Site-Directed Mutagenesis of Conserved Cysteine Residues in NqrD and NqrE Subunits of Na+-Translocating NADH:quinone Oxidoreductase
M. S. Fadeeva1, Y. V. Bertsova2, M. I. Verkhovsky3, and A. V. Bogachev2*
1Department of Bioinformatics and Bioengineering, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-0338; E-mail: masha@genebee.msu.ru2Department of Molecular Energetics of Microorganisms, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-0338; E-mail: bogachev@genebee.msu.su
3Helsinki Bioenergetics Group, Institute of Biotechnology, P. O. Box 65, (Viikinkaari 1), 00014 University of Helsinki, Helsinki, Finland; fax: (3589) 191-58001; E-mail: michael.verkhovsky@helsinki.fi
* To whom correspondence should be addressed.
Received September 6, 2007
Each of two hydrophobic subunits of Na+-translocating NADH:quinone oxidoreductase (NQR), NqrD and NqrE, contain a pair of strictly conserved cysteine residues within their transmembrane alpha-helices. Site-directed mutagenesis showed that substitutions of these residues in NQR of Vibrio harveyi blocked the Na+-dependent and 2-n-heptyl-4-hydroxyquinoline N-oxide-sensitive quinone reductase activity of the enzyme. However, these mutations did not affect the interaction of NQR with NADH and menadione. It was demonstrated that these conserved cysteine residues are necessary for the correct folding and/or the stability of the NQR complex. Mass and EPR spectroscopy showed that NQR from V. harveyi bears only a 2Fe-2S cluster as a metal-containing prosthetic group.
KEY WORDS: Na+-translocating NADH:quinone oxidoreductase, sodium potential, Vibrio, respiratory chainDOI: 10.1134/S0006297908020028
Abbreviations: Ap) ampicillin; dNADH) reduced nicotinamide hypoxanthine dinucleotide; Em) midpoint redox potential; EPR) electronic paramagnetic resonance; HQNO) 2-n-heptyl-4-hydroxyquinoline N-oxide; K3) menadione; NQR) Na+-translocating NADH:quinone oxidoreductase; Rf) rifampicin; SBP) sub-bacterial particles; Sm) streptomycin.
The Na+-translocating NADH:quinone oxidoreductases (NQRs) are
widely distributed among various bacteria and function to generate a
redox-driven transmembrane electrochemical Na+ potential
across the cytoplasmic membrane [1, 2]. The NQR-type enzymes consist of six subunits
(NqrA-F) encoded by the six genes of the nqr operon [3-5] and contain the following set
of prosthetic groups: one noncovalently bound FAD [1], two covalently bound FMN residues [6-8], one 2Fe-2S cluster [9], and possibly also one tightly bound ubiquinone-8.
Subunit NqrF possesses binding motifs for NADH, FAD, and 2Fe-2S cluster
and mediates the NADH dehydrogenase activity of the enzyme [4, 10, 11].
Two covalently bound FMN residues are attached by phosphoester bonds to
threonine residues in subunits NqrB and NqrC [7, 8].
Using EPR and optical spectroscopy, it was shown that oxidized as well as fully reduced NQR preparations contain flavin radicals with different properties. One of them was observed in the oxidized enzyme and assigned to a neutral flavosemiquinone. The second radical was observed in the reduced enzyme and assigned to the anionic form of flavosemiquinone [9, 12, 13]. These two different flavin radicals are stabilized on different NQR prosthetic groups (most likely on the two covalently bound FMN residues) [12].
The efficiency of Na+ translocation by NQR was shown to be 1 Na+/e- [14], but the mechanism of energy conversion between the redox transitions and the transmembrane translocation of a sodium ion is still unknown for this enzyme. The kinetics of NQR reduction by NADH displays three distinct phases corresponding to reduction of three different flavin species, as shown by optical spectroscopy studies using the stopped-flow approach [9, 12]. This reaction can be dissected into two parts with respect to the coupling site location. The first part (before the coupling site) is the sodium-independent fast reduction of FAD to FADH2 at the NADH dehydrogenating domain of NQR. The second part (after the coupling site) consists of the two slower phases that are strongly dependent on the Na+ concentration. It was shown that the two sodium-dependent phases reflect the reduction of the neutral flavosemiquinone to the fully reduced flavin, and the oxidized flavin to the anionic flavosemiquinone, respectively [12].
