Molecular Systematics of Lachancea Yeasts
E. S. Naumova1, E. V. Serpova1, and G. I. Naumov1,2*
1State Research Institute of Genetics and Selection of Industrial Microorganisms, 1-yi Dorozhnyi proezd 1, 117545 Moscow, Russia; E-mail: gnaumov@yahoo.com2Voronezh State University, Universitetskaya pl. 1, 394006 Voronezh, Russia
* To whom correspondence should be addressed.
Received August 10, 2007
This work presents the results of molecular-genetic investigation of a new yeast genus, Lachancea Kurtzman (2003). Analysis of rRNA sequences and molecular karyotyping have shown genetic homogeneity of the genus Lachancea. Yeasts of this genus have an identical haploid number of chromosomes equal to eight, whereas limiting chromosome sizes significantly differ in various species. The largest range of chromosome bands was registered in L. cidri strains (400-2800 kb), while the smallest was found in L. waltii (1400-2800 kb). The intra- and interspecies polymorphism of Lachancea chromosomes is discussed.
KEY WORDS: Lachancea yeasts, 26S rRNA, 5.8S-ITS, chromosomal DNA, molecular karyotyping, phylogenetic analysisDOI: 10.1134/S0006297907120097
Many problems of evolution, phylogeny, and systematics (taxonomy) of organisms can be solved by studying nucleic acids [1, 2]. The yeast Saccharomyces cerevisiae, widely used in science and practice, was the first eukaryotic organism for which the complete nucleotide sequence of the genome was determined [3]. During the past decade, the full-size genomes of another 20 yeast species have been sequenced. Fifty-five duplicated blocks incorporating several hundreds of homologous gene pairs were found in the genome of S. cerevisiae [4, 5]. The identical order and orientation of paralogic genes in duplicated blocks proved that the genome of S. cerevisiae was formed during ancestor duplication and following mass chromosome rearrangements and losses of individual genes. Comparative analysis of genomic sequences in a number of yeast species has shown that the complete duplication of eight ancestral chromosomes in S. cerevisiae happened after their divergence from Kluyveromyces waltii [6]. The haploid number of chromosomes in S. cerevisiae is 16, whereas in K. waltii it is 8.
Molecular-genetic investigation of one of the nearest relatives of Saccharomyces, genus Kluyveromyces van der Walt emend. van der Walt, has shown that the latter is very heterogeneous and includes a number of species that are not closely related to each other and to the type species of the genus K. polysporus [7-10]. At the same time, a small group of species from different genera (Zygosaccharomyces cidri, Z. fermentati, K. thermotolerans, K. waltii, and S. kluyveri) were found whose rRNA sequences are close to each other to different extent. The new genus Lachancea was formed on the basis of these five species, and L. thermotolerans was chosen as a type species [8]. Recently the new species L. meyersii was described [11], and a complex population structure of the type species L. thermotolerans has been shown [12]. The latter species includes one population represented by the type culture, and a second genetically divergent population of yeasts that assimilate melibiose.
Progress in molecular biology has resulted in the use of new methods in yeast taxonomy and stimulated active development of molecular phylogenetic classification [13-15]. Taxonomists are now trying to design a classification that will indicate the genetic relatedness between yeast species and genera. Analysis of rRNA genes, enabling evaluation of yeast relationship both at the species and higher taxonomical levels, is used to separate yeast taxons and establish their phylogenetic relationship. The computer database used for determination of taxonomic position of new strains was created according to the results of sequencing of the D1/D2 domain of the 28S rRNA gene (about 600 bp) in type cultures of over 600 species of ascomycete yeasts [8, 16]. To determine phylogenetic relationship of close species, analysis of the 5.8S-ITS fragment including the 5.8S rRNA gene and internal transcribed spacers ITS1 and ITS2 is used additionally. This region of rRNA is characterized by a significant interspecies divergence and a low level of intraspecies polymorphism. The length of ITS fragment is constant in the strains of the same species [17], but its sequence may vary [18, 19]. The principally important information about chromosomal composition of yeast genomes can be obtained using pulsed-field gel electrophoresis of native chromosomal DNA. This method, considered as a kind of “molecular karyology”, is widely used for studies in the field of evolution and taxonomy of various yeasts [20-22].
