Do We Need Many Genes for Phylogenetic Inference?
V. V. Aleshin1*, A. V. Konstantinova1, K. V. Mikhailov2, M. A. Nikitin2, and N. B. Petrov1
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: Aleshin@genebee.msu.su2Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received September 10, 2007
Fifty-six nuclear protein coding genes from Taxonomically Broad EST Database and other databases were selected for phylogenomic-based examination of alternative phylogenetic hypotheses concerning intergroup relationship between multicellular animals (Metazoa) and other representatives of Opisthokonta. The results of this work support sister group relationship between Metazoa and Choanoflagellata. Both of these groups form the taxon Holozoa along with the monophyletic Ichthyosporea or Mesomycetozoea (a group that includes Amoebidium parasiticum, Sphaeroforma arctica, and Capsaspora owczarzaki). These phylogenetic hypotheses receive high statistical support both when utilizing whole alignment and when only 5000 randomly selected alignment positions are used. The presented results suggest subdivision of Fungi into Eumycota and lower fungi, Chytridiomycota. The latter form a monophyletic group that comprises Chytridiales + Spizellomycetales + Blastocladiales (Batrachochytrium, Spizellomyces, Allomyces, Blastocladiella), contrary to the earlier reports based on the analysis of 18S rRNA and a limited set of protein coding genes. The phylogenetic distribution of genes coding for a ubiquitin-fused ribosomal protein S30 implies at least three independent cases of gene fusion: in the ancestors of Holozoa, in heterotrophic Heterokonta (Oomycetes and Blastocystis) and in the ancestors of Cryptophyta and Glaucophyta. Ubiquitin-like sequences fused with ribosomal protein S30 outside of Holozoa are not FUBI orthologs. Two independent events of FUBI replacement by the ubiquitin sequence were detected in the lineage of C. owczarzaki and in the monophyletic group of nematode worms Tylenchomorpha + Cephalobidae. Bursaphelenchus xylophilus (Aphelenchoidoidea) retains a state typical of the rest of the Metazoa. The data emphasize the fact that the reliability of phylogenetic reconstructions depends on the number of analyzed genes to a lesser extent than on our ability to recognize reconstruction artifacts.
KEY WORDS: molecular evolution, homoplasy, origin of multicellular organisms, secondary structure of SSU rRNA, TylenchidaDOI: 10.1134/000629790712005X
Abbreviations: eEF1A) eukaryotic elongation factor 1A; EFL) elongation factor-like (a paralog of elongation factor 1A); FUBI) ubiquitin-like peptide fused with S30 ribosomal protein of animals and other Holozoa; Hsp) heat shock protein, chaperon; RpL) ribosomal protein of large ribosomal subunit; RpS) ribosomal protein of small ribosomal subunit; tef) gene of elongation factor 1A.
This issue of Biochemistry (Moscow) is dedicated to the fiftieth
anniversary of the article [1] in which species
specificity of nucleic acids was first applied to solution of a
concrete taxonomic problem--revision of the systematic position of
bacterium Morganella (“Proteus”)
morganii. That article had an extraordinary fate: it opened a
new scientific discipline that is presently used for special courses of
lectures in universities, hundreds of researchers work in laboratories
all over the world, special journals are published, scientific
achievements of this field are used in express diagnosis of pathogens,
selective breeding, criminalistics, and debates on fishing quotas.
During a single scientific generation methods of comparative analysis
have made progress from determination of base composition to comparison
of complete genomes. Such quick progress significantly raises the level
of requirements for methodical aspects of works in this field, without
paying much attention to methodology by re-addressing it to the
conventional norm, the mode dictated by the methodical arsenal. Just
the formulation of the question submitted in the title of this article
was inconceivable 50 years ago: the structure of no gene was known at
the time. Now primary structures of all genes of approximately one
thousand species of prokaryotes and many eukaryotes are known and
phylogenetic conclusions “by a single gene” (for example, of
18S rRNA) are losing their attractiveness for the general public,
reports about them raise the usual question of whether these
conclusions will be supported by analyses of different genes. Such a
question is based simultaneously on the sensitivity to the dynamic
development of modern science (that raises high the methodical
requirements) and unfamiliarity with methodology of systematics and
phylogenetics. Systematics is intended to describe most concisely the
whole known variety [2]. Its subject is expanded
with every large and small discovery--it is as endless as knowledge
itself. For concise description, genomic structures should be
formalized and systematized like structures visible in the light and
electron microscopes, like types of respiration and embryonic
development, pathways of gene regulation and details of behavior, the
caddis-fly houses, ant's and bird's nests--everything that is already
discovered and will be discovered in the future by researchers of
biodiversity; all this should be considered in diagnoses of taxa,
created by systematics and influencing their composition. The mission
of phylogenetics is much more concrete, it is ultimate--to establish
kinship ties irrespective of the similarity and distinction as a whole
[3-5]. The first and simplest
theorem of phylogenetics gives a paradoxical answer to the question
whether phylogenetic hypotheses will be confirmed by analysis of other
features (genes): “They will not be confirmed, but this may be of
no significance” (Fig. 1).
It is admissible in theory that the monophyletic taxon emerging during rapid radiation or having an extremely old age is characterized by only a single feature. Even if we discover this feature and will be able to somehow prove its reliability, this discovery will grant us nearly nothing. The justification of phylogenetics is in the hope of discovering more features in species of a monophyletic taxon, including those that have not been studied yet. There are grounds for such a hope: these species are more closely related to each other than to other species. Thus, phylogenetics is expected to be prognostic but, in the strict sense, this is beyond its framework. More important is that such expectation is opposed by a strict prediction following from evolution itself: individual species, evolving from an ancestor, inevitably acquire distinctions in some features, which distort their ancestral resemblance. As a result, species that evolve at a slower rate, in relation to at least some features, inevitably will be in these features more similar to each other than to their fast-evolving nearest relatives. This does not matter for phylogenetics (but cannot be indifferent for systematics or practical applications). The uncertainty of biological evolution implies impossibility of predicting by what features the ancestral similarity will be lost during evolution, in other words, which genes carry phylogenetic signal for a given node, and which carry only noise or a “false” signal such as symplesiomorphy (Fig. 1).Fig. 1. Distinguishing monophyletic groups by shared evolutionary acquirements (synapomorphies) and the forbidden in phylogenetics procedure of combination by the ancestral (symplesiomorphic) similarity for a given tree. A, B, C, D, E are modern species (or races, classes, phyla--any operational taxonomical units); I-V features or groups of features (e.g. genes): ancestral state is denoted by filled symbols, open symbols show the apomorphic state; nodes where corresponding apomorphy was inherited by descendant species are marked on the tree; species sharing common features are boxed (some variants). Only combinations of open symbols mean monophyletic taxon. The grouping by feature IV is methodologically false, though it coincides in volume with taxon defined by synapomorphy III; plesiomorphic similarity by features I and V also does not distinguish monophyletic groups.