The simplest mechanism of redox-electrochemical coupling implies that the reduction of an NQR cofactor should be connected with the capture of the sodium ion from the cytoplasmic side of the membrane. The subsequent oxidation of this cofactor has to be accompanied by the ejection of the cation from the other side of the membrane. If this mechanism is employed by NQR, the Em value of at least one of its prosthetic groups should depend on the Na+ concentration [2]. However, the midpoint potentials of all the redox transitions determined in the enzyme were found to be independent of Na+ concentration [15].
Nevertheless, studying of sodium-binding properties of NQR by the 23Na NMR technique revealed that the dissociation constant of Na+ for the oxidized enzyme is 24 mM and for the reduced enzyme about 30 µM. Thus, reduction of NQR increases the affinity of the enzyme to the coupling ion by about three orders of magnitude [16]. Moreover, redox titration of NQR monitored by changes in linewidth of the 23Na NMR signal at 2 mM Na+ showed that the enzyme affinity to sodium ions follows the Nernst law for a one-electron carrier with Em about -300 mV [16]. These data indicate that the energy conservation by NQR involves a mechanism modulating ion affinity by the redox state of an enzyme redox cofactor, which contradicts the abovementioned results of spectroelectrochemical redox titration of the prosthetic groups of NQR [15].
To explain these observations, we proposed that NQR contains some additional redox prosthetic group having no or negligible absorption in the visible region, but which has the expected sodium dependence of its Em [16]. First of all, such spectral properties could be observed for some metals with transitional valence, which are usually bound to a protein via cysteine residues, and for cysteine residues capable for the formation of a disulfide bond. Therefore, in the present work we tried to identify the conservative cysteine residues in NQR subunits and to reveal their role in the functioning of the enzyme.
MATERIALS AND METHODS
Bacterial strains and growth conditions. The Vibrio harveyi and V. cholerae strains used in this study are listed in Table 1. Bacterial cells were grown at 32°C in FM medium [17]. To isolate sub-bacterial particles (SBP) from uninduced cells of V. cholerae, the different strains of this bacterium were grown for 18 h. For induction of nqr operon the cells were grown to the middle of exponential phase (OD600 = 0.3-0.4), and then the growth medium was supplemented with 0.05% of L-arabinose and the cells were grown in this medium for 3 h. Antibiotics were used at the following concentrations: ampicillin (Ap), 150 µg/ml; rifampicin (Rf), 100 µg/ml; streptomycin (Sm), 50 µg/ml.
Table 1. Bacterial strains, plasmids, and
primers used in this study

*Nucleotides substituted during mutagenesis are designated by
lower case letters and artificially introduced restriction sites are
underlined.
Sub-bacterial particles were isolated from V. cholerae cells as described in [17].
Purification of recombinant His-tagged NQR from V. cholerae cells and purification of NQR from V. harveyi cells were performed as described previously [15].
Various metals in NQR preparations were determined by mass spectrometry. Purified NQR preparations were diluted with medium containing 20 mM KCl, 5 mM Hepes-Tris (pH 7.5), 0.1 mM EDTA, and 0.05% dodecyl maltoside and concentrated by ultrafiltration to protein concentration of 0.32-0.65 mg/ml. To extract metals, HNO3 (final concentration 2%) was added to the concentrated protein samples (experiment) and to the filtrates passed through the ultrafiltration membrane (control for metal content in protein-free reaction medium). The resulting mixtures were centrifuged, and supernatants were used for metal determination using an Agilent 7500C ICP-MS apparatus.
