Cytoskeleton Inhibitors Combined with TRAIL Induce Apoptosis in HeLa Carcinoma Cells Overexpressing Antiapoptotic Protein Bcl-2
M. E. Gasparian1, L. V. Domnina2, O. Yu. Ivanova2, D. S. Izyumov2, A. Yu. Lomakin2, E. N. Popova2, A. V. Yagolovich1, O. Yu. Pletjushkina2, D. A. Dolgikh1, and B. V. Chernyak2*
1Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (495) 335-0100; E-mail: office@ibch.ru2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119899 Moscow, Russia; fax: (495) 939-3181; E-mail: bchernyak@yahoo.com
* To whom correspondence should be addressed.
Received September 13, 2007; Revision received November 9, 2007
TRAIL (Apo2L), a cytokine from the family of tumor necrosis factors (TNF), causes apoptosis in various types of tumor cells but is not toxic for normal cells. Recombinant TRAIL obtained using an original method stimulates the release of cytochrome c from mitochondria into the cytoplasm and apoptosis in HeLa carcinoma cells. Expression of oncoprotein Bcl-2 in these cells blocks both processes. The microtubule inhibitors taxol, nocodazole, and colcemid, as well as an inhibitor of actin microfilaments cytochalasin D, enhance the action of TRAIL and allow it to overcome protection caused by overexpression of Bcl-2. This effect is not associated with enhancement of early steps of TRAIL-dependent apoptosis leading to activation of caspase-8 and Bid protein. The inactivation of Bcl-2 also does not define the effect of cytoskeleton inhibitors. It is supposed that destruction of cytoskeleton alters the mechanism of the TRAIL- (or TNF)-dependent cytochrome c release from mitochondria by making it resistant to Bcl-2. The combined use of cytoskeleton inhibitors, which are antitumor drugs, with the recombinant TRAIL preparations may be efficient in therapy of tumors resistant to traditional chemotherapy.
KEY WORDS: apoptosis, cytokines, mitochondria, cytoskeleton, Bcl-2DOI: 10.1134/S000629790803019X
Abbreviations: TFP) trifluoperazine; TNF) tumor necrosis factor; TRAIL) TNF-related apoptosis-inducing ligand.
TRAIL (Apo2L) is a cytokine from the family of tumor necrosis factors
(TNF), and its name is the abbreviation of the words “tumor
necrosis factor-related apoptosis-inducing
ligand” [1]. The extremely high
interest in this protein is due to its high and selective antitumor
activity. Other cytokines of this family (in particular,
TNFalpha and Fas) were first also considered as perspective
antitumor agents, but their high toxicity made their broad use
impossible. Trials of recombinant human TRAIL on cell cultures have
revealed its high ability to cause apoptosis in tumor cells of
different origin and low toxicity towards normal cells [2]. One alarming exception was human hepatocytes, but
later it was shown that the TRAIL toxicity was associated with
artificial protein fragments (his-tags) introduced for easier
purification of recombinant TRAIL [3]. The later
obtained recombinant TRAIL preparation free of artificial fragments was
not toxic either for normal cells in culture and upon introduction into
primates [4]. At the present time it is under
clinical trials.
Investigation of the antitumor effect of TRAIL on cells in culture and on xenographic models of human tumors inoculated into mice has shown that some cell lines exhibited enhanced resistance to TRAIL. In most cases the nature of such resistance was not clear, but it was shown that a number of traditional chemical antitumor drugs (like cisplatin, etoposide, taxol, etc.) make it possible to partially overcome this problem by enhancing the effect of TRAIL (see review [5]). The resistance to TRAIL could be due to the lowered expression of its receptors, to specific alterations of intracellular signaling, or to alterations of the general system of apoptosis. The latter effect can be realized, in particular, owing to the expression of antiapoptotic proteins of the Bcl-2 family. The high level of these proteins is characteristic of many tumor types and correlates with their resistance to chemotherapy (see review [6]). The main mechanism of action of these proteins is the inhibition of cytochrome c (and other apoptosis-stimulating proteins) release from mitochondria into the cytoplasm, which is a necessary step in realization of most kinds of apoptosis (see review [7]).