The goal of this work was to study molecular polymorphism and evolution of Lachancea in strains of different ecological and geographic origin.
MATERIALS AND METHODS
Yeast strains used in this work are listed in the table. The yeasts were grown at 28°C on complete YPD medium containing (g/liter): bactoagar, 20; glucose, 20; yeast extract, 10; peptone, 20.
PCR is carried out on a Tercik DNA amplifier (DNA Technology, Russia) directly on yeast cells with primers NL-1 (5´-GCATATCAATAAGCGGAGGAAAG-3´) and NL-4 (5´-GGTCCGTGTTTCAAGACGG-3´) for domain D1/D2 or ITS1 (5´-TCCGTAGGTGAACCTGCG-3´) and ITS4 (5´-TCCTCCGCTTATTGATATGC-3´) for 5.8S fragment of ITS. To do this, a small amount of yeast biomass (on the tip of a microbiological loop) was suspended in 30 µl buffer containing 3 mM MgCl2, 0.3 mM dNTP, and 50 pmol of each primer. The mixture was kept at 95°C for 15 min for cell lysis, then 2.5 units of Taq polymerase (Syntol, Russia) was added. For amplification in the D1/D2 region, PCR was carried out under the following conditions: initial denaturation for 1 min at 94°C, then 36 cycles of 1 min at 94°C, 1 min at 52°C, 1 min at 72°C, and final completion for 1 min at 72°C. For amplification of 5.8S-ITS fragments, initial denaturation was performed at 94°C for 3 min, then 30 cycles as follows: 30 sec at 94°C, 30 sec at 56°C, 1 min at 72°C, and final completion for 10 min at 72°C. Amplification products were separated by electrophoresis in 1% agarose gel at 60-65 V in 0.5-fold TBE (Tris-borate buffer containing 45 mM Tris, 10 mM EDTA, 45 mM boric acid) for 2 h and stained with ethidium bromide.
Sequencing. Base sequences of the D1/D2 domain and internal transcribed spacers ITS1 and ITS2 were determined by two strands using direct sequencing on an Beckman-Coulter automatic sequencer (Beckman, USA) according to Sanger. Base sequences of 5.8S ITS region of type cultures of L. cidri, L. kluyveri, L. fermentati, L. meyersii, L. thermotolerans, and L. waltii were taken from the GenBank computer database. A search for homology to known base sequences was carried out under the BLAST program. Multiple alignment of obtained and already known base sequences was carried out manually using version 5.0.9 of the BioEdit program [23]. Phylogenetic relationship was detected and phylogenetic trees were built using the Neighbor-Joining and UPGMA algorithms from the computer package MEGA 3 [24]. Base sequence of 5.8S ITS fragment of yeast K. marxianus CBS 712 was used as an outgroup.
Pulsed-field gel electrophoresis of native chromosomal DNA. Conditions for chromosomal DNA preparation are described elsewhere [25]. Samples were placed in wells of 0.8-1.2% agarose gel. TBE (0.5-fold) cooled to 12-14°C was used. Electrophoresis was carried out in a CHEF-DR III apparatus (Bio-Rad, USA). Four different karyotyping regimes were used for optimal separation of the yeast chromosome bands. The two-step running was conducted at 200 V for 15 h with the field switching time of 70 sec and for 9 h with field switching time of 120 sec. The three-step running consisted of electrophoresis for 10 h at 170 V with field switching time of 40-120 sec, for 28 h at 130 V with field switching time of 120-360 sec, and for 9 h at 100 V with field switching time of 360-1200 sec. The four-step running procedure No. 1 consisted of electrophoresis for 10 h at 150 V with field switching time of 240 sec, 10 h with field switching time of 160 sec, 15 h with field switching time of 90 sec, and 8 h with field switching time of 60 sec. The four-step procedure No. 2 consisted of electrophoresis for 12 h at 150 V with field switching time of 240 sec, 16 h with field switching time of 160 sec, 16 h with field switching time of 90 sec, and 8 h with field switching time of 60 sec. After electrophoresis the gel was stained with ethidium bromide, then washed in distilled water, and photographed under ultraviolet light on a Vilber Lourmat transilluminator (France). Strains S. cerevisiae YNN 295 and Pichia canadensis (syn. Hasenula wingei) YB-4662-VIA (Bio-Rad) with known chromosome sizes and order were used as karyotyping standards.