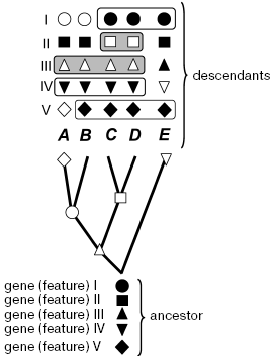
Thus, a phylogeneticist should be interested in the reliability of the phylogenetic hypothesis but not in the way the monophyletic origin was established--by single- or multigene analysis. In any case, it is possible to find features which might not be directly contradicting the drawn conclusion, but at least are not supporting it. The case of a limited set of features is a different matter. Sequences of a single 18S rRNA gene may have no features suitable for detection of a monophyletic origin of a given taxon, either due to an insignificant difference between its nearest common ancestor and contemporary species (a short time of the stem group existence) or because of the long-term independent evolution of daughter clades. Such features may be detected in future on larger samples. Without such samples, it is difficult to recognize homoplasies and exact reversion of evolution (violations of Dollo's law). Below we shall consider examples of phylogenetic conclusions drawn on gene sets and single genes.
UNICELLULAR RELATIVES OF MULTICELLULAR ANIMALS
Molecular synapomorphies of eEF1A. The search for unicellular relatives of animals is an important problem, the solution of which may elucidate evolution of cell communication, mechanisms of historical formation of ontogenesis of multicellular organisms, their growth, and cellular differentiation. Modern view on this problem start from the discovery of a specific insertion of twelve triplets in one protein-coding genes (elongation factor 1A, eEF1A--tef) [6]. In this case, the simple similarity of insertions is not what makes it significant; similar traits can be easily found in animals with other organisms too. It is important in the case of eEF1A that an alternative state observed in plants, ciliates, myxomycetes, dysentery amoeba, and Euglena gracilis is also characteristic of EF-Tu, an orthologous protein of prokaryotes (archeans and bacteria)--an outgroup of eukaryotes. This fact allows us to choose between two alternative evolutionary hypotheses: insertions or deletions of 12 triplets. According to the first hypothesis, the evolution of EF1A can be presented simply as proceeding from bacteria to eukaryotes without changes in length. In this case, it is sufficient to admit monophyletic origin of fungi and animals and a single acquirement of the insertion in their common ancestor. The alternative hypothesis of an initially long gene will require more than one independent precise deletion of the mentioned fragment in different evolutionary lineages of prokaryotes and eukaryotes, and is rejected as a less parsimonious assumption. Thus, the polarization of eEF1A evolution is achieved; in this case fungi and animals as having not simply “a common feature” but an evolutionarily new common feature (synapomorphy) are declared as relatives (Fig. 1). The insertion under consideration is localized in the unstructured region on the protein surface, forming a pair of antiparallel beta-sheets. It is unknown whether its conservativeness is associated with any functional features of fungal and animal eEF1A or only with intra- and intermolecular co-variations slowing-down the rate of molecular evolution [7].
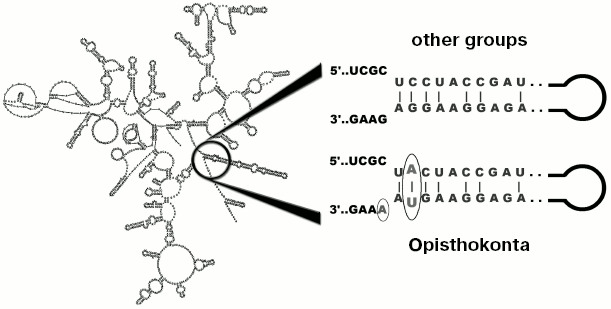
Results of analysis of different popular markers like 18S and 28S rRNA genes do not contradict the hypothesis of close relationship between fungi and animals, supplementing the group (with moderate level of statistical support) with unicellular organisms: Choanoflagellata, Mesomycetozoea, nucleariids, some other protists, and “trichomycetes” [8]. All these groups were defined as the supertaxon Opisthokonta. Along with general similarity of 18S rRNA sequences, all studied species (over 3000) have a common motif which is located at the base of helix 49 (Fig. 2) [9]. Outside of Opisthokonta this motif exists as a rare exception (line Cercozoa and in genera Goniomonas and Telonema, one species in each genus).
Although not every gene allows to reveal the monophyletic origin of Opisthokonta, at the present time the existence of this group is practically unquestionable [10, 11] (alternatives are considered very rarely [12, 13]). Cladistic methodology, used for its justification [3, 6], is used in clarification of other issues, such as rooting of phylogenetic tree of eukaryotes. Thus, most eukaryotes are characterized by fused genes of dihydrofolate reductase and thymidylate synthetase (two genes of pyrimidine metabolism), encoding a bifunctional protein [14], whereas in Opisthokonta these genes are transcribed individually. However this similarity cannot serve as an argument in favor of close relationship between fungi and animals since split state of these genes is also in common with the outgroup (bacteria) [12], but fusion of dihydrofolate reductase and thymidylate synthetase can be considered as an indication of relatedness (monophyletic origin) of the majority of other eukaryotes, thus forming a monophyletic group.Fig. 2. Localization of a nucleotide motif characteristic of Opisthokonta in 18S rRNA.
Present day situation. Fifteen years that passed after publication [6] have confirmed the heuristic and a certain prognostic power of phylogenetic hypothesis proposed on the basis of a single gene. Since that time, the predicted insertion was found in eEF1A of numerous species of Opisthokonta including microsporidia, choanoflagellates, and members of the later described “class” Mesomycetozoea [11], although its exact sequence and length were not as constant as it seemed from analysis of a smaller sample. Despite manifold enlargement of the database no species outside of Opisthokonta were found to possess a specific insertion in eEF1A. However the present day situation appears to be more complicated due to the discovery of a paralog of eEF1A, EFL [15], which is conserved to the same degree, but strongly differs from eEF1A in primary structure and always contains an insertion of similar size and localization. In some species of Opisthokonta and members of other groups of eukaryotes, EFL replaces eEF1A. For example, in the Monosiga brevicollis genome, now completely sequenced, only the EFL coding gene is present [16] and the gene for eEF1A is absent. On the contrary, the cDNA library (http://tbestdb.bcm.umontreal.ca/searches/login.php) of another choanoflagellate, Monosiga ovata, contains 104 clones of eEF1A cDNA, whereas not a single clone of EFL cDNA is found. EFL also replaces eEF1A in many chytrid fungi. Additional investigations are necessary to elucidate the distribution of EFL among different groups of eukaryotes. If in 1993 a limited set of eEF1A included their paralogs, then the hypothesis of fungal and animal relatedness would not have been formulated at that time.