It is shown in this work that cytoskeleton-damaging agents enhance the effect of TRAIL and enable overcoming the protection caused by Bcl-2 overexpression. The mechanism of this effect is evidently defined not by the inactivation of Bcl-2, but rather by alterations in the process of cytochrome c release from mitochondria into the cytoplasm.
MATERIALS AND METHODS
Human TRAIL (amino acid residues 114-281) gene was synthesized by PCR from 20 overlapping oligonucleotides. The gene was cloned into the pET-32a plasmid containing the thioredoxin gene, with formation of a fused gene. The protein was expressed in Escherichia coli BL21(DE3). Inclusion bodies were dissolved in buffer containing 0.1 M Tris-HCl, pH 8.6, 30 mM dithiothreitol, and 6 M guanidine-HCl. The fused protein was renatured by dialysis against buffer containing 50 mM NaPi (pH 7.5), 300 mM NaCl, and 1 mM dithiothreitol and purified on Ni-NTA agarose. The protein was cleaved by the recombinant light chain of human enteropeptidase [8]. Thioredoxin was removed on Ni-NTA agarose. The obtained TRAIL was stored at 4°C for 12 months without losses in its biological activity. The yield of purified protein was 41 mg per 100 ml of E. coli culture (150 mg of inclusion bodies). The details of the TRAIL preparation and its characteristics are described in [9]. Cells of HeLa and HeLa-Bcl-2 lines were grown in DMEM (Gibco, USA) supplemented with 10% fetal calf serum (HyClone, USA). HeLa-Bcl-2 cells were obtained and characterized in the laboratory of V. I. Agol (Institute of Poliomyelitis, Russian Academy of Medical Sciences) [10]. The apoptosis percent was determined by the number of cells with fragmented nuclei in preparations treated with Hoechst 33322 (1 µg/ml, 30 min) as described previously [11]. At least 500 cells were analyzed for each preparation. Cytochrome c release from mitochondria into the cytoplasm was registered by indirect immunofluorescence. After fixing the cells with methanol (at -20°C), preparations were treated with monoclonal antibodies to cytochrome c (7H8.2C12; BD Biosciences, USA), then with mouse antibodies conjugated with Alexa488 (Axxora, USA). Samples for western blotting were prepared 4 h after cell treatment with apoptosis inducers. Cells were disrupted in 62.5 mM Tris-HCl sample buffer, pH 6.8, containing 2% SDS (w/v), 10% glycerol (w/v), 0.01% bromophenol blue, 5% 2-mercaptoethanol (v/v), and a mixture of protease inhibitors (Sigma, USA). After incubation for 5 min at 95°C, the total protein content was determined in the resulting samples using the RCDC kit (Bio-Rad, USA). Proteins were separated by electrophoresis in 12% SDS-polyacrylamide gel and transferred onto Hybond-P PVDF membranes (Amersham Biosciences, USA). The membranes were treated with polyclonal rabbit antibodies to caspase-8 (BD Biosciences) or Bid (BD Biosciences) or by monoclonal mouse antibodies to tubulin (Sigma), and then by appropriate anti-idiotypic antibodies conjugated with horseradish peroxidase (Sigma). Enzyme activity of horseradish peroxidase was detected by the chemiluminescence technique using an ECL kit (Amersham Biosciences) and X-ray film (Kodak, USA).
RESULTS AND DISCUSSION
The recombinant TRAIL used in this work was obtained by an original method [9]. Structural analysis confirmed the identity of the N-terminal sequence of the protein, its homogeneity, and the high extent of trimer formation (over 90%), which corresponds to the natural form of soluble TRAIL [2]. The TRAIL preparation caused the death of no more than 40% of HeLa cells (Fig. 1a), the half-maximal effect being observed at a concentration of 20 ng/ml, which corresponds to results obtained with different TRAIL preparations [12]. In the presence of the protein synthesis inhibitor emetine, TRAIL caused apoptosis in 90-100% of cells (Fig. 1b). A similar effect is characteristic of the TNF-dependent apoptosis in HeLa cells [11] and is associated with enhanced expression of antiapoptotic proteins due to activation of transcription factor NFkappaB upon binding of TNF and TRAIL to their cellular receptors [13]. As in the case of TNF [11], apoptosis caused by TRAIL was completely inhibited upon overexpression of Bcl-2 in HeLa cells both in the presence and the absence of emetine (Fig. 1).