SDS-polyacrylamide gel electrophoresis and detection of covalently bound flavins in NQR were performed according to [6].
EPR spectra of NQR were determined on a Bruker ESP-300 X-band EPR spectrometer at field modulation frequency of 100 kHz. The temperature of the sample was controlled with an ESR 900 liquid helium cryostat with an ITC4 temperature controller (Oxford Instruments, Great Britain).
Site-directed mutagenesis. The plasmid pBADnqr4 bearing the full nqr operon from V. harveyi under control of arabinose promoter [15] was used as the template for mutagenesis. For each mutation, a pair of primers was designed; the sequence of the forward primer from each pair is given in Table 1. Additional restriction sites were built into the three primer pairs to facilitate identification of mutated plasmids by restriction enzyme digestion. Mutagenesis was carried out using the QuikChange II XL kit (Stratagene, USA). The mutations were verified by restriction enzyme digestion or by DNA sequencing. The V. cholerae O395N1 toxT::lacZ DeltanqrA-F cells were then transformed with the plasmids bearing the mutated gene by electroporation.
NADH and dNADH oxidation by SBP were measured using a Hitachi 557 spectrophotometer at 340 nm, 30°C. The reaction medium contained 20 mM Hepes-Tris, 5 mM MgSO4, 100 mM KCl or NaCl, pH 8.0. To measure dNADH:K3 oxidoreductase activity, the reaction medium was supplemented with menadione (50 µM). The extinction coefficient epsilon340 taken for NADH and dNADH was 6.22 mM-1·cm-1.
Protein concentration was determined by means of the bicinchoninic acid method [18] using bovine serum albumin as a standard.
RESULTS AND DISCUSSION
Analysis of amino acid sequences of NQR subunits and proteins homologous to NQR. NqrF subunit of NQR from V. harveyi contains eight cysteine residues. It is well known that four of them (Cys69, Cys75, Cys78, and Cys110) are involved in formation of a 2Fe-2S cluster [11]. Of the other four cysteine residues, only one (Cys377) is strictly conserved. Since this NQR subunit mediates only sodium-independent NADH-dehydrogenase function of the enzyme [10, 11], we did not study the role of this conserved cysteine residue. NqrA, NqrB, and NqrC subunits of NQR from V. harveyi contain five, two, and one cysteine residue, respectively. However, none of them are conserved. However, the NqrD and NqrE subunits contain two completely conserved cysteine residues (Cys29 and Cys112 in NqrD, Cys26 and Cys120 in NqrE for the enzyme from V. harveyi) (Fig. 1). Moreover, these residues are also conserved in all known protein complexes homologous to NQR. As seen in Fig. 1, these residues are present in the RnfE and RnfA subunits of the RNF complex from various bacteria as well as in MA0662 and MA0663 subunits of sodium-dependant ferredoxin:methanophenazine oxidoreductase from the archaeon Methanosarcina acetivorans [19].
According to proposed topological models of the RnfE and RnfA subunits [20] as well as of NqrD and NqrE subunits [21], all four conserved cysteine residues are located within the transmembrane alpha-helices; moreover, the cysteines were predicted to reside in the middle of the membrane. Thus in vivo these residues could be adjacent to each other and be able to bind some metal ion or form a disulfide bond(s).Fig. 1. Sequence alignment of the NqrD and NqrE subunits of NQR. Vh_NqrD and Vh_NqrE - V. harveyi (accession numbers Q9RFV8 and Q9RFV7, respectively), Av_NqrD and Av_NqrE - Azotobacter vinelandii (accession numbers ZP_00416258 and ZP_00416259), Cp_NqrD and Cp_NqrE - Chlamydophila pneumoniae (accession numbers NP_224629 and NP_224630), Tm_NqrD and Tm_NqrE - Thermotoga maritima (accession numbers NP_228061 and NP_228062), Ec_RnfE and Ec_RnfA - Escherichia coli (accession numbers P58344 and P76181), Rc_RnfE and Rc_RnfA - Rhodobacter capsulatus (accession numbers P97055 and Q07396), Ma_0662 and Ma_0663 - Methanosarcina acetivorans (accession numbers NP_615624 and NP_615625). Conservative cysteine residues are marked by asterisks, and proposed transmembrane helices (TM) are underlined (TM positions are shown according to [20, 21]).

