Interaction of bd-Type Quinol Oxidase from Escherichia coli and Carbon Monoxide: Heme d Binds CO with High Affinity
V. B. Borisov
Department of Molecular Energetics of Microorganisms, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: bor@genebee.msu.su
Received September 5, 2007; Revision received September 25, 2007
Comparative studies on the interaction of the membrane-bound and detergent-solubilized forms of the enzyme in the fully reduced state with carbon monoxide at room temperature have been carried out. CO brings about a bathochromic shift of the heme d band with a maximum at 644 nm and a minimum at 624 nm, and a peak at 540 nm. In the Soret band, CO binding to cytochrome bd results in absorption decrease and minima at 430 and 445 nm. Absorption perturbations in the Soret band and at 540 nm occur in parallel with the changes at 630 nm and reach saturation at 3-5 µM CO. The peak at 540 nm is probably either beta-band of the heme d-CO complex or part of its split alpha-band. In both forms of cytochrome bd, CO reacts predominantly with heme d. Addition of high CO concentrations to the solubilized cytochrome bd results in additional spectral changes in the gamma-band attributable to the reaction of the ligand with 10-15% of low-spin heme b558. High-spin heme b595 does not bind CO even at high concentrations of the ligand. The apparent dissociation constant values for the heme d-CO complex of the membrane-bound and detergent-solubilized forms of the fully reduced enzyme are about 70 and 80 nM, respectively.
KEY WORDS: respiratory chain, terminal oxidase, cytochrome bd, ligand binding, carbon monoxide, energy conservation, Escherichia coliDOI: 10.1134/S0006297908010021
Abbreviations: AEBSF) 4-(2-aminoethyl)benzenesulfonyl fluoride; kon) second order binding rate constant; koff) dissociation rate constant; Mb) myoglobin from sperm whale skeletal muscle; MCD) magnetic circular dichroism.
Cytochrome bd is a terminal quinol oxidase of the respiratory
chain of aerobic and facultatively anaerobic bacteria catalyzing the
four-electron reduction of molecular oxygen to water.
To date, the two classes of the respiratory oxidases have been discovered. The first class comprises a numerous family of the heme/Cu-containing enzymes, which function as proton pumps and contain a binuclear oxygen-reducing active site that includes a high-spin heme and a copper ion. A typical example of the heme-copper oxidases is an aa3-type cytochrome c oxidase; its structural and functional defects cause serious human pathologies [1-3]. The second class of terminal respiratory enzymes comprises the bd-type oxidases. The heme-copper oxidases are already quite well studied. In particular, with the use of X-ray analysis it was possible to resolve a three-dimensional structure of the aa3-type cytochrome c oxidases from beef heart and from bacteria Paracoccus denitrificans and Rhodobacter sphaeroides [4-9], a ba3-type cytochrome oxidase from a thermophilic bacterium Thermus thermophilus [10], and also a bo3-type ubiquinol oxidase from Escherichia coli [11]. In comparison with the typical representatives of the heme-copper class such as aa3- and bo3-type oxidases, the cytochrome bd complex remains poorly studied [12, 13].
Cytochrome bd has no apparent homology with other known oxidases, such as cytochrome c oxidase and bo3-type ubiquinol oxidase [13-15], does not contain copper and non-heme iron [16, 17], and does not function as a transmembrane proton pump though it generates a membrane potential [18-22]. Besides, cytochrome bd has unusually low sensitivity to cyanide, azide, and divalent metal cations [23]. The isolated enzyme is a stable oxygenated complex [24-26], which is possibly due to a high affinity of heme d for oxygen [27, 28].
The bd-type terminal oxidase is a key energy-transducing respiratory enzyme both in microorganisms harmless for humans and animals and in bacteria causing various diseases such as dysentery [29], pneumonia [30], salmonellosis [31, 32], periodontitis [33], brucellosis [34], typhoid [32], and tuberculosis [35]. A positive correlation between virulence of bacterial pathogens responsible for these diseases and level of cytochrome bd expression was reported.
The cytochrome bd contents increase under unfavorable conditions, such as lower oxygen concentration [36], the presence of poisons in the environment (for example, cyanide), uncouplers-protonophores [37], sharp change in ambient temperature, and in a number of other cases when the traditional terminal oxidases are not able to function, whereas the “alternative” cytochrome bd successfully works [38, 39]. In nitrogen-fixing bacteria, cytochrome bd takes part in respiratory protection of nitrogenase against oxygen [40]. Cytochrome bd from E. coli is involved in regulation of the disulfide (-S-S-) bond formation upon protein folding in a cell [41]. The data on unusually high NO dissociation from the cytochrome bd active site (in comparison with that of cytochrome c oxidase and other heme-copper oxidases) elucidate how a bd-type enzyme can help a pathogenic enterobacterium to overcome immune protection of the host cell [42-44].
The accepted scheme for conversions of the oxygen intermediates in the cytochrome bd active site [21, 22, 45-47] is the following: O → R + oxygen → A → P → F → O, where O, R, A, P, F are oxidized, reduced, oxygenated, peroxy, and oxo-ferryl forms of cytochrome d, respectively.
The enzyme consists of the two subunits (58 and 43 kD), each being a typical integral membrane protein. These subunits carry three redox centers: one low-spin (heme b558) and two high-spin (hemes b595 and d) [48, 49]. Heme b558 is located on subunit I, whereas hemes b595 and d are likely to be in the area of the subunit contact [50]. According to current thinking, all the three heme redox groups are located closer to the outer (periplasmic) side of the membrane [51].
The low-spin heme b558 apparently takes part in the oxidation of ubiquinol. The high-spin heme d binds oxygen and likely takes part in the oxygen-reducing reaction. The role of the high-spin heme b595 remains obscure. Some authors propose that heme b595 participates in the reduction of oxygen forming together with heme d a binuclear oxygen-reducing site, similar to the heme/Cu oxygen-reducing site in the aa3-and bo3-type oxidases [22, 52-57]. In the opinion of other researchers, the function of heme b595 is limited to transferring an electron from heme b558 to heme d [58, 59]. Some authors believe that heme b595 can form a second site capable of reacting with oxygen [60, 61].