RESULTS
First, species belonging of 38 collection strains and 10 strains isolated by us was determined by sequencing the D1/D2 region of the 26S rRNA gene. The base sequences were compared with those available in the GenBank database sequences of D1/D2 region of type cultures L. cidri, L. kluyveri, L. fermentati, L. meyersii, L. thermotolerans, and L. waltii. According to this analysis, all of the strains belong to the genus Lachancea (table). Strains of the same species had identical D1/D2 sequences. Among strains isolated by our laboratory, seven (DV 87-18, Yal. 87-1, Yal. 87-2, Yal. 87-5, Yal. 87-13, Fin. 89-2, and Fin. 89-11) belong to the species L. thermotolerans, whereas three others (CBS 10367, CBS 10368, and CBS 10369) belong to L. kluyveri. Strains UWO PS 79-139, UWO PS 82-231, NBRC 10066, and NBRC 10067 having identical D1/D2 sequences differ by this region from all known species of Lachancea and evidently belong to a new species, Lachancea sp.
Origin of studied Lachancea strains

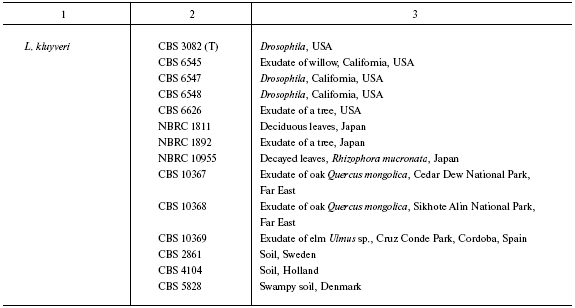
Note: Abbreviated collection names: CBS) Centraalbureau voor
Schimmelcultures, Utrecht, Holland; NBRC) NITE Biological Resource
Center, Osaka, Japan; UWO PS) University of Western Ontario, Department
of Biology (formerly Plant Sciences) culture collection, Canada; UCD)
Herman J. Phaff Yeast Culture Collection, Department of Food Science
and Technology, University of California, Davis, USA; NRRL) Northern
Region Research Center, Peoria Ill; VKPM (ARCIM)) All-Russian
Collection of Industrial Microorganisms, Moscow. The other strains are
from G. I. Naumov's collection. Strain correspondence in different
collections: CBS 6340 = NRRL Y-8284, DV 87-18 = CBS 10520,
Yal. 87-1 = CBS 10516, Yal. 87-2 = CBS 10517, Yal.
87-5 = CBS 10518, Yal. 87-13 = CBS 10519, Fin. 89-11 =
CBS 10521, CBS 797 = VKPM (ARCIM) Y-965, CBS 8951 = NRRL
Y-27269, CBS 9924 = NRRL Y-27762, CBS 9958 = NRRL Y-27411,
CBS 10367 = NBRC 10847 = N52; CBS 10368 = NBRC
10848 = N53; CBS 10369 = N54. T is a type culture.
Lachancea was investigated further by analysis of base sequences of the 5.8S ITS region and by molecular karyotyping.
Comparative analysis of base sequences of the 5.8S ITS region. The 5.8S ITS region was amplified using primers ITS1 and ITS4. The PCR product size was identical in all studied strains and made up approximately 600 bp. A phylogenetic tree was constructed based on comparative analysis of base sequences of this region (Fig. 1). The type culture of K. marxianus CBS 712 was used as an outgroup. Strains of the same species usually had identical ITS1 and ITS2 sequences. Single nucleotide substitutions were found only among strains L. thermotolerans, L. kluyveri, and L. fermentati. Three main clusters are distinguished on the phylogenetic tree. The first was formed by the yeasts L. thermotolerans, Lachancea sp., L. waltii, and L. meyersii. The ITS sequences in these yeasts differ by 2-33 nucleotides. The most closely related are L. thermotolerans and Lachancea sp., whereas the L. meyersii species is the most diverged one.