Cytoskeleton inhibitors cytochalasin D (inhibitor of actin microfilaments), taxol, nocodazole, and colcemid (inhibitors of microtubules) did not cause apoptosis in HeLa cells at least during 24-48 h and to a different extent stimulated apoptosis caused by TRAIL (Fig. 1a). The enhancement of the effect of TRAIL by cytoskeleton inhibitors was also maintained under conditions of protein synthesis inhibition by emetine (Fig. 1b). Thus, the ability of cytoskeleton inhibitors to increase expression of many pro-apoptotic proteins [14] could not play any significant role in this case. In the presence of cytoskeleton inhibitors, TRAIL caused apoptosis in HeLa-Bcl-2 cells, the level of which was comparable with that in the initial HeLa cell line (Fig. 1).Fig. 1. TRAIL-stimulated apoptosis in HeLa (light columns) and HeLa-Bcl-2 (dark columns) cells. Cells were incubated with TRAIL (500 ng/ml) for 24 h (a) or for 8 h in combination with the protein synthesis inhibitor emetine (0.3 µg/ml) (b). Microtubule inhibitors colcemid (0.5 µg/ml) and taxol (10 µM) as well as inhibitor of actin microfilaments cytochalasin D (1 µg/ml) were added simultaneously with TRAIL. Apoptosis was determined by counting cell nuclei with condensed chromatin after staining with Hoechst 33342 (1 µg/ml). Mean values obtained in 3-5 experiments are shown. Designations here and in Figs. 3 and 4: c, control; cyt. D, cytochalasin D; colc., colcemid; tax., taxol.
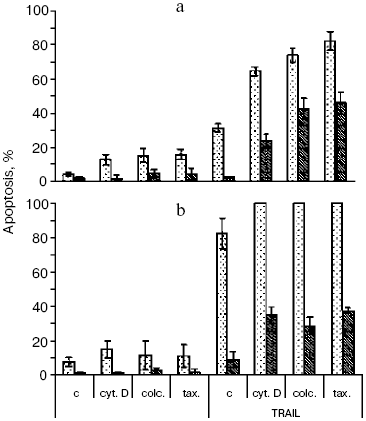
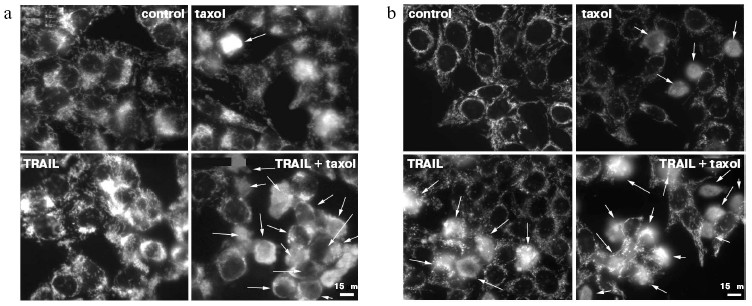
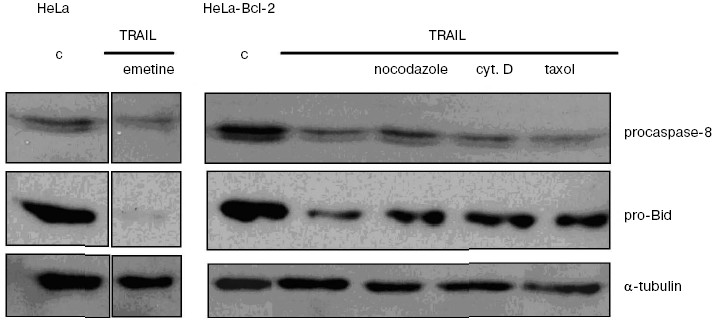
The signal pathway in the TRAIL-dependent apoptosis includes formation of a ligand-receptor complex that incorporates a number of cytoplasmic proteins, caspase-8 being among them. Further, there are proteolytic self-activation of caspase-8, cleavage of Bid protein, its translocation into mitochondria, the release of cytochrome c (and different pro-apoptotic proteins) from mitochondria into the cytoplasm, formation of apoptosomes, in which activation of caspase-9, caspases -3, -6, and -7, responsible for final stages of apoptosis take place [15, 16]. Recording the TRAIL-induced cytochrome c release from mitochondria in HeLa and HeLa-Bcl-2 cells (Fig. 2) showed that Bcl-2 blocks this process. The cytochrome c release was observed only in the case of treatment of HeLa-Bcl-2 cells by TRAIL combined with cytoskeleton inhibitors. The overcoming of the Bcl-2 blocking effect by cytoskeleton inhibitors can be associated with activation of earlier stages of apoptosis, Bcl-2 inactivation, or with alteration of the cytochrome c release mechanism. Besides, caspase-8 is capable of direct processing and activation of caspase-3 that attacks mitochondria and stimulates the Bcl-2-independent release of cytochrome c. However, in the presence of cytoskeleton inhibitors we found neither enhancement of the TRAIL-induced processing of procaspase-8, nor proteolysis of its immediate substrate Bid protein (Fig. 3). These data show that cytoskeleton inhibitors do not activate early steps of the TRAIL-dependent signaling. It seems unlikely that apoptosis in this model is defined by direct caspase-8-dependent activation of caspase-3 by passing over mitochondrial steps. This mechanism is realized in some cell types upon apoptosis caused by TNF [17].