It is important to understand whether the presence of a binuclear redox site in terminal oxidases is a prerequisite for the reduction of oxygen to water and, if so, for which of the steps of the catalytic cycle. An alternative could be the necessity for the presence of the second redox site to pump protons through the membrane. It is worth noting that terminal oxidases lacking the binuclear oxygen-reducing site have still not been found. Furthermore, the cytochrome bo3 mutants lacking CuB (the second component of the oxygen-reducing site) are not able to bind and reduce oxygen [62]. However, the data available in the literature on the presence of such a site in cytochrome bd can hardly be considered as sufficiently valid. Formally the reaction of reduction of oxygen to water can be divided into two steps: the actually oxidase step where O2 is reduced to H2O2, and the peroxidase step where the bound peroxide undergoes further reduction to water. The enzymes carrying out either the first part of the reaction (a quinol oxidase from the sea bacterium Pseudomonas nautica 617 [63-65]) or the second one (many peroxidases) [66-68] but not containing a binuclear site have been found. Furthermore, cytochrome cd1-nitrite reductase found in many denitrifying bacteria can also catalyze the four-electron reduction of oxygen to water [69]. However, cytochrome cd1-nitrite reductase does not contain a binuclear bimetallic site; only one redox center (heme d1) is involved in the oxygen-reducing reaction [69]. Thus, deciphering the role of high-spin heme b595 is of particular interest.
Studying the interaction of heme-containing enzymes with ligands is one of the efficient and frequently used approaches for establishing the arrangement and functioning of the catalytic sites of these proteins. In the present work, a comparative study on the reaction of carbon monoxide with cytochrome bd incorporated into the E. coli membrane and the purified enzyme solubilized in the detergent micelles has been carried out.
MATERIALS AND METHODS
Reagents. Yeast extract and tryptone was purchased from Difco (USA); ampicillin, DNase I, kanamycin, and sodium N-lauroyl-sarcosinate were obtained from Fluka (Switzerland); carbon monoxide, dimethylsulfoxide, sodium dithionite, KCl, and MgSO4 from Merck (Germany); myoglobin from sperm whale skeletal muscle (Mb), EDTA, Ches, and Hepes from Serva (Germany); DEAE-Sepharose Fast Flow, 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), leupeptin, and benzamidine from Sigma (USA); sucrose monolaurate from Anatrace (USA). Other reagents were of chemically pure grade produced in Russia. All aqueous solutions were prepared with distilled water that was additionally purified by means of a Milli-Q System (Millipore, USA).
Bacterial strain. The work was performed using the bd-type oxidase from E. coli (strain GO105/pTK1, kindly provided by Prof. R. Gennis from the University of Illinois at Urbana-Champaign, USA). This strain is convenient due to overexpression of the enzyme achieved by introduction of a plasmid containing the cytochrome bd gene. Also, cytochrome bo3 (the second terminal oxidase in E. coli) has been deleted in this strain, which allows obtaining the cytochrome bd preparation without impurity of another oxidase.
Growth of cells. The cells were grown aerobically in a fermenter (with controlled feed of O2) or in flasks on a shaker (at 200 rpm) at 37°C. The medium contained 80 mM sodium phosphate, 2.5 mM sodium citrate, 19 mM ammonium sulfate, 1% tryptone, 0.5% yeast extract, 0.5% casamino acids, 0.01% L-tryptophan, 2% (v/v) glycerol, 0.8 mM MgSO4, 0.18 mM FeSO4·7H2O, 0.1 mM CuSO4·5H2O, 0.005% kanamycin, and 0.01% ampicillin, pH 7.2. The cell growth was tracked by monitoring increase in turbidity at 600 nm. The cultivation typically took 22-24 h. On the late logarithmic growth phase, the cells were sedimented (5500g, 10 min, 4°C) and washed twice with medium containing 172 mM NaCl, 5 mM sodium phosphate, and AEBSF (on a spatula tip), pH 7.5.
Membrane preparation. The washed cells were suspended in medium containing 20 mM Tris, 0.5 mM EDTA, 5 mM MgSO4, 20 mM benzamidine, 1 µM leupeptin, pH 8.3. Just before the cell disruption, AEBSF (on a spatula tip), 1 mM 1,4-dithiothreitol, and DNase I (0.01 mg/ml) were added to the suspension. The suspension was passed twice through a French press with 35 ml portions. Intact and partially destroyed cells were removed by centrifugation (14,500g, 15 min, 4°C). Subcellular vesicles were sedimented from the supernatant by ultracentrifugation (160,000g, 2 h, 4°C) and then homogenized with a desired medium. The membranes were stored at -70 to -80°C or in liquid nitrogen.
Isolation and purification of the enzyme. Cytochrome bd was solubilized and purified as described earlier by Miller and Gennis [70] with some modifications.
The thawed E. coli membranes were homogenized with a medium for equilibration of a chromatography column without detergent (50 mM potassium phosphate, 25 mM KCl, 5 mM EDTA, AEBSF, pH 6.5). The homogenate was supplemented with sucrose monolaurate (1.8% final concentration) and incubated in an ice bath upon stirring for 2 h. The suspension was centrifuged (160,000g, 60 min, 4°C), the pellet was discarded and the supernatant loaded on a DEAE-Sepharose Fast Flow column equilibrated with 50 mM potassium phosphate buffer also containing 25 mM KCl, 5 mM EDTA, and 0.1% sucrose monolaurate, pH 6.5. The column was washed with 100 ml of the above buffer at 100-120 ml/h flow rate and 4°C (the flow rate was set with a peristaltic pump (Pharmacia, Sweden)), then the elution was performed with a KCl gradient (25-350 mM) at 90-100 ml/h flow rate. Fractions of 7 ml were collected (LKB 2070 Ultrorac II Fraction Collector), monitoring absorption at 405 nm (spectrophotometer UA-5 with a flow-through cell, ISCO). The fractions with an absorbance ratio of A412/A280 >= 0.7 were collected for further work. These fractions were pooled and concentrated by ultrafiltration with the aid of an Amicon cell and a YM-30 membrane filter. The resulting solution was dialyzed at 4°C against 50 mM sodium phosphate buffer containing 5 mM EDTA and 0.05% sodium N-lauroyl-sarcosinate, pH 7.0, with buffer change every 5 h for two times. Sometimes gel filtration on a PD10 column prepacked with Sephadex G-25 (Pharmacia) was used instead of dialysis. The enzyme was stored with small portions (100-200 µl) in liquid nitrogen.