Analysis of metal content in NQR from V. harveyi. In the present work, the content of various metal ions in different preparations of NQR from V. harveyi was determined by mass spectrometry. In accordance with the presence of a 2Fe-2S cluster in NQR, different preparations of this enzyme contained 650-780 ng Fe per mg protein, which corresponds to 2.5-3.0 atoms of Fe per NQR complex. All other metals tested (Co, Ni, Cr, Zn, Ti, Cd, Mn, Mo, Ag, W, V, Sc) were present in NQR preparations at very low levels (<=0.05 atom of a metal per NQR complex). The content of copper ions was higher, but also substoichiometric, 0.2-0.3 Cu atom per NQR complex. It is also noteworthy that Cu was the main metal impurity in our reaction medium (see “Materials and Methods” section). This indicates that copper ions detected in NQR are probably a contamination.
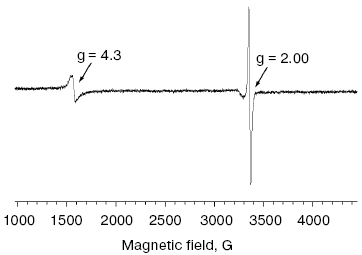
It is well known that the Fe-S cluster of NQR contains two Fe atoms [10, 11], while in the current work we have determined 2.5-3.0 Fe atoms per NQR complex. Thus, theoretically NQR may bear an additional prosthetic group containing one Fe atom (like the Fe-S center of rubredoxin). This center exhibits a characteristic EPR spectrum in the oxidized state with the main rhombic signal at g = 4.3 and also lines at g ~ 9 [22]. As seen in Fig. 2, the EPR spectrum of oxidized NQR along with radical signal at g = 2.00 contains a rhombic signal at g = 4.3 (but not at g = 9, data not shown). However, the spin concentration corresponding the signal at g = 4.3 was significantly lower than the spin concentration calculated from the radical signal (for the spectrum shown in Fig. 2, the ratio of spin concentrations was less than 1 : 10; spin concentration for radical signal was determined at microwave power of 10 µW). It is also noteworthy that this ratio varied in different NQR preparations. These data let us assign the signal at g = 4.3 to a “junk iron” and conclude that NQR bears only the 2Fe-2S cluster as the sole metal-containing prosthetic group.
Site-directed mutagenesis of the conserved cysteine residues in NqrD and NqrE subunits of NQR from V. harveyi. To test the functional role of the four conserved cysteine residues of NqrD and NqrE, these residues were replaced using site-directed mutagenesis. The respiratory chain of marine bacteria of genus Vibrio contains two different NADH:quinone oxidoreductases: NQR and NDH-2 [23, 24]. It is known that various Na+-translocating NADH:quinone oxidoreductases are able to oxidize either NADH or its analog dNADH (reduced nicotinamide hypoxanthine dinucleotide), while noncoupled NADH:quinone oxidoreductases (NDH-2) oxidize only NADH, but not dNADH [24, 25]. Thus, it is possible to use dNADH oxidase and dNADH:K3 oxidoreductase activities of SBP from Vibrio to determine quinone reductase and NADH dehydrogenase activities of NQR, respectively [24].Fig. 2. EPR spectrum of oxidized preparation of wild type NQR. EPR conditions: microwave frequency, 9.409 GHz; microwave power, 2 mW; temperature, 10 K; modulation amplitude, 10 G.