Fig. 2. Cytochrome c release from mitochondria into the cytoplasm of HeLa-Bcl-2 (a) and HeLa (b) cells in the case of TRAIL-dependent apoptosis. Cells were incubated for 24 h with TRAIL (500 ng/ml) and emetine (0.3 µg/ml) and in noted experiments with taxol (10 µM). Cytochrome c was revealed by specific monoclonal antibodies. Arrows point to cells in which cytochrome c was released into the cytoplasm.
An alternative possibility associated with Bcl-2 inactivation was described in some cell models where inhibitors of microtubules (especially taxol) stimulated Bcl-2 phosphorylation [18]. To check this possibility, different methods of apoptosis induction in HeLa cells were investigated. It was shown previously that the line of HeLa-Bcl-2 cells used in this work is resistant to staurosporine (nonselective inhibitor of protein kinases) [11] and to transient exhaustion of ATP [19]. In both cases, protective effect of Bcl-2 was not reduced in the presence of cytoskeleton inhibitors (data not shown), so the inactivation of Bcl-2 seems unlikely.Fig. 3. Caspase-8 and Bid proteolysis induced by TRAIL in HeLa-Bcl-2 cells. HeLa cells were incubated with TRAIL (500 ng/ml) and emetine (0.3 µg/ml) for 4 h. HeLa-Bcl-2 cells were incubated for 1 h with TRAIL (500 ng/ml) in combination with nocodazole (1 µg/ml), cytochalasin D (1 µg/ml), or taxol (10 µM). Procaspase-8, pro-Bid, and tubulin (loading control) were revealed by Western blotting using specific antibodies.
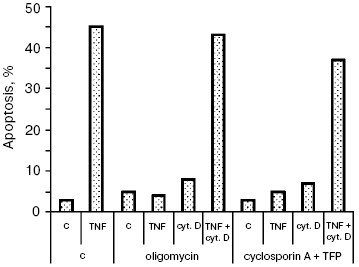
Additional data supporting this conclusion were obtained in experiments on the TNF-dependent apoptosis. We showed previously [11] that the cytochrome c release and apoptosis caused by TNF in HeLa cells were blocked by the mitochondrial ATPase inhibitor oligomycin and by cyclosporin A combined with trifluoperazine (mitochondrial pore inhibitors). The actin microfilament inhibitor (Fig. 4) and those of microtubules (data not shown) made it possible to overcome the TNF-dependent apoptosis block caused by oligomycin or cyclosporin A. So, there is a different (not associated with Bcl-2 inactivation) mechanism of the simulating effect of cytoskeleton inhibitors on the cytochrome c release from mitochondria.