The second cluster with 100% reliability combines species L. cidri and L. fermentati, whose strains differ by 10 nucleotide substitutions in the ITS1 region and 5-6 nucleotide substitutions in the ITS2 region. The distinctions between strains of the two clusters made up over 50 nucleotide substitutions.Fig. 1. Phylogenetic analysis of nucleotide sequences of the 5.8S ITS region of Lachancea rRNA. The bootstrap values > 50% are given. The bar corresponds to 20 nucleotide substitutions per 1000 nucleotide positions. The nucleotide sequence of the 5.8S ITS fragment of Kluyveromyces marxianus CBS 712 was used as an outgroup. T, type culture. Figures in parentheses correspond to groups of strains with identical nucleotide sequences: 1) Yal. 87-2, Yal. 87-5, Yal. 87-13, DV 87-18, UWOPS 83-1097, Fin. 89-2, Fin. 89-11; 2) UWOPS 82-231, NBRC 10067; 3) CBS 8527, CBS 8528, CBS 7703; 4) CBS 9958, CBS 9923, CBS 9924, CBS 9925; 5) CBS 2950, CBS 5666; 6) CBS 797, CBS 4686, CBS 6544, CBS 6711, CBS 7004, CBS 7005, CBS 7007; 7) CBS 6545; 8) CBS 6547, CBS 6548, CBS 6626, CBS 2861, CBS 4104, CBS 10369, NBRC 1811, CBS 10368.
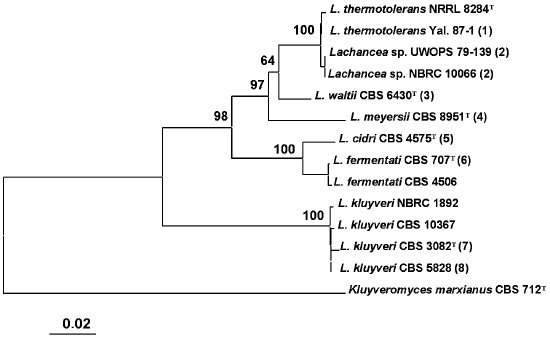
A special position on the phylogenetic tree is occupied by the L. kluyveri strains that have identical ITS sequences or those differing by 1-3 nucleotides. This is the most divergent species of the Lachancea genus. Differences by ITS1/ITS2 sequences between L. kluyveri and other species of this genus make up over 80 nucleotides.
Pulsed-field gel electrophoresis of native chromosomal DNA. The genomes of Lachancea species were compared by karyotypic analysis. The order and size of chromosome bands were detected according to karyotypic standards of strains S. cerevisiae and P. canadensis. To achieve optimal separation of chromosome bands of Lachancea, several karyotyping procedures were used.
Figure 2 shows a scheme of molecular karyotypes of Lachancea based on analysis of several electrophoretic gels. Comparative analysis of 48 strain patterns revealed a significant polymorphism of Lachancea. Chromosomal DNA of different strains was separated into 7-9 electrophoretic bands from 400 to 2800 kb. Most strains have patterns with eight chromosomal bands. The most homogeneous by molecular karyotypes are L. meyersii, L. waltii, and L. kluyveri. In the strains of the last two species, a slight polymorphism was noted only for chromosomal bands 1500-2400 kb in length. Most studied strains of L. kluyveri were identical in molecular karyotypes to the type culture CBS 3082. Figure 2 shows only L. kluyveri strains that differ in their karyotypes, whereas karyotypes of all 14 studied strains were published earlier [26]. The L. meyersii strains have patterns practically identical to eight chromosomal bands of 1000-2800 kb. A high similarity between the L. meyersii karyotypes can be explained by the same source of isolation (table). All six studied strains were isolated from sea water in a mangrove bay. Despite different geographical origin (table), strains of Lachancea sp. also have practically identical karyotypes with eight chromosomal bands of 900-2800 kb.