It is interesting that the routine application of calculation procedures, only partially based on cladistic principles, easily leads to erroneous results irrespective of the number of compared genes. Thus, a tree based on concatenated sequences of 780 genes chosen from eight fully sequenced genomes and a supertree based on 780 individual trees give an erroneous result that rejects monophyletic origin of Opisthokonta at the statistically reliable level [12]. It appears that the mistake in this case is caused by an artifact of “long branch attraction” [11, 17]. Factors, responsible for enhancement of such artifacts upon mechanistic increase of the number of analyzed genes were discussed elsewhere [7, 17, 18].
Despite numerous attempts to establish phylogenetic relationship within the Opisthokonta clade employing a single or a set of genes, modern hypotheses in this respect are rather controversial. Comparison of the mitochondrial proteome supports the traditional view that Choanoflagellata are nearest relatives of multicellular animals [19]; it is also supported by the presence of a specific amino acid motif STEPPYS in eEF1A of choanoflagellates and metazoans [11]. On the other hand, trees based on 18S rRNA analysis sometimes place choanoflagellates as a sister group of Mesomycetozoea [20], and recently it has been suggested that a symbiotic amoeba Capsaspora owczarzaki comprises their sister group on the basis of combined analysis of 18S and 28S rRNA [8]. Other authors consider the position of this amoeba within Opisthokonta as uncertain [21, 22] or qualify this organism as Mesomycetozoea [23, 24]. The newest consensus suggestion is to recognize the uncertainty of relatedness between animals, Choanoflagellata, Mesomycetozoea, Ministeria, Corallochytrium, and Capsaspora, which is graphically shown in the tree as multifurcation [25].
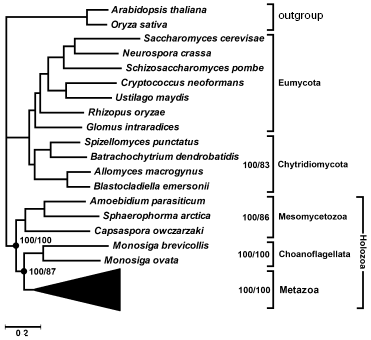
Multigene analysis. We have attempted to solve the above-mentioned uncertainty using genomic data. Recently different laboratories [25] have obtained representative cDNA libraries of several unicellular Opisthokonta species and the genome of one species, Monosiga brevicollis, was completely sequenced in the Joint Genome Institute (http://genome.jgi-psf.org/Monbr1/Monbr1.home.html). We have selected 56 orthologs from available databases that had their representatives in all Opisthokonta groups. Thirty-five of them encode ribosomal proteins, the rest were genes encoding transcription factors, chaperons, cytoskeletal proteins, components of translation apparatus and energy metabolism. Their amino acid sequences were manually aligned and concatenated, the tree was built by the maximum parsimony and Bayesian methods. In the latter case, parameters were optimized for each gene (a model of amino acid substitutions; rate heterogeneity (alpha parameter of gamma distribution); portion of invariable sites). The total length of alignment covered 10,678 positions. The consensus tree (Fig. 3) is consistent with the hypothesis of sister group relationship between animals and choanoflagellates. Its posterior probability is 100%. The posterior probability of the hypothesis of monophyletic origin of Mesomycetozoea including Capsaspora owczarzaki is also 100%. Both these groups also receive 100% support in bootstrap analysis. In the maximally parsimonious and Bayesian trees all Opisthokonta are separated into two evolutionary branches. One of them combines Metazoa + Choanoflagellata and a clade of Mesomycetozoea in a taxon, for which the name Holozoa was proposed earlier [19], whereas the other is represented in our set by fungi. A monophyletic group of the lower, chytrid fungi, characterized by flagellated stages in the life cycle, receives 100% posterior probability. It is notable that analysis of 18S rRNA sequences [26] and six protein-coding fungal genes [27] did not reveal monophyletic origin of chytrid fungi--species of the order Blastocladiales were separated from them. Analysis of concatenated sequences of 56 proteins favors monophyletic origin of chytrid fungi (Fig. 3).
The advantage of topologies containing the above-mentioned groups is confirmed by KH- [28], SH- [29], and AU-tests [30]. According to the criteria used, alternative topologies, obtained from the tree in Fig. 3 by displacement of individual branches, are worse than the consensus one (table). KH and AU tests show, that in all cases except alterations distorting monophyletic origin of chytrid fungi, distinctions are statistically significant. A more liberal SH-test rejects with the probability of 0.995 both the hypothesis of closer relatedness of animals with Mesomycetozoea rather than with Choanoflagellata and the hypothesis of non-monophyly of Holozoa.Fig. 3. Phylogenetic tree of Opisthokonta (56 proteins-10,678 alignment positions) reconstructed by MrBayes 3.1.2 [58] with parameters optimized for each protein and by protpars program of the PHYLIP package [31] (100 bootstrap replicas). General parameters of MrBayes 3.1.2: nchains = 4; nruns = 2; rates = invgamma; ngammacat = 8; ngen = 2,000,000; burnin = 1,000,000. Values of posterior probability and bootstrap support of above-discussed taxa are shown.
Statistical estimation of alternative hypotheses of phylogenetic
relationships within Opisthokonta
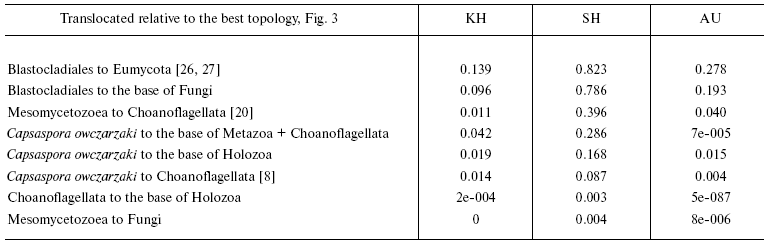
Note: Testing was carried out using programs TREE-PUZZLE 5.2 [57] and CONSEL [30]; the
likelihoods for sites were calculated according to the model WAG +
Gamma (6 categories).
