Res., 29, 5017-5028.
68.Stackebrandt, E., Tindall, B., Ludwig, W., and
Goodfellow, M. (1999) in Biology of the Prokaryotes (Lengeler,
J. W., et al., eds.) Georg Thieme Verlag, Stuttgart.
69.Slesarev, A. I., Mezhevaya, K. V., Makarova, K.
S., Polushin, N. N., Shcherbinina, O. V., Shakhova, V. V., Belova, G.
I., Aravind, L., Natale, D. A., Rogozin, I. B., Tatusov, R. L., Wolf,
Y. I., Stetter, K. O., Malykh, A. G., Koonin, E. V., and Kozyavkin, S.
A. (2002) Proc. Natl. Acad. Sci. USA, 99, 4644-4649.
70.Omelchenko, M. V., Wolf, Y. I., Gaidamakova, E.
K., Matrosova, V. Y., Vasilenko, A., Zhai, M., Daly, M. J., Koonin, E.
V., and Makarova, K. S. (2005) BMC Evol. Biol., 5,
57.
71.Barns, S. M., Delwiche, C. F., Palmer, J. D., and
Pace, N. R. (1996) Proc. Natl. Acad. Sci. USA, 93,
9188-9193.
72.Takai, K., and Horikoshi, K. (1999)
Genetics, 152, 1285-1297.
73.Takai, K., and Sako, Y. (1999) FEMS Microbiol.
Ecol., 28, 177-188.
74.Makarova, K. S., and Koonin, E. V. (2003)
Genome Biol., 4, 115.
75.Brochier, C., Forterre, P., and Gribaldo, S.
(2004) Genome Biol., 5, R17.
76.Brock, T. D., Brock, K. M., Belly, R. T., and
Weiss, R. L. (1972) Arch. Microbiol., 84, 54-68.
77.Bonch-Osmolovskaya, E. A. (1994) FEMS
Microbiol. Rev., 15, 65-77.
78.Prokofeva, M. I., Miroshnichenko, M. L.,
Kostrikina, N. A., Chernyh, N. A., Kuznetsov, B. B., Tourova, T. P.,
and Bonch-Osmolovskaya, E. A. (2000) Int. J. Syst. Bacteriol.,
50, 2001-2008.
79.Bertoldo, C., and Antranikian, G. (2006) in
The Prokaryotes (Dworkin, M., Falkow, S., Rosenberg, E.,
Scleifer, K.-H., and Stakebrandt, E., eds.) Vol. 3, Springer-Verlag,
New York, pp. 69-81.
80.Kurr, M., Huber, R., Konig, H., Jannasch, H. W.,
Fricke, H., Trincone, A., Krstjansson, J. K., and Stetter, K. O. (1991)
Arch. Microbiol., 156, 239-247.
81.Wasserfallen, A., Nolling, J., Pfister, P.,
Reeve, J., and Conway de Macario, E. (2000) Int. J. Syst. Evol.
Microbiol., 50, 43-53.
82.Whitman, W. B., Boone, D. R., and Koga, Y. (2001)
in Bergey's Manual of Systematic Bacteriology, 2nd Edn.
(Garrity, G. M., et al., eds.) Vol. 1, Springer-Verlag, New
York-Berlin-Heidelberg, p. 236.
83.Golyshina, O. V., Pivovarova, T. A., Karavaiko,
G. I., Kondrat'eva, T. F., Moore, E. R. B., Abraham, W. R., Lunsdorf,
H., Timmis, K. N., Yakimov, M. M., and Golyshin, P. N. (2000) Int.
J. Syst. Evol. Microbiol., 50, 997-1006.
84.Inagaki, Y., Susko, E., and Roger, A. J. (2006)
Proc. Natl. Acad. Sci. USA, 103, 4528-4533.
85.Gao, B., and Gupta, R. S. (2007) BMC
Genomics, 8, 86.