Spectroscopy. Absorption changes were recorded in an Aminco-SLM DW 2000 spectrophotometer in split beam mode in standard cuvettes at room temperature. The medium composition used in the experiments is described in the figure legends.
Protein concentration was measured by the methods of Lowry et al. [71] and Bradford [72] using bovine serum albumin as a standard. Cytochrome bd concentration was determined from the difference absorption spectra (dithionite-reduced minus “air-oxidized”) using a millimolar extinction coefficient value of Deltaepsilon628-607 = 10.8 mM-1·cm-1 [54].
CO concentration was estimated assuming a solubility of this gas in water at 20°C and 1 atm to be 1 mM [73].
Treatment of the experimental data. Computer analysis of the experimental data was carried out with the aid of the software package GIM (Scientific Graphic Interactive Management System) developed by A. L. Drachev in the A. N. Belozersky Institute of Physico-Chemical Biology, M. V. Lomonosov Moscow State University.
Analysis of carbon monoxide titration curves of cytochrome bd spectral changes. CO titration curves of spectral changes of the dithionite-reduced cytochrome bd were processed using the GIM package according to Eq. (1):
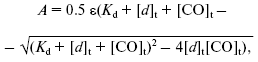
where A is DeltaA644-624 estimated from the absorption difference of the dithionite-reduced cytochrome bd in the presence and absence of CO, epsilon is extinction coefficient for the heme d-CO complex (Deltaepsilon644-624), Kd is an apparent dissociation constant for the heme d-CO complex, [d]t is total concentration of cytochrome d, [CO]t is concentration of added CO.
Upon processing of the titration curves, epsilon and Kd were given as sought values.
Equation (1) was derived from the following equations:
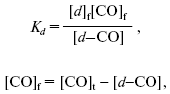
where L is light path length.
Determination of apparent dissociation constant (Kd) for the heme d-CO complex. An apparent Kd for the heme d-CO complex was determined according to Eq. (2):
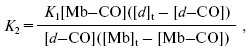
where K1 is an apparent dissociation constant for the Mb-CO complex; K2 is an apparent dissociation constant for the heme d-CO complex; [d]t is total concentration of cytochrome d; [Mb]t is total concentration of Mb determined from Deltaepsilon579-594 = 6.4 mM-1·cm-1 for difference spectra (carboxymyoglobin (Fe2+-CO) minus deoxymyoglobin (Fe2+)), from Deltaepsilon435-480 = 121 mM-1·cm-1 for absolute spectra of reduced myoglobin (deoxymyoglobin), and from Deltaepsilon502-600 = 10.2 mM-1·cm-1 for absolute spectra of oxidized myoglobin (Fe3+) (Deltaepsilon values for Mb were taken from the report of Wood [74]); [Mb-CO] is concentration of the Mb-CO complex determined as [Mb]t·(DeltaA579-594x µM CO)/(DeltaA579-5941 mM CO) (DeltaA was taken from the difference spectra (CO + reduced minus reduced)); [d-CO] is concentration of the heme d-CO complex determined as [d]t·(DeltaA643-623x µM CO)/(DeltaA643-6231 mM CO) (DeltaA was taken from the difference spectra (CO + reduced minus reduced)); [CO]t is concentration of added CO.
Equation (2) was derived from the following equations:
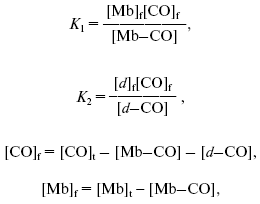
where [CO]f is concentration of free carbon monoxide, [Mb]f is concentration of free Mb, [d]f is concentration of free cytochrome d.
RESULTS
CO-induced changes of reduced cytochrome bd absorption spectra. Membrane form of cytochrome bd. Addition of CO to the membrane-bound reduced cytochrome bd results in spectral changes shown in Fig. 1a. Difference (CO + dithionite-reduced minus dithionite-reduced) spectra at low (17 µM) and high (1 mM) concentrations of the ligand are almost identical except for slight differences in the Soret region (see Fig. 1c, spectrum 2) and are in agreement with the published data [48, 75]. In the visible region, a band with lambdamax = 644 nm and lambdamin = 624 nm (Deltaepsilon644-624 ~ 13-14 mM-1·cm-1), a peak at 540 nm (Deltaepsilon540-500 ~ 4.4 mM-1·cm-1), and a “W”-shaped trough in the Soret region with two minima, at 430 and 445 nm, and a local maximum near 437 nm (Deltaepsilon470-445 ~ 23-25 mM-1·cm-1) are observed.