As seen from Table 2, SBP from V. cholerae of wild type catalyzed dNADH:K3 oxidoreductase and dNADH oxidase reactions, and the latter activity was stimulated by sodium ions and highly sensitive to 2-n-heptyl-4-hydroxyquinoline N-oxide (HQNO), a specific inhibitor of NQR. The ratio of dNADH:K3 oxidoreductase and dNADH oxidase activities was close to 1.8 : 1. At the same time, SBP from mutant V. cholerae strain lacking genes of the nqr operon were unable to catalyze dNADH oxidation, while these SBP exhibit a high level of NADH oxidase activity. Transformation of this strain by pBADnqr4 plasmid bearing all six nqr genes from V. harveyi led to partial recovery of the dNADH:K3 oxidoreductase activity as well as the Na+-dependent and HQNO-sensitive dNADH oxidase activity. Since the nqr operon on pBADnqr4 plasmid is under control of the arabinose promoter, addition of L-arabinose to the growth medium led to significant enhancement of these dNADH:K3 oxidoreductase and dNADH oxidase activities. However, the ratio between these two activities in this case became too high (about 9.2 : 1), which indicates that overexpression of nqr operon results in only partial correct folding of the NQR complex.
Table 2. Rate of NADH and dNADH oxidation by
sub-bacterial particles isolated from different V. cholerae
strains
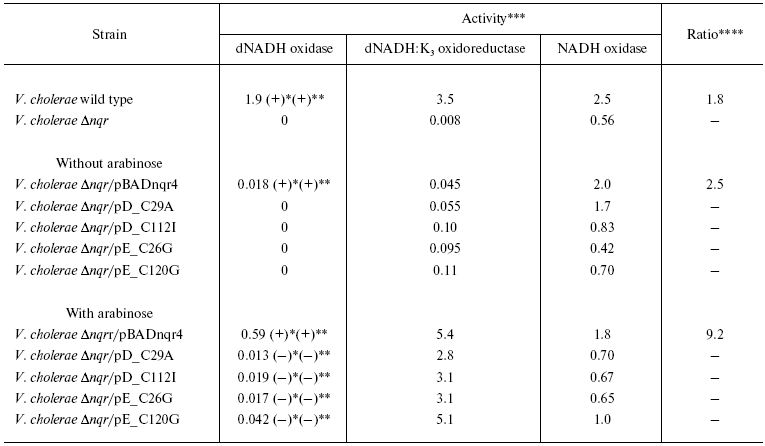
*Stimulation of the activity by sodium ions.
**Sensitivity of the activity to HQNO (4 µM).
***Activities are given in µmol (d)NADH oxidized per
min per mg protein.
****Ratio of dNADH:K3 oxidoreductase to dNADH oxidase
activities.
As seen from Table 2, SBP from Deltanqr strain of V. cholerae transformed by plasmids pD_C29A, pD_C112I, pE_C26G, or pE_C120G (bearing nqr operon of V. harveyi with mutated cysteine residues: Cys29 and Cys112 in NqrD; Cys26 and Cys120 in NqrE, respectively) demonstrated dNADH:K3 oxidoreductase activity, but not dNADH oxidase activity. Growing of these strains in the presence of L-arabinose resulted in significant enhancement of the dNADH:K3 oxidoreductase activity. After induction by L-arabinose only insignificant dNADH oxidase activity could also be observed. Moreover, as seen from Table 2, this dNADH oxidase activity was not stimulated by sodium ions and was not sensitive to HQNO. Thus, the substitutions of the conserved cysteine residues in NqrD and NqrE subunits of NQR from V. harveyi block the Na+-dependant and HQNO-sensitive quinone reductase activity of the enzyme, being without effect on the interaction of NQR with (d)NADH and K3.