To estimate the correlation between the alignment length and statistical support of phylogenetic hypotheses, sets of partial alignments were generated from the concatenated alignment of 56 proteins using the jackknife procedure of random site removal [31]. A hundred replicas of 75 and 50% of the length of the original alignment were generated; 20 sets were generated for each alignment of 25, 12.5, 6.3, and 3.1% of the length of the original alignment and for each set 100 replicas were obtained, i.e. a total of 10,200 partial alignments were obtained. Maximally parsimonious trees were built using the protpars program of the PHYLIP package [31], and the level of support of the nodes of interest was found to be linearly dependent on the logarithm of the alignment length (Fig. 4, a and b). A similar procedure was proposed [32] and applied [33] earlier. With the alignment length of 5000 and more randomly chosen sites, the hypothesis of sister relationship of multicellular animals and choanoflagellates receives high statistical support, so as the hypothesis of monophyletic origin of Mesomycetozoea including Capsaspora. When utilizing alignments of 668 to 2670 sites, these hypotheses receive moderate statistical support, and at the lower border of this interval, these groups fall out of the consensus majority tree in many sets. When utilizing shorter alignments, the considered groups are not revealed by the maximum parsimony method.
The performed rough estimation shows that the length of most individual proteins is not sufficient to support the groups under consideration. This estimation agrees with the results of analysis of four proteins: eEF1A, Hsp70, actin, and beta-tubulin, none of which individually provides high support for a group uniting animals and choanoflagellates, whereas the four proteins taken together provide moderate support for this group (also including a unicellular organism Ministeria vibrans) (data from Table 2 of [11]). Only four proteins from our set of 56 confirm monophyletic origin of animals and choanoflagellates with bootstrap support of over 50% in maximum parsimony trees, and only two proteins support monophyletic origin of Mesomycetozoea.Fig. 4. Support for monophyletic origin of some Opisthokonta groups in the maximum parsimonious trees with respect to the alignment length: a) animals + choanoflagellates; b) Mesomycetozoea; c) Holozoa; d) chytrid fungi (Chytridiales + Spizellomycetales + Blastocladiales).
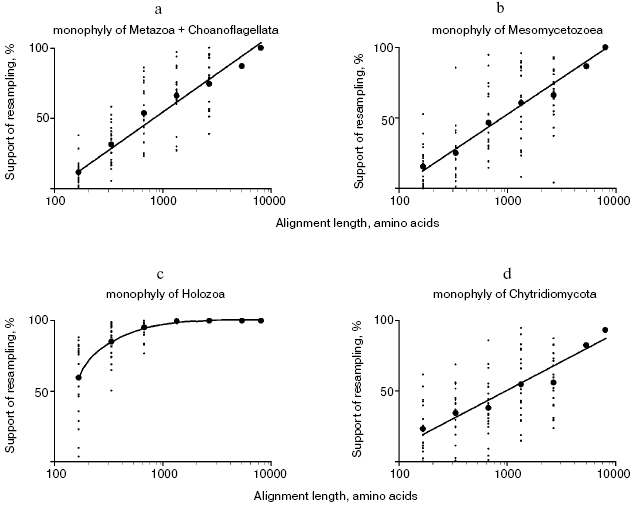
Certainly, 5000 alignment sites cannot be considered as a universal sampling size suitable for statistical evaluation of phylogenetic hypotheses. For example, monophyletic origin of Holozoa receives support in over 85% of pseudo-samplings with the length of analyzed alignment of 333 random sites and over 95% with 668 sites (Fig. 3). Apparently the stem group of Holozoa existed much longer and accumulated more distinctions from the sister group leading to fungi compared with stem groups of daughter clades Choanoflagellata + Metazoa and Mesomycetozoea, which were separated, respectively, to the lines of choanoflagellates and animals, on one side, and lines of Capsaspora and Amoebidium + Sphaeroforma, on the other, soon after the divergence of two lineages of Holozoa.
In order to reveal alignment positions that agree or disagree with the tree in Fig. 3, the protpars program of the PHYLIP package [31] was used to estimate amino acid sequences for the nodes of the tree in Fig. 3 and for the trees of alternative topology. The hypothetical synapomorphies of Mesomycetozoea are amino acid substitutions in protein 2 of the potential-dependent anion channel (porin 2 of external mitochondrial membrane, VDAC-2) (G94→N), heat shock protein Hsp86 (HS90A_HUMAN) (S165→T), ribosomal proteins RpS2 (P208→G, V209→T), RpS25 (G13→T, K17→E), RpL2 (V62→H, R184→A), RpL6 (V121→T), RpL10 (D55→L), etc. (here and below numbering is given by the human proteins). Potential synapomorphies of chytrid fungi are amino acid substitutions in methionine adenylate transferase II (A68→L, H122→L, D129→K, V195→P, L261→V), processivity factor of DNA polymerase delta (PCNA) (E93→G, Q184→P), 1-3 C-terminal amino acid residues of ubiquitin ligase E2 (UBC9), etc. These features are localized in the evolutionarily conserved regions of the macromolecules. Nevertheless, almost all of them are susceptible to homoplasy. Thus, in all vertebrates from actinopterygian fish to mammals there are two paralogs: methionine adenylate transferase II with a “usual” residue L261 characteristic of numerous invertebrates, fungi, and plants, and S-adenosylmethionine synthetase I with residue V261 like in chytrid fungi. Judging by the genealogical tree of these proteins (data not shown), duplication happened in the last common ancestor of vertebrates and was followed by the L261→V substitution in one of the paralogs. After recognition of two independent cases of L261→V substitution: in the ancestral S-adenosylmethionine synthetase I of vertebrates and in the ancestors of chytrid fungi, one cannot deny the possibility of even a larger number of independent transitions to V261 in different lines of chytrid fungi. Thus, this and other discovered features seem to be insufficient to prove monophyletic origin of the groups under consideration individually. Among potentially significant phylogenetic markers, ribosomal protein RpS30 is particularly noteworthy, which in all Holozoa differs from the corresponding fungal protein by the presence of a specific peptide at the NH2 terminus.