Solubilized enzyme. Addition of CO to the solubilized bd complex gives rise to a band with lambdamax = 644 nm and lambdamin = 624 nm, a peak at 540 nm, and a characteristic “W”-shaped curve in the Soret region with the two minima, at 430 and 444 nm, and a local maximum at ~437 nm in the difference absorption spectrum of the enzyme (Fig. 1b). The spectrum of cytochrome bd at low CO concentration (Fig. 1b, curve 1) is analogous to that of the CO complex with the membrane enzyme (Fig. 1a). However, in case of the isolated enzyme, the differences between the spectra at high (1 mM) and low (17 µM) CO concentrations are much more pronounced (compare spectra 1 and 2 in Fig. 1c): the difference spectrum shows a band with a maximum at 422 nm, a minimum at 434 nm, and a small shoulder at ~440 nm in the Soret band (Deltaepsilon422-434 ~ 19 mM-1·cm-1), and the two minima, at 562 and 531 nm, in the visible region, Deltaepsilon578-562 ~ 4 mM-1·cm-1 (Fig. 1c, curve 1).Fig. 1. CO-induced spectral changes of cytochrome bd from E. coli. Panels (a) and (b) show difference absorption spectra of the cytochrome d-containing membranes (a) and the solubilized enzyme (b): treated with 17 µM CO (curves 1) and 1 mM CO (curves 2) after reduction with dithionite versus dithionite-reduced. Panel (c) shows difference between the spectra at high and low CO concentrations for the solubilized enzyme (curve 1) and the membrane form of cytochrome bd (curve 2). Spectra were recorded in the split-beam mode. The optical cuvette contains cytochrome bd (1.6 µM) in medium containing 50 mM Hepes, 50 mM Ches, 0.1 mM EDTA, pH 8.0. In case of the solubilized enzyme, the detergent 0.025% sodium N-lauroyl-sarcosinate was also present.
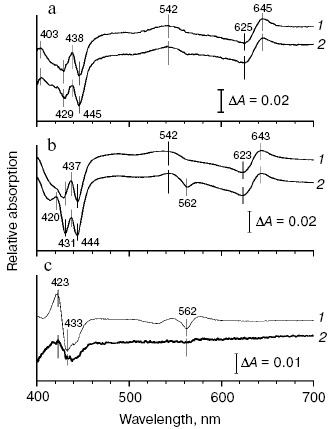
The CO-induced shift of the absorption band near 630 nm is usually assigned to heme d. Absorption changes at about 436 nm could be due to contribution from heme b595 [76]. We have tried to test homogeneity of the CO-induced absorption changes in different parts of the spectrum of the membrane and solubilized forms of the dithionite-reduced cytochrome bd.
CO concentration dependence of the reduced cytochrome bd absorption changes. Membrane form of cytochrome bd. Figure 2a shows CO concentration dependence of cytochrome bd absorption changes at different wavelengths. It can be seen that the changes in the Soret band (curve 1), as well as the development of a peak at 540 nm (curve 3) titrate with CO in parallel with the absorption increase in the alpha-band of the heme d-CO complex (curve 2) reaching saturation at 3-5 µM CO, which is not much higher than the concentration of the enzyme (1.6 µM). A hardly appreciable second phase of the spectral changes is observed in the Soret region, which continues to grow even at 1 mM CO and does not contribute substantially to the dependence shown in Fig. 2a. The difference spectrum of the second phase (curve 2 in Fig. 1c) shows a band with lambdamax = 423 nm, lambdamin = 436 nm (Deltaepsilon423-436 ~ 5-6 mM-1·cm-1) and a very small minimum at 561 nm in the visible (Deltaepsilon561 ~ 0.3-0.5 mM-1·cm-1). This spectrum possibly reflects the interaction of the ligand with a very small part of heme b558 (1-3%). The saturation curves observed at different wavelengths fit well the Eq. (1) (see “Materials and Methods” section). This allows making only an upper estimate of the Kd value (0.1-0.2 µM) since the latter is much lower than the concentration of the enzyme (1.6 µM): the theoretical curves for this Kd range are virtually indistinguishable from each other.
Solubilized enzyme. As for the membrane form of cytochrome bd, the spectral changes of the solubilized enzyme at 540 nm and 644-624 nm titrate with CO in parallel, look like saturation curves, and apparently reflect the same process--CO binding to heme d (Fig. 2b, curves 2 and 3). The peak at 540 nm may be either beta-band of the heme d-CO complex or part of its split alpha-band. These curves can also be described by the equation used for the Kd determination. However, the Kd values obtained (0.1-0.25 µM CO) are only an upper estimate, as for the membrane form of cytochrome bd. CO concentration dependence of the spectral changes in the Soret band is clearly biphasic: the first phase corresponds to the changes in the visible region; the second phase does not saturate even at 1 mM CO and makes the greater contribution to the total amplitude of the changes in the gamma-band than in case of the membrane form of the enzyme (compare spectra 1 and 2 in Fig. 1c). It is worth noting that the amplitude of the second phase apparently corresponding to the interaction of the ligand with part of heme b558 (about 10-15%) is not a constant but preparation-dependent. Usually larger amplitude of the second phase is observed with the enzyme kept for a long time.Fig. 2. Dependence of the spectral changes of cytochrome bd from E. coli at different wavelength ranges on CO concentration: a) membrane form of the enzyme; b) solubilized enzyme. Shown are the curves obtained by processing of the corresponding difference absorption spectra (CO + dithionite-reduced minus dithionite-reduced) with the use of the software package GIM: 1) 470-444 nm; 2) 644-624 nm; 3) 540 nm versus an arbitrary baseline connecting the 500 and 590 nm points. Other conditions as in Fig. 1.
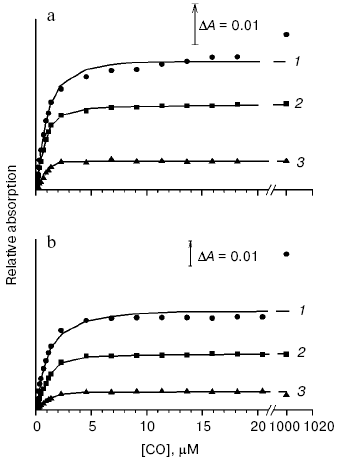
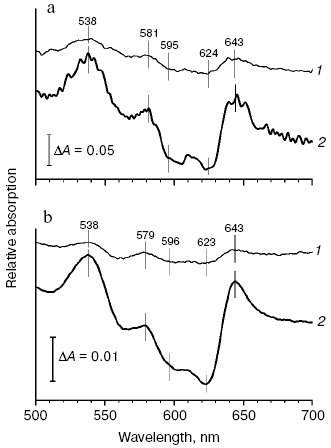
Determination of an apparent dissociation constant (Kd) for the CO complexes of the membrane and solubilized forms of the reduced cytochrome d. Membrane form of cytochrome bd. Since we failed to obtain the exact Kd value from the data on the concentration dependence of the CO-induced absorption changes of the reduced cytochrome bd, another approach has been used. To determine Kd for the CO complex with heme d of the bacterial membranes, CO titration of spectral changes of the mixture of cytochrome bd and Mb has been performed observing distribution of the ligand between these two proteins (Fig. 3a). Kd for heme d has been estimated according to Eq. (2) as described in “Materials and Methods” section taking the Kd value for the CO-Mb complex to be 37 nM [77]. This was possible to do because the spectral characteristics of the CO complexes with heme d and Mb in the visible region of the difference (CO + reduced minus reduced) absorption spectrum are different: the alpha-band of the heme d-CO complex shows a maximum at 644 nm and a minimum at 624 nm, whereas the Mb-CO complex displays a band with lambdamax = 579 nm and lambdamin = 594 nm. The apparent Kd value for the CO complex with heme d of the membrane form of the enzyme determined in such a way appeared to be ~70 nM.