Size polymorphism of chromosomal bands is found among strains of L. thermotolerans, L. fermentati, and L. cidri. Despite equal range of chromosome size (700-2800 kb), sizes of individual chromosomes were different in each of nine studied strains of L. thermotolerans. The type culture CBS 6340 has seven chromosomes, whereas chromosomal DNA of the other strains was separated into eight electrophoretic bands. Distinctions in the number of chromosomal bands were earlier registered in the L. thermotolerans strains isolated in Spain [27].Fig. 2. Overall scheme of Lachancea molecular karyotypes. Chromosomal band sizes (kb) are given for standard strains Saccharomyces cerevisiae YNN 295 and Pichia canadensis YB-4662-VIA. Different karyotyping conditions were used.
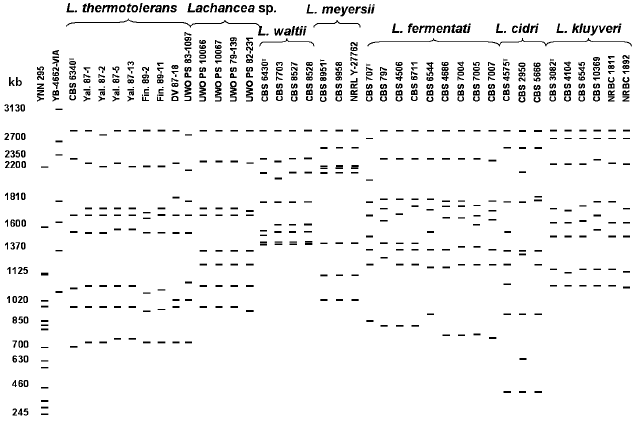
Chromosomal DNA of L. fermentati was separated into eight electrophoretic bands, but individual chromosomes varied in size in different strains. Only strains CBS 4686, CBS 7004, CBS 7005, and CBS 7007, isolated from alpechin (a waste of olive oil production) in Spain (table) and able to ferment melibiose, have similar patterns. Evidently, they make up a special ecological population of L. fermentati.
Lachancea cidri, whose karyotypes differed in the number and size of chromosome bands, were most heterogeneous. Chromosomal DNA of the type culture CBS 4575 and strain CBS 2950 was separated into nine electrophoretic bands, whereas DNA of strain CBS 5666 produced eight bands. All three strains had identical limiting size of chromosome bands (400-2800 kb). However, the size of individual chromosomes differed significantly. Only two upper chromosome bands and the two lowest ones were of similar size (400 and 850 kb, respectively). An additional small chromosome of about 580 kb was found in strain CBS 2950. It cannot be excluded that strains CBS 4575 and CBS 2950 are aneuploid and contain extra chromosomes.
DISCUSSION
Sequencing the full-size genome of S. cerevisiae not only stimulated comparative analysis of yeast genomes and creation of new methods for gene identification and functional analysis, but also contributed to investigation of molecular evolution of the genus Saccharomyces. Recently full genomes of biological species S. paradoxus, S. mikatae, S. bayanus [28], and S. kudriavzevii (registration number AACI01000133) have been sequenced along with partial sequencing the S. cariocanus genome. It has been found that the genome of S. cerevisiae significantly differs at the molecular level from the other five Saccharomyces species, especially in telomeric regions of chromosomes [28]. The Saccharomyces species are known to have similar basic karyotypic characteristics like haploid number of chromosomes equal to 16 and identical size range of chromosomal bands (250-2200 kb) [29]. At the same time, size variability of practically all homeologous chromosomes was found: only three out of 16 chromosomes are of equal size in all six Saccharomyces species [30].