HISTORY OF THE RIBOSOMAL PROTEIN RpS30 GENE
Opisthokonta. A potential marker suitable for recognition of nearest relatives of multicellular animals is ribosomal protein RpS30. In Metazoa, it is encoded by a single gene fau directing synthesis of a fused protein whose NH2 terminus, FUBI, resembles ubiquitin [34, 35]. Human FUBI differs from ubiquitin by substitutions of 47 amino acids out of 74 and in addition by deletion of two amino acids. Phylogenetic significance of FUBI is defined by the fact that in another member of Opisthokonta, fungi, RpS30 is not fused with other proteins [36], as in the outgroup--in plants and most protists. In other words, the RpS30 fusion is an evolutionary acquirement (apomorphy) of Metazoa ancestors. In order to find out at what level of phylogenetic tree this event took place, we have searched for the RpS30 homologs in available databases of different Opisthokonta species. It turned out that in cDNA libraries of all unicellular Holozoa (including choanoflagellates Monosiga ovata and M. brevicolls and Mesomycetozoea: Amoebidium parasiticum and Sphaeroforma arctica) the RpS30 protein is fused with the ubiquitin-like sequence; the only exception is species Capsaspora owczarzaki where it is fused with true ubiquitin (Fig. 4). Such distribution of features among present-day species is consistent with the hypothesis of single fusion event in a common ancestor of Holozoa, thus confirming their monophyletic origin. FUBI of multicellular and unicellular Holozoa, despite variability, has specific peculiarities supporting its common origin (deletion of two amino acid residues near the NH2 terminus, substitution I13→H, Q39→P, G53→E/D (mainly), H70→E/D (mainly), the predominant loss of a functional residue K48 of the site of ubiquitinylation).
According to the most parsimonious evolutionary scenario, the fusion of RpS30 with ubiquitin happened once in the last common ancestor of Holozoa, and in the line of C. owczarzaki this ancestral condition was maintained, whereas in the other Holozoa alterations, that accumulated in the ubiquitin half of the fusion protein, transformed it to FUBI. This is supported by the absence of FUBI orthologs beyond Holozoa. Numerous ubiquitin-like proteins of other eukaryotes and paralogs in genomes of animals differ from typical ubiquitin, but do not display specific similarity to FUBI. This means that FUBI emerged as a specific ubiquitin modification just in Holozoa and just in connection with RpS30. Another ribosomal protein, RpS27a, which is also fused with ubiquitin, reveals a high conservativeness of the ubiquitin part across all eukaryotes. For example, this region of human and Arabidopsis thaliana proteins differs by only three amino acids. However human sequences and those of nematode Caenorhabditis elegans differ in this region by 39 amino acid residues and three deletions [37]. Evidently some alteration happened in a distant ancestor of C. elegans that suppressed the requirement for preservation of a conserved structure of ubiquitin fused with RpS27a, and in different Rhabditida species (C. briggsae, Nippostrongylus brasiliensis, Pristionchus pacificus, etc.) it was transformed into a variable ubiquitin-like protein (distinct from FUBI). In nematodes Ascaris, Xiphinema, and others, not belonging to the Rhabditida clade, the conservative state of ubiquitin fused with RpS27a is retained. Evidently, ubiquitin fused with RpS30 in the Holozoa ancestor, repeated in general terms a more illustrative history of the nematode ubiquitin fused with RpS27a.
In spite of apparent plausibility, the described maximally parsimonious scenario of the Holozoa RpS30 evolution raises serious doubts. The above-described data of multigene analysis that favors monophyletic origin of Mesomycetozoea contradicts this scenario. This means that evolution of RpS30 deviated from the most parsimonious. Taking into account the pronounced similarity of FUBI of Amoebidium parasiticum and Sphaeroforma arctica with sequences of Metazoa and Choanoflagellata, we have to recognize that after formation of FUBI in the closest common ancestor of Holozoa, the line of C. owczarzaki experienced a replacement of FUBI by ubiquitin, a peculiar violation of the Dollo's law.
An attempt to find an analogous example in other opisthokonts revealed the replacement of FUBI by ubiquitin in nematodes Tylenchomorpha (Meloidogyne chitwoodi, M. incognita, M. javanica, Globodera rostochiensis, Heterodera glycines, Pratylenchus vulnus) and Cephalobidae (Zeldia punctata). Along with other species, Bursaphelenchus xylophilus traditionally considered within the Aphelenchoidoidea family of Tylenchida order retains the state typical of Metazoa. Analysis of 18S rRNA sequences is consistent with monophyletic origin of Cephalobidae and Tylenchomorpha and with autonomy of the Aphelenchoidoidea branch [38], although exceptionally varying branch lengths of the scaled tree precluded any taxonomical conclusions, owing to the recognition of “long branch attraction” artifact. The replacement of FUBI by ubiquitin represents a clear synapomorphy of Cephalobidae and Tylenchomorpha, which is indicative of their monophyletic origin and improbability of a group uniting Tylenchomorpha with Aphelenchoidoidea in a phylogenetic system. Thus, essential differences in early embryonic development of Aphelenchoides and Aphelenchus [39] as well as other distinctions between them receive natural explanations. From the technical point of view the FUBI replacement by ubiquitin could follow the mechanism of exon shuffling, because the sequences coding FUBI and RpS30 are separated by an intron [36].
Complex transformations of fused proteins during evolution of Holozoa raise a question concerning the functional role of the ubiquitin or FUBI combination with ribosomal proteins. Previous experiments have shown that ubiquitin tails of RpS27a and RpL40 (another ubiquitin-fused ribosomal protein) are not vitally important for yeasts, but their absence impedes their growth and can be compensated by increase in the copy number of these genes [40]. Ubiquitin may contribute to RpS27a and RpL40 folding and ribosome biogenesis [40]. Taking into account that the role of FUBI is presently unknown [41], these suppositions are certainly insufficient to reveal physiological grounds for the discovered convergence.
Other eukaryotes. Fusion of ubiquitin and RpS30 genes is a clear example of homoplasy--independent emergence of strictly homologous structures in ancestors of Holozoa and heterotrophic heterokonts (Blastocystis and Oomycetes), which are separated by a significant distance on the tree of eukaryotes (Fig. 5). The gene coding RpS30 is unique, and it is not difficult to distinguish ubiquitin-like paralogs from ubiquitin proper, which is characterized by extreme state of conservation. An identical fusion of orthologs results in independent emergence of fully homologous fused proteins in C. owczarzaki, Tylenchomorpha, and Heterokonta.
Interestingly, the tendency for modification of the ubiquitin part fused with RpS30 also emerges in heterokonts of genus Phytophthora. In this case, alterations that happened during evolution of RpS30 in Holozoa are not copied, instead a deletion of a larger part of ubiquitin takes place (P. infestans, P. sojae, P. ramorum, P. brassicae, P. parasitica). Despite significant changes, the remaining ubiquitin fragment of 21 amino acid residues still contains the conserved C-terminal motif. It is not known whether its preservation is due to recency of the truncated form or to covariations in RpS30 preventing the complete loss of the ubiquitin fragment. In Phytophthora citrophtora and other oomycetes (Pythium ultimum, Saprolegnia parasitica, Aphanomyces cochlioides) a complete ubiquitin sequence fused with RpS30 retains high conservativeness.Fig. 5. Distribution of ubiquitin fused with ribosomal S30 protein in a phylogenetic tree. The dark pictogram shows ubiquitin, the light pictogram shows FUBI or any ubiquitin-like peptide fused with ribosomal protein S30. Ubiquitin-like sequence in Phytophthora genus is truncated at the N-terminus; the light rectangle corresponds to RpS30.