Solubilized enzyme. The apparent Kd for the CO complex with the solubilized cytochrome d has been determined as for the membranes (Fig. 3b). Its value appeared to be ~80 nM. For comparison, the corresponding apparent Kd value for the R-state alpha-subunits of human hemoglobin is 2 nM [78], horseradish peroxidase - 30 nM [79], cytochrome aa3 - 0.29 µM [80], and cytochrome bo3 - 1.7 µM [81].
Thus, there is no evidence for the existence of a second (apart from heme d) CO-binding heme with Kd lower than 1 µM both in the membrane-bound cytochrome bd and in the solubilized enzyme.Fig. 3. Spectral changes of the mixture of cytochrome bd from E. coli and sperm whale skeletal muscle myoglobin induced by CO. a) Difference spectra of the cytochrome d-containing membranes (1.21 µM) with myoglobin (1.31 µM): treated with 0.4 and 4 µM CO (curves 1 and 2, respectively) after reduction with dithionite versus dithionite-reduced. b) Difference spectra of the solubilized enzyme (2.33 µM) with myoglobin (2.1 µM): treated with 0.7 and 5.5 µM CO (curves 1 and 2, respectively) after reduction with dithionite versus dithionite-reduced. Spectra were recorded in the split-beam mode in 10 mM potassium phosphate buffer, pH 7.0. In case of the solubilized enzyme, the detergent 0.025% sodium N-lauroyl-sarcosinate was also present.
DISCUSSION
We have compared the interaction of the native (membrane) form of the E. coli cytochrome bd and the solubilized enzyme with carbon monoxide. We aimed to determine which of the three heme centers of the bd-oxidase can bind CO. We showed that CO reacts predominantly with heme d. This is evident from the two observations:
(i) the amplitude of the CO-induced absorption changes in the visible region and in the Soret band is similar. Such a ratio is unusual for most hemoproteins but typical for cytochrome d, the Soret band of which is much less intense as compared to the corresponding gamma-bands of the iron-porphyrin complexes of the a-, b-, c-, or o-type [75];
(ii) CO concentration dependencies of the absorption changes of cytochrome bd at all wavelengths tested are very similar. Since the CO-induced spectral changes of cytochrome bd at 540 and 644-624 nm titrate identically, the peak at 540 nm may be beta-band of the heme d-CO complex or part of its split alpha-band.
It is important to emphasize that the CO-induced spectral changes of the membrane form of cytochrome bd are virtually identical to those observed with the solubilized enzyme at a low concentration of the ligand. In both cases, CO added at micromolar concentrations reacts predominantly with heme d. At the same time, the addition of high concentrations of carbon monoxide (1 mM) to the solubilized cytochrome bd results in additional spectral changes in the gamma absorption band of the enzyme that points to binding of the ligand to a b-type heme. The data are fully consistent with an earlier published work [54] where, upon studying the interaction of cyanide and CO with the solubilized form of the enzyme by the magnetic circular dichroism (MCD), it was established that these ligands, added at high concentration, react with 10-20% of low-spin heme b558.
At first glance, it seems surprising that CO and other ligands are capable of binding to part of the low-spin heme b558 of the solubilized enzyme. However, it is well-known that many soluble heme c-containing proteins can bind CO at atmospheric pressure [82]. Cyanide also reacts with cytochrome c under alkaline conditions. This is because the sixth axial ligand of cytochrome c is Met [74] bonding a polypeptide with heme iron rather poorly and therefore can be replaced with strong exogenous ligands. The studies carried out by the groups of Gennis and Thomson showed that amino acid residues Met393 and His186 of subunit I can serve as the axial ligands of heme b558 [83, 84]. It is interesting that a mutant, in which Met393 was replaced with Leu, retains heme b558 but in a high-spin form. Probably one His186 is enough for maintenance of the bond between heme b558 and the protein [83].
It cannot be excluded that binding of CO with part of heme b558 in the solubilized enzyme occurs due to partial denaturation of the protein upon solubilization. However, reconstitution of the enzyme into artificial phospholipid membranes (azolectin liposomes) reverts its CO-binding properties to those typical for cytochrome bd of the bacterial membranes, i.e. heme b558 no longer binds CO upon reconstitution of the enzyme into the liposomes (V. B. Borisov, unpublished data). Therefore, if weakening the bond of Met with the heme b558 iron is caused by protein denaturation, this denaturation is apparently reversible.