Neither oligomycin nor cyclosporin A combined with trifluoperazine (TFP) inhibits the cytochrome c release and apoptosis caused by TRAIL (data not shown). It seems that the signal pathways of the two cytokines are different, which results in different mechanisms of cytochrome c release from mitochondria. Additional pathways stimulating the cytochrome c release may be activated upon formation of the TRAIL complex with receptors. A convincing argument in favor of this hypothesis is data of experiments on cells with knockout of the bid gene. The TNF-dependent apoptosis was completely blocked in these cells [20], whereas the TRAIL-dependent one was only partially inhibited [21]. For example, it is possible that TNF and TRAIL to different extents activate stress-kinase JNK, able to play both pro- and anti-apoptotic roles [22], or the Bid-related proteins (so-called “BH3-only”) that are able to attack mitochondria by enhancing the effect of each other [23]. Both stress-kinase JNK [24] and Bid-like proteins (such as Bim and Bmf) [25, 26] may be activated upon cytoskeleton damage. It can be supposed that the combination of these factors weakens the Bcl-2 protective effect in the case of simultaneous action of TRAIL (or TNF) and cytoskeleton inhibitors.Fig. 4. Apoptosis of HeLa cells caused by TNF combined with cytochalasin D. Cells were incubated for 8 h with TNF (25 ng/ml) and emetine (0.3 µg/ml). Cytochalasin D was added as in Fig. 1. Oligomycin (5 µg/ml) and cyclosporin A (5 µM) combined with trifluoperazine (TFP) (20 µM) were added simultaneously with TNF and emetine. Apoptosis was determined as in Fig. 1. Inhibitors of microtubules caused a similar effect (data not shown).
Taking into account that taxol and colcemid are included in many present-day schemes of tumor chemotherapy, their combined application with recombinant TRAIL preparations opens broad perspectives for therapy of tumors resistant to traditional therapy.
The authors are grateful to Academicians V. P. Skulachev and M. P. Kirpichnikov for a useful discussion of our results. The authors are also indebted to O. K. Nepryakhina for technical assistance.
This work was supported by the Federal Program “Investigations and elaborations on priority trends in development of scientific-technical complex of Russia” (State Contract 02.522.11.2005) and by the Russian Foundation for Basic Research (grant Nos. 07-04-00335 and 05-04-39009).
REFERENCES
1.Pitti, R. M., Marsters, S. A., Ruppert, S.,
Donahue, C. J., Moore, A., and Ashkenazi, A. (1996) J. Biol.
Chem., 271, 12687-12690.
2.Ashkenazi, A., Pai, R. C., Fong, S., Leung, S.,
Lawrence, D. A., Marsters, S. A., Blackie, C., Chang, L., McMurtrey, A.
E., Hebert, A., DeForge, L., Koumenis, I. L., Lewis, D., Harris, L.,
Bussiere, J., Koeppen, H., Shahrokh, Z., and Schwall, R. H. (1999)
J. Clin. Invest., 104, 155-162.
3.Jo, M., Kim, T. H., Seol, D. W., Esplen, J. E.,
Dorko, K., Billiar, T. R., and Strom, S. C. (2000) Nat. Med.,
6, 564-567.
4.Lawrence, D., Shahrokh, Z., Marsters, S., Achilles,
K., Shih, D., Mounho, B., Hillan, K., Totpal, K., DeForge, L., Schow,
P., Hooley, J., Sherwood, S., Pai, R., Leung, S., Khan, L., Gliniak,
B., Bussiere, J., Smith, C. A., Strom, S. S., Kelley, S., Fox, J. A.,
Thomas, D., and Ashkenazi, A. (2001) Nat. Med., 7,
383-385.
5.Duiker, E. W., Mom, C. H., de Jong, S., Willemse,
P. H., Gietema, J. A., van der Zee, A. G., and de Vries, E. G. (2006)
Eur. J. Cancer, 42, 2233-2240.
6.Coultas, L., and Strasser, A. (2003) Semin.
Cancer Biol., 13, 115-123.
7.Kroemer, G., Galluzzi, L., and Brenner, C. (2007)
Physiol. Rev., 87, 99-163.
8.Gasparian, M. E., Ostapchenko, V. G., Schulga, A.
A., Dolgikh, D. A., and Kirpichnikov, M. P. (2003) Protein. Expr.
Purif., 31, 133-139.
9.Gasparian, M. E., Ostapchenko, V. G., Yagolovich,
A. V., Tsygannik, I. N., Chernyak, B. V., Dolgikh, D. A., and
Kirpichnikov, M. P. (2007) Biotechnol. Lett., 29