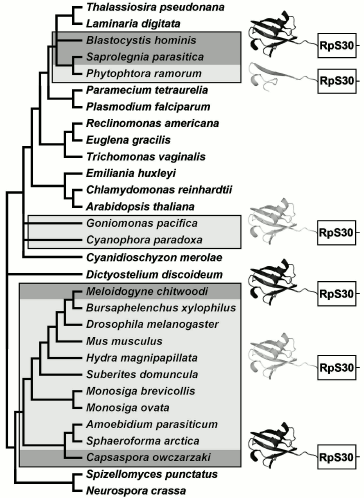
A role of fusion of these two proteins as a potential phylogenetic marker is expressed by the fact that autotrophic heterokonts (diatoms, brown algae) as well as close relatives of heterokonts--Alveolata [42, 43] and Excavata--bear no signs of fusion of RpS30 with ubiquitin. Thus, the fused protein of oomycetes and Blastocystis is an obvious synapomorphy. The position of Blastocystis, a nonflagellated animal parasite, in the system of heterokonts remains disputable. The tree of 18S rRNA sequences of Heterokonta [44] contains a crown of short branches, leading to contemporary phototrophic taxa, and a bundle of numerous long branches, independently branching off from the base of the tree towards heterotrophic taxa, in this case the order of their branching does not receive any significant statistical support (Fig. 3 in [44]). It would be tempting to combine them by synapomorphies in independent genes, as in the case of Oomycetes and Blastocystis by the presence of ubiquitin-fused RpS30. However, taking into account the recurring character of such fusions, it is hardly possible to recognize this feature as a decisive one. Features less susceptible to homoplasy are desirable for confirmation or disproof of the monophyletic origin of Oomycetes and Blastocystis.
Two additional taxa with RpS30 fused with the ubiquitin-like peptide have also been found. These are unicellular glaucophyte and cryptophyte algae, which include only a few species and form isolated groups. Glaucophytes are known to contain as photosynthesizing organelles cyanelles--obligate symbiotic cyanobacteria whose genome is close in size to that of chloroplasts, whereas the plastid of phototrophic species of cryptophytes is a eukaryotic endosymbiont with rudimental nucleus and chromosomes. It may be hypothesized that cryptophytes and glaucophytes occupy a central place in the history of emergence of photosynthesizing eukaryotes, while the phylogeny of “host” components of their composite cells is still poorly studied. On the nuclear 18S rRNA trees, cryptophytes and glaucophytes are combined in a single cluster [45-47], but the consistency of this grouping is usually considered as doubtful because of insufficient statistical support. The fusion of RpS30 with the ubiquitin-like peptide (Fig. 4) is an additional, though also hardly convincing, argument in favor of monophyletic origin of cryptophytes and glaucophytes. Unlike FUBI of animals, choanoflagellates, and Mesomycetozoea, no obvious similarity in primary structure is observed between ubiquitin-like sequences of cryptophytes and glaucophytes. This speaks in favor of an independent fusion and independent transformation of ubiquitin within fused protein in their ancestors. In this case, the fusion criterion is depreciated as a phylogenetic marker, but it makes more intriguing the question concerning factors causing independent loss of conservation by ubiquitin in different taxonomical groups just upon its fusion with RpS30.
DISCUSSION
Works on multigene phylogenetic inference, published during recent years under genomic projects, and analysis of whole cDNA libraries provide support for hypotheses that were formulated after analysis of individual genes, first of all that of rRNA of the small ribosomal subunit. It is hardly worth focusing on taxa that have appeared in textbooks since the XIX century. The relationships of species comprising them is not surprising. More interesting is the substantiation of groups that were distinguished on the basis of scarce molecular data, monophyletic origin of which is also unexpected in the light of morphological evolution and can not be very easily substantiated by material from the traditional deficient baggage of zoology and botany. They include a group of animals, fungi, and some unicellular Opisthokonta [43]; echinoderms, hemichordates, and Xenoturbella--in Ambulacraria [48]; nematodes, arthropods, and other animals--in Ecdysozoa; annelids, mollusks, and flat worms--in Lophotrochozoa [49]; vascular plants--with mosses of order Anthocerotales [50, 51]; displacement of Nymphaceae, Magnoliaceae, and Amborella to the base of flowering plants before the separation of dicotyledons and monocotyledons [33]; inclusion of myxosporidian “protists” in multicellular animals [24], and other nontrivial propositions. Nobody has doubts concerning the high taxonomical rank of Archaea originally established by comparison of the rRNA oligonucleotide maps. Generally, groups established “by a single gene” are preserved in the case of significant statistical support, sufficient taxonomical sampling, undisputable orthology of genes under comparison, and absence of long branch attraction artifacts. The authors have no data concerning groups that were established on the basis of 18S rRNA genes and meet the above-mentioned conditions and at the same time were rejected by later works on phylogenomics.
However, the epoch of “great phylogenetic discoveries by a single gene” is leaving: utilization of vast amounts of data becomes the norm for contemporary works. In many respects, this circumstance is the result of aspiration to put the proposed hypotheses onto a firm statistical basis. Besides, it is assumed that multigene analysis is able to enhance the phylogenetic signal that may eventually exceed the noise and achieve correct resolution of conflicts between different genes. We have found in this work that hypotheses of monophyletic origin of Choanoflagellata + Metazoa, Mesomycetozoea (including Capsaspora owczarzaki), and Chytridiales + Blastocladiales receive high statistical support only when over 5000 randomly chosen positions of amino acid alignment are used. The size of such alignment exceeds that of the majority of individual proteins. In analysis of a smaller number of sites, the support of these hypotheses becomes significantly lower. However, one should have in mind that the number of 5000 positions of alignment is the threshold for above-mentioned taxa only and cannot be considered as universal (e.g., [52]). For example, in order to reveal monophyly of Holozoa the number of sites that is lower by one order of magnitude than the above mentioned threshold is apparently enough. It is likely that this difference is due to rapid radiation in some cases and long-term phyletic evolution of the stem group in the other. Thus, the level of support depends both on the length of alignment and on other factors.