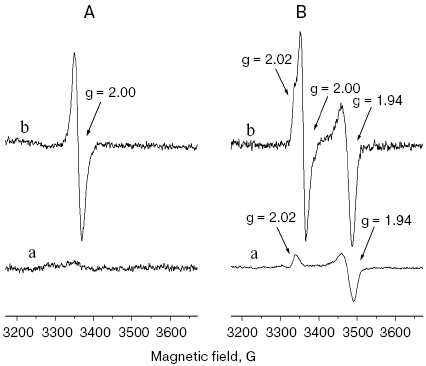
Characterization of purified NQR preparations with mutated conserved cysteine residues in the NqrD and NqrE subunits. Using affinity chromatography, the preparations of NQR of V. harveyi (either wild type or with substitutions of the conserved cysteine residues) were purified from corresponding V. cholerae strains grown in the absence as well as in the presence of L-arabinose. In accordance to earlier results [7, 26], the wild type NQR contained covalently bound flavins. As seen in Fig. 3, EPR spectra of this protein show two radical signals originating from different flavosemiquinones (one in oxidized, the other in reduced preparation) and the signal derived from 2Fe-2S cluster. It is noteworthy that after growth of V. cholerae/pBADnqr4 cells in the presence of arabinose the ratio of spin concentration in signals from 2Fe-2S cluster and the radical in purified NQR was >> 1, i.e. significant excess of Fe-S cluster over flavosemiquinones was observed. These data confirm the conclusion that overexpression of nqr operon results in only partial correct folding of the NQR complex.
After growing of V. cholerae cells bearing pD_C29A, pD_C112I, pE_C26G, or pE_C120G plasmids in the presence of L-arabinose, purified preparations of mutated NQRs contained covalently bound flavins, but their content was considerably smaller with respect to the wild type NQR. These preparations did not show any radical EPR signals either in oxidized or in reduced states but only lines from the 2Fe-2S cluster (Fig. 3). Thus, the substitutions of the conserved cysteine residues in NqrD and NqrE subunits result in inability of covalently bound flavins to stabilize flavosemiquinone states, i.e. lead to incorrect folding of the NQR complex.Fig. 3. EPR spectra of NQR with Cys112Ile substitution in the NqrD subunit (a) as well as wild type NQR (b). A) Oxidized; B) reduced by dithionite. NQR preparations were purified from V. cholerae Deltanqr/pD_C112I (a) and V. cholerae Deltanqr/pBADnqr4 (b) cells grown in the presence of L-arabinose. EPR conditions: microwave frequency, 9.414 GHz; microwave power, 100 µW; temperature, 45 K; modulation amplitude, 6.7 G. The spectra are normalized to protein concentration 5 mg/ml.
NQR preparations with mutated conserved cysteine residues in the NqrD and NqrE subunits purified from uninduced V. cholerae cells (grown in the absence of L-arabinose) did not contain any covalently bound flavins. Moreover, these preparations had lower content of NqrB and NqrC subunits. This means that these mutations also result in significant acceleration of NQR degradation.
Our data indicate that the conserved cysteine residues in NqrD and NqrE subunits are essential for the Na+-dependant NQR activity. However, at present it is impossible to establish whether this is due to only specific structural defects or also the involvement of these residues in the formation of a special redox cofactor. Further studies are needed to clarify the role of the conserved cysteine residues of NQR in electron transport and Na+ translocation.
We thank Dr. C. C. Häse for providing us with V. cholerae strains and Dr. M. A. Bolshov for his assistance with the mass spectrometry experiments.
This work was supported in part by the Russian Foundation for Basic Research (grant 07-04-00619).
REFERENCES
1.Hayashi, M., Nakayama, Y., and Unemoto, T. (2001)
Biochim. Biophys. Acta, 1505, 37-44.
2.Bogachev, A. V., and Verkhovsky, M. I. (2005)
Biochemistry (Moscow), 70, 143-149.
3.Hayashi, M., Hirai, K., and Unemoto, T. (1995)
FEBS Lett., 363, 75-77.
4.Rich, P. R., Meinier, B., and Ward, B. (1995)
FEBS Lett., 375, 5-10.
5.Nakayama, Y., Hayashi, M., and Unemoto, T. (1998)
FEBS Lett., 422, 240-242.