These results suggest that, upon addition of CO to both the cytochrome d-containing membranes and the purified solubilized enzyme, a majority of heme b595 (>95%) does not bind this ligand. This is in agreement with data according to which the spectral changes induced by dissociation of CO from its complex with the isolated reduced cytochrome bd from Azotobacter vinelandii at low temperatures (from -70 to -100°C) correspond to binding of the ligand to less than 5% of heme b595 [56]. It is likely that heme b595, although in the high-spin state, is resistant to the interaction with the classical ligands of the high-spin iron-porphyrin complexes. It cannot be excluded that despite the high-spin state of the iron-porphyrin group, the specific features of the protein environment are such that this redox center is protected from interaction with the ligands. In such case, the participation of b595 in the oxygen reduction in cooperation with heme d is unlikely and its role is limited to a transfer of an electron to heme d. Another possible explanation that we favor is the following: (i) both heme b595 and heme d potentially can bind ligands; (ii) the hemes are located very close to each other forming a binuclear active site; (iii) the spatial proximity of hemes b595 and d results in steric restrictions allowing such a di-heme site to bind only one ligand molecule; (iv) heme d has a higher affinity for ligands than heme b595. In that case, the final result observed upon addition of a ligand will be always the ligand binding to heme d, whereas heme b595 will remain mainly in the free state.
We determined an apparent Kd value for the CO complex with cytochrome d from E. coli. It appeared that heme d binds this ligand with a high affinity. The Kd values for the membrane and solubilized forms of the enzyme appeared to be about 70 and 80 nM, respectively. It has to be noted that the Kd values measured in this work are higher than Kd = 20 nM published by Hill et al. [85] for the CO complex with the isolated cytochrome bd from E. coli. However, the value of Kd = 20 nM was obtained indirectly, namely, calculated from the equation for Kd = koff/kon, taking into account the values of koff = 1.6 sec-1 and kon = 8·107 M-1·sec-1 measured experimentally [85]. Very recently, using the rapid mixing (stopped-flow) method, it was established that the koff value for the CO complex with the isolated cytochrome bd from E. coli is actually 6 sec-1 [42], i.e. several times higher than koff measured by Hill et al. If the specified value of koff = 6 sec-1 is introduced into the equation for Kd = koff/kon, the estimated Kd value appears to be 75 nM that coincides the apparent Kd values for the heme d-CO complex measured in this work.
It is of interest to consider a plausible structure of the heme d2+-CO complex. In reactions of the fully reduced oxidases of the heme-copper family comprising cytochrome c oxidase and cytochrome bo3 with carbon monoxide, the conversion of a high-spin pentacoordinate heme (a-, o-, or b-type) to its low-spin hexacoordinate CO complex occurs:
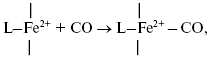
It is proposed that heme d in cytochrome bd is in the high-spin pentacoordinate state [87, 88]. Moreover, according to the data of Sun et el., heme d retains the high-spin pentacoordinate state in the oxygenated (Fe2+-O2) and oxo-ferryl (Fe4+=O2-) intermediates, as well as in the cyanide complex [87, 88]. Hence, one may propose that, when in the CO complex, heme d is also in the high-spin state. Recently, it has been reported that anaerobic reduction of cytochrome bd is followed by secondary absorption changes on a time scale of tens of minutes pointing to bonding of heme d iron to an endogenous protein ligand [89]. In the present study, CO was added to the sample immediately after anaerobic reduction of cytochrome bd, i.e. prior to a major part of the spontaneous spectral changes develops. According to a work of Azarkina et al. [89], anaerobic reduction of heme d converting a distal oxygen ligand to water can produce transiently either tetracoordinate state (without axial ligands) or pentacoordinate state (with a weak axial ligand of the heme iron on the proximal side of the heme plane). Thus, several possible ways of CO bonding to heme d may be considered.
Initially generated tetracoordinate heme d can be converted to the high-spin pentacoordinate complex with CO:
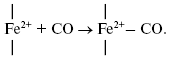
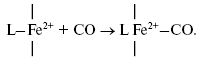
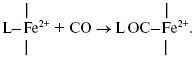
The author is grateful to Dr. A. A. Konstantinov for support and help in planning the experiments and analysis of the data, Dr. R. Gennis for the strain of E. coli GO105/pTK1, and Dr. V. P. Skulachev and Dr. A. D. Vinogradov for their interest in this work, useful discussions, and critical remarks.
This work was supported in part by grants from the Russian Foundation for Basic Research (project 05-04-48096-a), Howard Hughes Medical Institute (project 55005615), and the Civilian Fund for Research and Development (project RUB-1-2836-MO-06).
REFERENCES
1.Skulachev, V. P. (1999) Mol. Aspects Med.,
20, 139-184.
2.Borisov, V. B. (2002) Mol. Aspects Med.,
23, 385-412.
3.Borisov, V. B. (2004) Ital. J. Biochem.,
53, 34-40.
4.Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki,
T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, T., Yaono, R., and
Yoshikawa, S. (1995) Science, 269, 1069-1074.
5.Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki,
T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., and
Yoshikawa, S. (1996) Science, 272, 1136-1144.
6.Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R.,
Yaono, R., Yamashita, E., Inoue, N., Yao, M., Fei, M. J., Libeu, C. P.,
Mizushima, T., Yamaguchi, H., Tomizaki, T., and Tsukihara, T. (1998)
Science, 280, 1723-1729.
7.Iwata, S., Ostermeier, C., Ludwig, B., and Michel,
H. (1995) Nature, 376, 660-669.
8.Abramson, J., Svensson-Ek, M., Byrne, B., and
Iwata, S. (2001) Biochim. Biophys. Acta, 1544, 1-9.
9.Svensson-Ek, M., Abramson, J., Larsson, G.,
Tornroth, S., Brzezinski, P., and Iwata, S. (2002) J. Mol.
Biol., 321, 329-339.
10.Soulimane, T., Buse, G., Bourenkov, G. P.,
Bartunik, H. D., Huber, R., and Than, M. E. (2000) EMBO J.,
19, 1766-1776.
11.Abramson, J., Riistama, S., Larsson, G.,
Jasaitis, A., Svensson-Ek, M., Laakkonen, L., Puustinen, A., Iwata, S.,
and Wikstrom, M. (2000) Nat. Struct. Biol., 7,
910-917.
12.Borisov, V. B. (1996) Biochemistry
(Moscow), 61, 565-574.
13.Poole, R. K., and Cook, G. M. (2000) Adv.
Microb. Physiol., 43, 165-224.