86.Pace, N. R., Stahl, D., Lane, D. J., and Olsen,
G. (1986) J. Adv. Microb. Ecol., 9, 1-55.
87.Dawson, S., DeLong, E., and Pace, N. (2006) in
The Prokaryotes (Dworkin, M., Falkow, S., Rosenberg, E.,
Scleifer, K.-H., and Stackebrandt, E., eds.) Vol. 3, Springer-Verlag,
New York, pp. 281-289.
88.Konneke, M., Bernhard, A. E., De la Torre, J. R.,
Walker, C. B., Waterbury, J. B., and Stahl, D. A. (2005) Nature,
437, 543-546.
89.Preston, C. M., Wu, K. Y., Molinski, T. F., and
DeLong, E. F. (1996) Proc. Natl. Acad. Sci. USA, 93,
6241-6246.
90.Auchtung, T. A., Takacs-Vesbach, C. D., and
Cavanaugh, C. M. (2006) Appl. Environ. Microbiol., 72,
5077-5082.
91.Reysenbach, A.-L., Liu, Y., Bant, A. B.,
Beveridge, T. J., Kirshtein, J. D., Schouten, S., Tivey, M. K., von
Damm, K. L., and Voytek, M. A. (2006) Nature, 422,
444-447.
92.Huber, H., Hohn, M. J., Rachel, R., Fuchs, T.,
Wimmer, V. C., and Stetter, K. O. (2002) Nature, 417,
63-67.
93.Waters, E., Hohn, M. J., Ahel, I., Graham, D. E.,
Adams, M. D., Barnstead, M., Beeson, K. Y., Bibbs, L., Bolanos, R.,
Keller, M., Kretz, K., Lin, X., Mathur, E., Ni, J., Podar, M.,
Richardson, T., Sutton, G. G., Simon, M., Soll, D., Stetter, K. O.,
Short, J. M., and Noordewier, M. (2003) Proc. Natl. Acad. Sci.
USA, 100, 12984-12988.
94.Di Giulio, M. (2007) Gene, 401,
108-113; Epub 2007 Jul 17.
95.Brochier, C., Gribaldo, S., Zivanovic, Y.,
Confalonieri, F., and Forterre, P. (2005) Genome Biol.,
6, R42.
96.Brochier-Armanet, C., and Forterre, P. (2006)
Archaea, 2, 83-93.
97.Bonch-Osmolovskaya, E. A., Miroshnichenko, M. L.,
Lebedinsky, A. V., Chernyh, N. A., Nazina, T. N., Ivoilov, V. S.,
Belyaev, S. S., Boulygina, E. S., Lysov, Yu. P., Perov, A. N.,
Mirzabekov, A. D., Hippe, H., Stackebrandt, E., L'Haridon, S., and
Jeanthon, C. (2003) Appl. Environ. Microbiol., 69,
6143-6151.
98.Balk, M., Weijma, J., and Stams, A. J. (2002)
Int. J. Syst. Evol. Microbiol., 52, 1361-1368.
99.Wery, N., Lesongeur, F., Pignet, P., Derennes,
V., Cambon-Bonavita, M. A., Godfroy, A., and Barbier, G. (2001) Int.
J. Syst. Evol. Microbiol., 51, 495-504.
100.Alain, K., Marteinsson, V. T., Miroshnichenko,
M. L., Bonch-Osmolovskaya, E. A., Prieur, D., and Birrien, J. L. (2002)
Int. J. Syst. Evol. Microbiol., 52, 1331-1339.
101.Zeikus, J. G., Dawson, M. A., Thompson, T. E.,
Ingvorsen, K., and Hatchikian, E. C. (1983) J. Gen. Microbiol.,
129, 1159-1169.
102.Henry, E. A., Devereux, R., Maki, J. S.,
Gilmour, C. C., Woese, C. R., Mandelco, L., Schauder, R., Remsen, C.
C., and Mitchell, R. (1994) Arch. Microbiol., 161,
62-69.