Karyotypic analysis performed in this work revealed a significant size polymorphism of chromosomes in Lachancea. Chromosomal DNA of most studied strains was separated into eight electrophoretic bands. Thus, all species of Lachancea appear to have an identical number of chromosomes equal to eight. This points to the evolutionary relationship of L. thermotolerans, L. cidri, L. fermentati, L. kluyveri, L. meyersii, L. waltii, and Lachancea sp. Karyotypes of all these species contain a chromosome band of about 2800 kb. However, unlike Saccharomyces, chromosomes of Lachancea species significantly differ in their limiting size. This can be indicative of different sizes of the genomes in the studied species. The largest size range of chromosomal bands was registered in strains of L. cidri (400-2800 kb) and the smallest in L. waltii (1400-2800 kb). Since the range of the chromosomal band sizes is practically identical in strains of the same species, this feature is species-specific.
In addition to interspecies distinctions of karyotypic profiles, we have found intraspecies polymorphism of the chromosomal band sizes in some Lachancea species. Almost each studied strain of L. thermotolerans, L. fermentati, and L. cidri is characterized by a unique pattern. At the same time, independently of karyotypic profile, all strains of the same species have practically identical ITS1 and ITS2 sequences. For example, homogeneous by molecular karyotypes L. kluyverii appeared to be the most polymorphic in ITS sequences (up to three nucleotide substitutions).
The found size variability of individual chromosomes in Lachancea confirms the high dynamics of yeast chromosomes discovered during comparative analysis of complete genomes of S. cerevisiae and other species. In addition to the whole genome duplication, multiple duplications of individual chromosomes, short segments, and individual genes along with mass loss of approximately 90% of duplicate genes, mainly due to small deletions, happened in the course of evolution of S. cerevisiae [4, 31, 32]. Prominent chromosomal rearrangements (reciprocal translocations), found in Saccharomyces, significantly decrease the interspecies hybrid fertility and, probably, are responsible for reproductive isolation and formation of new biological species [30, 33]. It is known that reciprocal translocations are able to induce formation of multivalents in meiosis and to cause frequent formation of aneuploid gametes during chromosome divergence, which lower the hybrid fertility. Evidently, the ancestral number of chromosomes equal to eight survived in Lachancea during evolution. The karyotype variability found by us may be the result of different chromosomal rearrangements (duplications and deletions of short chromosome segments or individual genes, reciprocal translocations, etc.) during evolution of Lachancea.
Strains able to ferment melibiose and those negative in this feature are found among Lachancea yeasts. Mel+ strains were not found in two (L. meyersii and L. waltii) out of seven species of the genus Lachancea. In this connection, studying the alpha-galactosidase (MEL) and other genes will make possible the detailed comparison of the Lachancea genomes and determination of the extent of their divergence.
To summarize our results, we can state that comparative analyses of rRNA sequences and molecular karyotypes agree well with each other. The results are indicative of evolutionary relationship of Lachancea species, in other words, of genetic homogeneity of this genus.
The authors are grateful to A. S. Antonov for his attention to this work.
This work was supported by the Russian Foundation for Basic Research (grant 06-04-49051).
REFERENCES
1.Belozersky, A. N. (1969) Nucleic Acids and Their
Connection with Evolution, Phylogeny, and Systematics of
Organisms [in Russian], Publisher FAN SSSR.
2.Antonov, A. S. (2005) Mol. Biol. (Moscow),
38, 581-589.
3.Goffeau, A., Barrell, B. G., Bussey, H., Davis, R.
W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J. D., Jacq, C.,
Johnston, M., Louis, E. J., Mewes, H. W., Murakami, Y., Philippsen, P.,
Tettelin, H., and Oliver, S. G. (1996) Science, 274,
546-567.
4.Wolfe, K. H., and Shields, D. C. (1997)
Nature, 387, 708-713.
5.Wong, S., Butler, G., and Wolfe, K. H. (2002)
Proc. Natl. Acad. Sci. USA, 99, 9272-9277.
6.Seoighe, C., and Wolfe, K. H. (1998) Proc. Natl.
Acad. Sci. USA, 95, 4447-4452.
7.Cai, J., Roberts, I. N., and Collins, M. D. (1996)
Int. J. Syst. Bacteriol., 46, 542-549.