Moreover, a simple increase in the number of genes by no means assures reconstruction of a genuine tree. The above-mentioned example of an artifactual tree without monophyletic group Opisthokonta [12] is not unique. Thus, results of analysis of 146 protein-coding genes (35,346 amino acid residues), support alternative hypotheses of monophyletic origin either of coelomic animals or Ecdysozoa, depending on the outgroup composition (only yeasts or yeasts and choanoflagellates) [53]. At least one of these topologies is false. It may be one containing the celomate group and consistent with the tree from another work [54] built on the matrix of introns presence/absence in 3000 genes of 11 eukaryote species (a total of 25,676 features). The same tree can be obtained from a single 18S rRNA gene of the same set of species, if inadequate models of base sequence evolution are used. In a tree of 23 species, derived from concatenated alignment of 133 proteins, flatworms are combined with the nematode Caenorhabditis elegans, but this grouping dissociates if C. elegans is replaced by another nematode species, Xiphinema index, that forms a shorter branch in the tree [49]. The tree of complete chloroplast genomes (over 150 kb) and tree of 18S rRNA (1.7 kb) of the same species contain identical errors caused by insufficient taxonomical sampling [55]. All of this demonstrates the fact that the present day methods of phylogeny reconstruction from a large number of genes might not help one to get rid of artifacts known for single genes. In this case assumptions of evolution models, difference in the evolutionary rates of species, errors in alignment and ortholog choice, and insufficient taxonomical sampling may also show up in exactly the same way. Data selection [17] is proposed to help remove artifacts of multigene phylogenetic analysis, which certainly makes it less formal. Thus, practice of modern phylogenomics shows that statistical support of phylogenetic reconstructions increases as the number of genes under comparison becomes larger, but the high level of statistical support of the tree as a whole or its separate nodes cannot serve as an index of correctness of phylogenetic reconstruction.
While causes of possible errors when using computational methods for building a phylogenetic tree by nucleotide or amino acid sequences are rather numerous and not sufficiently studied, there are only three causes of errors in cladistic analysis: incorrect “polarization” of evolutionary transition (in this case symplesiomorphy is erroneously taken instead of synapomorphy); incorrect homologization (like comparison of paralogs); homoplasy and exact reversions of evolution. The first two are subjective and the third one is objective. It is obvious that only rare events are suitable for substantiation of phylogenetic hypotheses [7, 56], because the limited number of different states of molecular features, certainly, makes them vulnerable to homoplasy. Elimination of the third kind errors is possible only by comparison with independent markers and iterative refinement of our knowledge concerning the mechanisms of molecular evolution: what events may recur and what should be considered as unique. Taken separately, the fact of FUBI replacement by ubiquitin requires the grouping of the amoeba Capsaspora owczarzaki with nematodes Cephalobidae and Tylenchomorpha, which contradicts many other features. However, if such replacement was repeated many times during evolution, we already cannot consider it as an undisputable proof of monophyletic origin of these nematodes, but only as one argument (but still significant) among others. Thus, both the results of computational and cladistic reconstructions may come under suspicion. Their combination, i.e. verification of correctness of a tree for single or multiple genes by molecular synapomorphies seems to be the best way out.
How can we find a gene or a nucleotide deserving unlimited trust? The shorter is the geological time of the stem group existence, the lower is the probability that the chosen at random gene will carry a synapomorphy, not susceptible to homoplasy and reversions. The only way to obtain a lottery ticket for sure is to buy up the whole drawing. Taking into account the rate of development of sequencing technology and computer processing, this idea may seem not that ridiculous. On the other hand, if the level of similarity is high in related species, then it will come to light in the majority of genes chosen at random and probably even in a single gene that is long enough, like those of 18S or 28S rRNA. Such similarity will be revealed in many different features, and detection of the monophyletic origin of a group will equip us with numerous justified predictions. The question arises however that is completely beyond the scope of phylogenetics: how much we are ready to pay for synapomorphic indicators of relationship if they turn out to be the only shared features of species under comparison?
Authors are grateful to L. Yu. Rusin and P. V. Troshin for help in computer calculations.
The work was supported by the Russian Foundation for Basic Research (grants 05-04-49705 and 06-04-49288).
REFERENCES
1.Spirin, A. S., Belozersky, A. N., Shugaeva, N. V.,
and Vanyushin, B. F. (1957) Biokhimiya, 22, 744-754.
2.Beklemishev, V. N. (1994) Methodology of
Systematics [in Russian], KMK Publishing House, Moscow.
3.Hennig, W. (1966) Phylogenetic Systematics,
Illinois University Press, Urbana.
4.Shatalkin, A. I. (1988) Biological
Systematics [in Russian], Moscow State University Publishing House,
Moscow.
5.Pavlinov, I. Ya. (2005) Introduction into Modern
Phylogenetics (Cladogenetic Analysis) [in Russian], KMK Publishing
House, Moscow.
6.Baldauf, S. L., and Palmer, J. D. (1991) Proc.
Natl. Acad. Sci. USA, 90, 11558-11562.
7.Petrov, N. B., and Aleshin, V. V. (2002)
Genetika, 38, 1043-1062.
8.Moreira, D., von der Heyden, S., Bass, D.,
Lopez-García, P., Chao, E., and Cavalier-Smith, T. (2007) Mol.
Phylogenet. Evol., 44, 255-266.
9.Cavalier-Smith, T., and Chao, E. E.-Y. (2003)
Protist, 154, 341-358.
10.Shatalkin, A. I. (2005) Zh. Obshch. Biol.,
66, 389-415.
11.Steenkamp, E. T., Wright, J., and Baldauf, S. L.
(2006) Mol. Biol. Evol., 23, 93-106.
12.Philip, G. K., Creevey, C. J., and McInerney, J.
O. (2005) Mol. Biol. Evol., 22, 1175-1184.
13.Seravin, L. N., and Gudkov, A. V. (2005) Zh.
Obshch. Biol., 66, 212-223.
14.Stechmann, A., and Cavalier-Smith, T. (2002)
Science, 297, 89-91.
15.Keeling, P. J., and Inagaki, Y. (2004) Proc.
Natl. Acad. Sci. USA, 101, 15380-15385.
16.King, N., and Carroll, S. B. (2001) Proc.
Natl. Acad. Sci. USA, 98, 15032-15037.
17.Jeffroy, O., Brinkmann, H., Delsuc, F., and
Philippe, H. (2006) Trends Genet., 22, 225-231.
18.Felsenstein, J. (1978) Syst. Zool.,
27, 401-410.
19.Lang, B. F., O'Kelly, C., Nerad, T., Gray, M. W.,
and Burger, G. (2002) Curr. Biol., 12, 1773-1778.
20.Medina, M., Collins, A. G., Taylor, J. W.,
Valentine, J. W., Lipps, J. H., Amaral-Zettler, L., and Sogin, M. L.