103.Kashefi, K., Holmes, D. E., Reysenbach, A.-L.,
and Lovley, D. R. (2002) Appl. Environ. Microbiol., 68,
1735-1742.
104.Brock, T. D., and Freeze, H. (1969) J.
Bacteriol., 98, 289-297.
105.Miroshnichenko, M. L., L'Haridon, S., Jeanthon,
C., Antipov, A. N., Kostrikina, N. A., Tindall, B. J., Schumann, P.,
Spring, S., Stackebrandt, E., and Bonch-Osmolovskaya, E. A. (2003)
Int. J. Syst. Evol. Microbiol., 53, 747-752.
106.Miroshnichenko, M. L., L'Haridon, S.,
Nersessian, O., Antipov, A. N., Kostrikina, N. A., Tindall, B. J.,
Schumann, P., Spring, S., Stackebrandt, E., Bonch-Osmolovskaya, E. A.,
and Jeanthon, C. (2003) Int. J. Syst. Evol. Microbiol.,
53, 1143-1148.
107.Chumakov, K. M. (1987) Sov. Sci. Rev. Sect.
D Biol. Rev., 7, 51-94.
108.Hensel, R., Demharter, W., Kandler, O.,
Kroppenstedt, R. M., and Stackebrandt, E. (1986) Int. J. Syst.
Bacteriol., 36, 444-453.
109.Hartmann, R. K., Wolters, J., Kroger, B.,
Schultz, S., Sprecht, T., and Erdmann, V. A. (1989) Syst. Appl.
Microbiol., 11, 243-249.
110.Embly, T. M., Thomas, R. H., and Williams, R.
A. D. (1993) Syst. Appl. Microbiol., 16, 25-29.
111.Brown, W. M., Prager, E. M., Wang, A., and
Wilson, A. C. (1982) J. Mol. Evol., 18, 225-286.
112.Eisen, J. A. (1995) J. Mol. Evol.,
41, 1105-1123.
113.Jackson, T. J., Ramaley, R. F., and Meinschein,
W. G. (1973) Int. J. Syst. Bacteriol., 23, 28-36.
114.Saiki, T., Kobayashi, Y., Kawagoe, K., and
Beppu, T. (1985) Int. J. Syst. Bacteriol.,
35, 253-259.
115.Greene, A. C., Patel, B. K. C., and Sheehy, A.
J. (1997) Int. J. Syst. Bacteriol.,
47, 505-509.
116.Miroshnichenko, M. L., Slobodkin, A. I.,
Kostrikina, N. A., L'Haridon, S., Nercessian, O., Spring, S.,
Stackebrandt, E., Bonch-Osmolovskaya, E. A., and Jeanthon, C. (2003)
Int. J. Syst. Evol. Microbiol., 53, 1637-1641.
117.Caldwell, D. E., Caldwell, S. J., and Laycock,
J. P. (1976) Can. J. Microbiol., 22,
1509-1517.
118.Bonch-Osmolovskaya, E. A., Sokolova, T. G.,
Kostrikina, N. A., and Zavarzin, G. A. (1990) Arch. Microbiol.,
153, 151-155.
119.Nazina, T. N., Tourova, T. P., Poltaraus, A.
B., Novikova, E. V., Grigoryan, A. A., Ivanova, A. E., Lysenko, A. M.,
Petrunyaka, V. V., Osipov, G. A., Belyaev, S. S., and Ivanov, M. V.
(2001) Int. J. Syst. Evol. Microbiol.,
51, 433-446.
120.Rainey, F. A., Ward, N. L., Morgan, H. W.,
Toalster, R., and Stackebrandt, E. (1993) J. Bacteriol.,
175, 4772-4779.
121.Sokolova, T., Hanel, J., Onyenwoke, R. U.,
Reysenbach, A. L., Banta, A., Geyer, R., Gonza, J. M., Whitman, W. B.,
and Wiegel, J. (2007) Extremophiles, 11, 145-157.