14.Van der Oost, J., deBoer, A. P. N., deGier, J.-W.
L., Zumft, W. G., Stouthamer, A. H., and van Spanning, R. J. M. (1994)
FEMS Microbiol. Lett., 121, 1-10.
15.Green, G. N., Fang, H., Lin, R.-J., Newton, G.,
Mather, M., Georgiou, C. D., and Gennis, R. B. (1988) J. Biol.
Chem., 263, 13138-13143.
16.Miller, M. J., and Gennis, R. B. (1983) J.
Biol. Chem., 258, 9159-9165.
17.Kita, K., Konishi, K., and Anraku, Y. (1984)
J. Biol. Chem., 259, 3375-3381.
18.Puustinen, A., Finel, M., Haltia, T., Gennis, R.
B., and Wikstrom, M. (1991) Biochemistry, 30,
3936-3942.
19.Bertsova, Y. V., Bogachev, A. V., and Skulachev,
V. P. (1997) FEBS Lett., 414, 369-372.
20.Kolonay, J. F., Jr., and Maier, R. J. (1997)
J. Bacteriol., 179, 3813-3817.
21.Jasaitis, A., Borisov, V. B., Belevich, N. P.,
Morgan, J. E., Konstantinov, A. A., and Verkhovsky, M. I. (2000)
Biochemistry, 39, 13800-13809.
22.Belevich, I., Borisov, V. B., Zhang, J., Yang,
K., Konstantinov, A. A., Gennis, R. B., and Verkhovsky, M. I. (2005)
Proc. Natl. Acad. Sci. USA, 102, 3657-3662.
23.Poole, R. K., Williams, H. D., Downie, J. A., and
Gibson, F. (1989) J. Gen. Microbiol., 135, 1865-1874.
24.Poole, R. K., Kumar, C., Salmon, I., and Chance,
B. (1983) J. Gen. Microbiol., 129, 1335-1344.
25.Kahlow, M. A., Loehr, T. M., Zuberi, T. M., and
Gennis, R. B. (1993) J. Am. Chem. Soc., 115,
5845-5846.
26.Borisov, V. B., Smirnova, I. A., Krasnoselskaya,
I. A., and Konstantinov, A. A. (1994) Biochemistry (Moscow),
59, 437-443.
27.Belevich, I., Borisov, V. B., Konstantinov, A.
A., and Verkhovsky, M. I. (2005) FEBS Lett., 579,
4567-4570.
28.Belevich, I., Borisov, V. B., Bloch, D. A.,
Konstantinov, A. A., and Verkhovsky, M. I. (2007) Biochemistry,
46, 11177-11184.
29.Way, S. S., Sallustio, S., Magliozzo, R. S., and
Goldberg, M. B. (1999) J. Bacteriol., 181, 1229-1237.
30.Smith, A., Hill, S., and Anthony, C. (1990) J.
Gen. Microbiol., 136, 171-180.
31.Osborne, J. P., and Gennis, R. B. (1999)
Biochim. Biophys. Acta, 1410, 32-50.
32.Turner, A. K., Barber, L. Z., Wigley, P.,
Muhammad, S., Jones, M. A., Lovell, M. A., Hulme, S., and Barrow, P. A.
(2003) Infect. Immun., 71, 3392-3401.
33.Baughn, A. D., and Malamy, M. H. (2004)
Nature, 427, 441-444.
34.Endley, S., McMurray, D., and Ficht, T. A. (2001)
J. Bacteriol., 183, 2454-2462.
35.Kana, B. D., Weinstein, E. A., Avarbock, D.,
Dawes, S. S., Rubin, H., and Mizrahi, V. (2001) J. Bacteriol.,
183, 7076-7086.
36.Trutko, S. M., Evtushenko, L. I., Dorofeeva, L.
V., Shlyapnikov, M. G., Gavrish, E. Y., Suzina, N. E., and Akimenko, V.
K. (2003) Microbiology (Moscow), 72, 301-307.
37.Avetisyan, A. V., Bogachev, A. V., Murtasina, R.
A., and Skulachev, V. P. (1992) FEBS Lett., 306,
199-202.
38.Hill, S., Viollet, S., Smith, A. T., and Anthony,
C. (1990) J. Bacteriol., 172, 2071-2078.
39.Kelly, M. J. S., Poole, R. K., Yates, M. G., and
Kennedy, C. (1990) J. Bacteriol., 172, 6010-6019.
40.Poole, R. K., and Hill, S. (1997) Biosci.
Rep., 17, 307-317.
41.Bader, M., Muse, W., Ballou, D. P., Gassner, C.,
and Bardwell, J. C. A. (1999) Cell, 98, 217-227.
42.Borisov, V. B., Forte, E., Sarti, P., Brunori,
M., Konstantinov, A. A., and Giuffre, A. (2007) Biochem. Biophys.
Res. Commun., 355, 97-102.
43.Borisov, V. B., Forte, E., Konstantinov, A. A.,
Poole, R. K., Sarti, P., and Giuffre, A. (2004) FEBS Lett.,
576, 201-204.
44.Borisov, V. B., Forte, E., Sarti, P., Brunori,
M., Konstantinov, A. A., and Giuffre, A. (2006) FEBS Lett.,
580, 4823-4826.
45.Borisov, V. B., Gennis, R. B., and Konstantinov,
A. A. (1995) Biochemistry (Moscow), 60, 231-239.
46.Borisov, V., Gennis, R., and Konstantinov, A. A.
(1995) Biochem. Mol. Biol. Int., 37, 975-982.
47.Belevich, I., Borisov, V. B., and Verkhovsky, M.
I. (2007) J. Biol. Chem., 282, 28514-28519.
48.Lorence, R. M., Koland, J. G., and Gennis, R. B.
(1986) Biochemistry, 25, 2314-2321.
49.Miller, M. J., Hermodson, M., and Gennis, R. B.
(1988) J. Biol. Chem., 263, 5235-5240.
50.Newton, G., and Gennis, R. B. (1991) Biochim.
Biophys. Acta, 1089, 8-12.