(2003) Int. J. Astrobiol., 2, 203-211.
21.Ruiz-Trillo, I., Inagaki, Y., Davis, L. A.,
Sperstad, S., Landfald, B., and Roger, A. J. (2004) Curr. Biol.,
14, R946-947.
22.Ruiz-Trillo, I., Lane, C. E., Archibald, J. M.,
and Roger A. J. (2006) J. Eukaryot. Microbiol., 53,
379-384.
23.Hertel, L. A., Bayne, C. J., and Loker, E. S.
(2003) Int. J. Parasitol., 32, 1183-1191.
24.Jimenez-Guri, E., Philippe, H., Okamura, B., and
Holland, P. W. (2007) Science, 317, 116-118.
25.Ruiz-Trillo, I., Burger, G., Holland, P. W.,
King, N., Lang, B. F., Roger, A. J., and Gray, M. W. (2007) Trends
Genet., 23, 113-118.
26.Lutzoni, F., Kauff, F., Cox, C. J., McLaughlin,
D., et al. (2004) Am. J. Bot., 91, 1446-1480.
27.James, T. Y., Kauff, F., Schoch, C., Matheny, P.
B., et al. (2006) Nature, 443, 818-822.
28.Kishino, H., and Hasegawa, M. (1989) J. Mol.
Evol., 29, 170-179.
29.Shimodaira, H., and Hasegawa, M. (1999) Mol.
Biol. Evol., 16, 1114-1116.
30.Shimodaira, H., and Hasegawa, M. (2001)
Bioinformatics, 17, 1246-1247.
31.Felsenstein, J. (1993) PHYLIP (Phylogeny
Inference Package) version 3.5c, Department of Genetics, University
of Washington, Seattle.
32.Philippe, H. (1993) Nucleic Acids Res.,
21, 5264-5272.
33.Logacheva, M. D., Penin, A. A., Samigullin, T.
Y., Vallejo-Roman, C. M., and Antonov, A. S. (2007) Biochemistry
(Moscow), 72, 1324-1330.
34.Kas, K., Michiels, L., and Merregaert, J. (1992)
Biochem. Biophys. Res. Commun., 187, 927-933.
35.Perina, D., Cetkovic, H., Harcet, M., Premzl, M.,
Lukic-Bilela, L., Müller, W. E., and Gamulin, V. (2006)
Gene, 366, 275-284.
36.Baker, R. T., Williamson, N. A., and Wettenhall,
R. E. (1996) J. Biol. Chem., 271, 13549-13555.
37.Jones, D., and Candido, E. P. (1993) J. Biol.
Chem., 268, 19545-19551.
38.Meldal, B. H., Debenham, N. J., De Ley, P., De
Ley, I. T., Vanfleteren, J. R., Vierstraete, A. R., Bert, W., Borgonie,
G., Moens, T., Tyler, P. A., Austen, M. C., Blaxter, M. L., Rogers, A.
D., and Lambshead, P. J. (2007) Mol. Phylogenet. Evol.,
42, 622-636.
39.Drozdovskii, E. M. (1968) Dokl. AN SSSR,
180, 750-753.
40.Finley, D., Bartel, B., and Varshavsky, A. (1989)
Nature, 338, 394-401.
41.Rossman, T. G., Visalli, M. A., and Komissarova,
E. V. (2003) Oncogene, 22, 1817-1821.
42.Burki, F., Shalchian-Tabrizi, K., Minge, M.,
Skjćveland, A., Nikolaev, S. I., Jakobsen, K. S., and Pawlowski,
J. (2007) PLoS ONE, 2, e790.
43.Rodriguez-Ezpeleta, N., Brinkmann, H., Burger,
G., Roger, A. J., Gray, M. W., Philippe, H., and Lang, B. F. (2007)
Curr Biol., 17, 1420-1425.
44.Cavalier-Smith, T., and Chao, E. E-Y. (2006)
J. Mol. Evol., 62, 388-420.
45.Bhattacharya, D., Helmchen, T., Bibeau, C., and
Melkonian, M. (1995) Mol. Biol. Evol., 12, 415-420.
46.Hoef-Emden, K., Marin, B., and Melkonian, M.
(2002) J. Mol. Evol., 55, 161-179.
47.Deane, J. A., Strachan, I. M., Saunders, G. W.,
Hill, D. R. A., and McFadden, G. I. (2002) J. Phycol.,
38, 1529-8817.
48.Bourlat, S. J., Juliusdottir, T., Lowe, C. J.,
Freeman, R., Aronowicz, J., Kirschner, M., Lander, E. S., Thorndyke,
M., Nakano, H., Kohn, A. B., Heyland, A., Moroz, L. L., Copley, R. R.,
and Telford, M. J. (2006) Nature, 444, 85-88.
49.Baurain, D., Brinkmann, H., and Philippe, H.
(2006) Mol. Biol. Evol., 24, 6-9.
50.Samigullin, T. K., Yacentyuk, S. P., Degtyaryeva,
G. V., Valiehoroman, K. M., Bobrova, V. K., Capesius, I., Martin, W.
F., Troitsky, A. V., Filin, V. R., and Antonov, A. S. (2002)
Arctoa, 11, 31-43.
51.Qiu, Y. L., Li, L., Wang, B., Chen, Z., Knoop,
V., Groth-Malonek, M., Dombrovska, O., Lee, J., Kent, L., Rest, J.,
Estabrook, G. F., Hendry, T. A., Taylor, D. W., Testa, C. M., Ambros,
M., Crandall-Stotler, B., Duff, R. J., Stech, M., Frey, W., Quandt, D.,
and Davis, C. C. (2006) Proc. Natl. Acad. Sci. USA, 103,
15511-15516.
52.Rokas, A., Williams, B. L., King, N., and
Carroll, S. B. (2003) Nature, 425, 798-804.
53.Philippe, H., Lartillot, N., and Brinkmann, H.
(2005) Mol. Biol. Evol., 22, 1246-1253.
54.Dopazo, H., Santoyo, J., and Dopazo, J. (2004)
Bioinformatics, 20 (Suppl. 1), i116-i121.
55.Degtjareva, G. V., Samigullin, T. Y., Sokoloff,
D. D., and Vallejo-Roman, C. M. (2004) Botan. J., 89,
896-907.
56.Rokas, A., and Holland, P. W. H. (2000) Trends
Ecol. Evol., 15, 454-459.
57.Schmidt, H. A., Strimmer, K., Vingron, M., and
von Haeseler, A. (2002) Bioinformatics, 18, 502-504.
58.Huelsenbeck, J. P., and Ronquist, F. (2001)
Bioinformatics, 17, 754-755.