8.Kurtzman, C. P. (2003) FEMS Yeast Res.,
4, 233-245.
9.Kurtzman, C. P., and Robnett, C. J. (2003)
FEMS Yeast Res., 3, 417-432.
10.Naumov, G. I., and Naumova, E. S. (2002) FEMS
Yeast Res., 2, 39-46.
11.Fell, J. W., Statzell-Tallman, A., and Kurtzman,
C. P. (2004) Stud. Mycol., 50, 359-363.
12.Naumova, E. S., Serpova, E. V., and Naumov, G. I.
(2005) Dokl. Akad. Nauk, 405, 711-714.
13.Valente, P., Ramos, J. P., and Leoncini, O.
(1999) Can. J. Microbiol., 45, 949-958.
14.Kurtzman, C. P. (2000) in Dimorphism in Human
Pathogenic and Apathogenic Yeasts (Ernst, J. F., and Schmidt, A.,
eds.) Contrib. Microbiol., Basel-Karger, pp. 1-14.
15.Suh, S. O., Blackwell, M., Kurtzman, C. P., and
Lachance, M. A. (2006) Mycologia, 98, 1006-1017.
16.Kurtzman, C. P., and Robnett, C. J. (1998)
Antonie van Leeuwenhoek, 73, 331-371.
17.Valente, P., Gouveia, F. C., de Lemos, G. A.,
Pimentel, D., van Elsas, J. D., Mendonca-Hagler, L. C., and Hagler, A.
N. (1996) FEMS Microbiol. Lett., 137, 253-256.
18.James, S. A., Collins, M. D., and Roberts, I. N.
(1996) Int. J. Syst. Bacteriol., 46, 189-194.
19.James, S. A., Roberts, I. N., and Collins, M. D.
(1998) Int. J. Syst. Bacteriol., 48, 591-596.
20.Naumov, G. I., Naumova, E. S., Lantto, R. A.,
Louis, E. J., and Korhola, M. (1992) Yeast, 8,
599-612.
21.Boekhout, T., and Naumova, E. S. (2003) in
Experimental Approaches for Assessing Genetic Diversity among
Microbial Pathogens (van Belkum, A., et al., eds.) Wageningen,
Netherlands, pp. 93-103.
22.Boekhout, T., Renting, M., Scheffers, W. A., and
Bosboom, R. (1993) Antonie van Leeuwenhoek, 63,
157-167.
23.Hall, T. A. (1999) Nucleic Acids Symp.
Ser., 41, 95-98.
24.Kumar, S., Tamura, K., and Nei, M. (2004)
Briefings in Bioinformatics, 5, 150-163.
25.Naumova, E. S., Zholudeva, M. V., Martynenko, N.
N., and Naumov, G. I. (2005) Mikrobiologiya, 74,
179-187.
26.Naumova, E. S., Serpova, E. V., Korshunova, I.
V., and Naumov, G. I. (2007) Mikrobiologiya, 76,
361-368.
27.Belloch, C., Barrio, E., Garcia, M. D., and
Querol, A. (1998) Yeast, 14, 1341-1353.
28.Kellis, M., Patterson, N., Endrizzi, M., Birren,
B., and Lander, E. S. (2003) Nature, 423, 241-254.
29.Naumov, G. I., James, S. A., Naumova, E. S.,
Louis, E. J., and Roberts, I. N. (2000) Int. J. Syst. Evol.
Microbiol., 50, 1931-1942.
30.Fischer, G., James, S. A., Roberts, I. N.,
Oliver, S. G., and Louis, E. J. (2000) Nature, 405,
451-454.
31.Langkjer, R. B., Nielsen, M. L., Daugaard, P. R.,
Liu, W., and Piskur, J. (2000) J. Mol. Biol., 304,
271-288.
32.Piskur, J., and Langkjer, R. B. (2004) Mol.
Microbiol., 52, 381-389.
33.Delneri, D., Colson, I., Grammenoudi, S.,
Roberts, I. N., Louis, E. J., and Oliver, S. G. (2003) Nature,
422, 68-72.