6.Zhou, W., Bertsova, Y. V., Feng, B., Tsatsos, P.,
Verkhovskaya, M. L., Gennis, R. B., Bogachev, A. V., and Barquera, B.
(1999) Biochemistry, 38, 16246-16252.
7.Nakayama, Y., Yasui, M., Sugahara, K., Hayashi, M.,
and Unemoto, T. (2000) FEBS Lett., 474, 165-168.
8.Hayashi, M., Nakayama, Y., Yasui, M., Maeda, M.,
Furuishi, K., and Unemoto, T. (2001) FEBS Lett., 488,
5-8.
9.Bogachev, A. V., Bertsova, Y. V., Barquera, B., and
Verkhovsky, M. I. (2001) Biochemistry, 40, 7318-7323.
10.Turk, K., Puhar, A., Neese, F., Bill, E., Fritz,
G., and Steuber, J. (2004) J. Biol. Chem., 279,
21349-21355.
11.Barquera, B., Nilges, M. J., Morgan, J. E.,
Ramirez-Silva, L., Zhou, W., and Gennis, R. B. (2004)
Biochemistry, 43, 12322-12330.
12.Bogachev, A. V., Bertsova, Y. V., Ruuge, E. K.,
Wikstrom, M., and Verkhovsky, M. I. (2002) Biochim. Biophys.
Acta, 1556, 113-120.
13.Barquera, B., Morgan, J. E., Lukoyanov, D.,
Scholes, C. P., Gennis, R. B., and Nilges, M. J. (2003) J. Am. Chem.
Soc., 125, 265-275.
14.Bogachev, A. V., Murtazina, R. A., and Skulachev,
V. P. (1997) FEBS Lett., 409, 475-477.
15.Bogachev, A. V., Bertsova, Y. V., Bloch, D. A.,
and Verkhovsky, M. I. (2006) Biochemistry, 45,
3421-3428.
16.Bogachev, A. V., Bertsova, Y. V., Aitio, O.,
Permi, P., and Verkhovsky, M. I. (2007) Biochemistry, 46,
10186-10191.
17.Fadeeva, M. S., Yakovtseva, E. A., Belevich, G.
A., Bertsova, Y. V., and Bogachev, A. V. (2007) Arch.
Microbiol., 188, 341-348.
18.Smith, P. K., Krohn, R. I., Hermanson, G. T.,
Mallia, A. K., Gartner, F. H., Provenzano, M. D., Fujimoto, E. K.,
Goeke, N. M., Olson, B. J., and Klenk, D. C. (1985) Analyt.
Biochem., 150, 76-85.
19.Li, Q., Li, L., Rejtar, T., Karger, B. L., and
Ferry, J. G. (2005) J. Proteome Res., 4, 112-128.
20.Saaf, A., Johansson, M., Wallin, E., and Heijne,
G. (1999) Proc. Natl. Acad. Sci. USA, 96, 8540-8544.
21.Duffy, E. B., and Barquera, B. (2006) J.
Bacteriol., 188, 8343-8351.
22.Peisach, J., Blumberg, W. E., Lode, E. T., and
Coon, M. J. (1971) J. Biol. Chem., 246, 5877-5881.
23.Hayashi, M., Miyoshi, T., Sato, M., and Unemoto,
T. (1992) Biochim. Biophys. Acta, 1099, 145-151.
24.Fadeeva, M. S., Nunez, C., Bertsova, Y. V.,
Espin, G., and Bogachev, A. V. (2008) FEMS Microbiol. Lett.,
279, 116-123.
25.Bertsova, Y. V., and Bogachev, A. V. (2004)
FEBS Lett., 563, 207-212.
26.Barquera, B., Hellwig, P., Zhou, W., Morgan, J.
E., Hase, C. C., Gosink, K. K., Nilges, M., Bruesehoff, P. J., Roth,
A., Lancaster, C. R., and Gennis, R. B. (2002) Biochemistry,
41, 3781-3789.