51.Zhang, J., Barquera, B., and Gennis, R. B. (2004)
FEBS Lett., 561, 58-62.
52.Hill, J. J., Alben, J. O., and Gennis, R. B.
(1993) Proc. Natl. Acad. Sci. USA, 90, 5863-5867.
53.Tsubaki, M., Hori, H., Mogi, T., and Anraku, Y.
(1995) J. Biol. Chem., 270, 28565-28569.
54.Borisov, V., Arutyunyan, A. M., Osborne, J. P.,
Gennis, R. B., and Konstantinov, A. A. (1999) Biochemistry,
38, 740-750.
55.Vos, M. H., Borisov, V. B., Liebl, U., Martin,
J.-L., and Konstantinov, A. A. (2000) Proc. Natl. Acad. Sci.
USA, 97, 1554-1559.
56.Borisov, V. B., Sedelnikova, S. E., Poole, R. K.,
and Konstantinov, A. A. (2001) J. Biol. Chem., 276,
22095-22099.
57.Borisov, V. B., Liebl, U., Rappaport, F., Martin,
J.-L., Zhang, J., Gennis, R. B., Konstantinov, A. A., and Vos, M. H.
(2002) Biochemistry, 41, 1654-1662.
58.Poole, R. K., and Williams, H. D. (1987) FEBS
Lett., 217, 49-52.
59.Hata-Tanaka, A., Matsuura, K., Itoh, S., and
Anraku, Y. (1987) Biochim. Biophys. Acta, 893,
289-295.
60.D'mello, R., Hill, S., and Poole, R. K. (1996)
Microbiology, 142, 755-763.
61.Rothery, R. A., Houston, A. M., and Ingledew, W.
J. (1987) J. Gen. Microbiol., 133, 3247-3255.
62.Mogi, T., Hirano, T., Nakamura, H., Anraku, Y.,
and Orii, Y. (1995) FEBS Lett., 370, 259-263.
63.Denis, M., Arnaud, S., and Malatesta, F. (1989)
FEBS Lett., 247, 475-479.
64.Arnaud, S., Malatesta, F., Guigliarelli, B.,
Gayda, J.-P., Bertrand, P., Miraglio, R., and Denis, M. (1991) Eur.
J. Biochem., 198, 349-356.
65.Arnaud, S., Malatesta, F., and Denis, M. (1992)
FEBS Lett., 296, 259-262.
66.Vitello, L. B., Erman, J. E., Miller, M. A.,
Wang, J., and Kraut, J. (1993) Biochemistry, 32,
9807-9818.
67.Marquez, L. A., Huang, J. T., and Dunford, H. B.
(1994) Biochemistry, 33, 1447-1454.
68.Miller, M. A. (1996) Biochemistry,
35, 15791-15799.
69.Koppenhofer, A., Little, R. H., Lowe, D. J.,
Ferguson, S. J., and Watmough, N. J. (2000) Biochemistry,
39, 4028-4036.
70.Miller, M. J., and Gennis, R. B. (1986) Meth.
Enzymol., 126, 87-94.
71.Lowry, O. H., Rosebrough, N. J., Farr, A. L., and
Randall, R. J. (1951) J. Biol. Chem., 193, 265-275.
72.Bradford, M. M. (1976) Analyt. Biochem.,
72, 248-254.
73.Anderson, S. R., and Anthononi, E. (1968) J.
Biol. Chem., 243, 2918-2920.
74.Wood, P. M. (1984) Biochim. Biophys. Acta,
768, 293-317.
75.Poole, R. K. (1988) in Bacterial Energy
Transduction (Anthony, C., ed.) Academic Press, London, pp.
231-291.
76.Poole, R. K. (1994) Anthonie van
Leeuwenhoek, 65, 289-310.
77.Li, T., Quillin, M. L., Phillips, G. N., and
Olson, J. S. (1994) Biochemistry, 33, 1433-1446.
78.Mims, M. P., Porras, A. G., Olson, J. S., Noble,
R. W., and Peterson, J. A. (1983) J. Biol. Chem., 258,
14219-14232.
79.Coletta, M., Ascoli, F., Brunori, M., and
Traylor, T. G. (1986) J. Biol. Chem., 261, 9811-9814.
80.Gibson, Q., and Greenwood, C. (1963) Biochem.
J., 86, 541-555.
81.Cheesman, M. R., Watmough, N. J., Pires, C. A.,
Turner, R., Brittain, T., Gennis, R. B., Greenwood, C., and Thomson, A.
J. (1993) Biochem. J., 289, 709-718.
82.Pettigrew, G. W., and Moore, G. R. (1987)
Cytochromes c. Biological Aspects. Springer Series in
Molecular Biology (Rich, A., ed.) Springer-Verlag, Cambridge,
Massachusetts.
83.Fang, H., Lin, R.-J., and Gennis, R. B. (1989)
J. Biol. Chem., 264, 8026-8032.
84.Spinner, F., Cheesman, M. R., Thomson, A. J.,
Kaysser, T., Gennis, R. B., Peng, Q., and Peterson, J. (1995)
Biochem. J., 308, 641-644.
85.Hill, B. C., Hill, J. J., and Gennis, R. B.
(1994) Biochemistry, 33, 15110-15115.
86.Garcia-Horsman, J. A., Barquera, B., Rumbley, J.,
Ma, J., and Gennis, R. B. (1994) J. Bacteriol., 176,
5587-5600.
87.Sun, J., Osborne, J. P., Kahlow, M. A., Kaysser,
T. M., Gennis, R. B., and Loehr, T. M. (1995) Biochemistry,
34, 12144-12151.
88.Sun, J., Kahlow, M. A., Kaysser, T. M., Osborne,
J. P., Hill, J. J., Rohlfs, R. J., Hille, R., Gennis, R. B., and Loehr,
T. M. (1996) Biochemistry, 35, 2403-2412.
89.Azarkina, N., Borisov, V., and Konstantinov, A.
A. (1997) FEBS Lett., 416, 171-174.