122.Hugenholtz, P., Pitulle, C., Hershberger, K.
L., and Pace, N. R. (1998) J. Bacteriol., 180,
366-376.
123.Sievert, S. M., Kuever, J. M., and Muyzer, G.
(2000) Appl. Environ. Microbiol., 66, 3102-3109.
124.Miroshnichenko, M. L., Kostrikina, N. A.,
Chernyh, N. A., Pimenov, N. V., Tourova, T. P., Antipov, A. N.,
Spring, S., Stackebrandt, E., and Bonch-Osmolovskaya, E. A. (2003)
Int. J. Syst. Evol. Microbiol., 53, 323-329.
125.Miroshnichenko, M. L., Kostrikina, N. A.,
L'Haridon, S., Jeanthon, S., Hippe, S., Stackebrandt, E., and
Bonch-Osmolovskaya, E. A. (2002) Int. J. Syst. Evol. Microbiol.,
52, 1299-1304.
126.Miroshnichenko, M. L., L'Haridon, S., Schumann,
P., Spring, S., Bonch-Osmolovskaya, E. A., Jeanthon, C., and
Stackebrandt, E. (2004) Int. J. Syst. Evol. Microbiol.,
54, 41-45.
127.Islam, T., Jensen, S., Reigstad, L. J., Larsen,
O., and Birkeland, N.-K. (2007) Thermophiles 2007, 9th Int.
Conf. on Thermophiles Research, Bergen, 24-27 September, 2007.
128.Pace, N. R. (1991) Cell, 65,
531-533.
129.Pace, N. R. (1997) Science, 276,
734-40.
130.Schwartzman, D. W., and Lineweaver, C. H.
(2004) Biochem. Soc. Trans., 32, 168-171.
131.Klenk, H.-P., Spitzer, M., Ochsenreiter, T.,
and Fuellen, G. (2004) Biochem. Soc. Trans., 32,
175-178.
132.Gogarten-Boekels, M., Hilario, E., and
Gogarten, J. P. (1995) Orig. Life Evol. Biosph., 25,
251-264.
133.Makarova, K. S., Aravind, L., Grishin, N. V.,
Rogozin, I. B., and Koonin, E. V. (2002) Nucleic Acids Res.,
30, 482-496.
134.Makarova, K. S., Wolf, Y. I., and Koonin, E. V.
(2003) Trends Genet., 19, 172-176.
135.Vieille, C., and Zeikus, G. J. (2001)
Microbiol. Mol. Biol. Rev., 65, 1-43.
136.Hickey, D. A., and Singer, G. A. (2004)
Genome Biol., 5, 117.
137.Nesbo, C. L., L'Haridon, S., Stetter, K. O.,
and Doolittle, W. F. (2001) Mol. Biol. Evol., 18,
362-375.
138.Aravind, L., Tatusov, R. L., Wolf, Y. I.,
Walker, D. R., and Koonin, E. V. (1998) Trends Genet.,
14, 442-444.
139.Koonin, E., Makarova, K. S., and Aravind, L.
(2001) Annu. Rev. Microbiol., 55, 709-742.
140.Forterre, P., Bouthier De La Tour, C.,
Philippe, H., and Duguet, M. (2000) Trends Genet., 16,
152-154.
141.Galtier, N., Tourasse, N., and Gouy, M. (1999)
Science, 283, 220-221.
142.Di Giulio, M. (2000) J. Theor. Biol.,
203, 203-213.
143.Di Giulio, M. (2003) J. Mol. Evol.,
57, 721-730.
144.Shimizu, H., Yokobori, S., Ohkuri, T.,
Yokogawa, T., Nishikawa, K., and Yamagishi, A. (2007) J. Mol.
Biol., 369, 1060-1069.
145.Harris, J. K., Kelley, S. T., and Pace, N. R.
(2004) Appl. Environ. Microbiol., 70, 845-849.