
|
REVIEW: Papillomaviruses--to Vaccination and BeyondH. zur HausenDeutsches Krebsforschungszentrum Im Neuenheimer Feld 280, 69120 Heidelberg, Germany; fax: 49-6221-423851; E-mail: zurhausen@dkfz-heidelberg.de |
Received December 19, 2007
High risk human papillomavirus (HPV) types 16 and 18 DNAs were initially identified in 1983-1984. Subsequently the DNA of several other high risk HPV types has been identified. HPV 16 is present in more than 50% of cervical cancer biopsies, and HPV 18 is close to 20%. Some geographic variations exist in the prevalence of HPV high risk types: e.g. HPV 45 is more frequently observed in equatorial Africa, whereas types 58 and 52 have been more often found in East Asia. Molecular as well as epidemiological studies demonstrate that high risk HPV are indeed the causative agents for cervical cancer, they are also involved in other anogenital cancers, and in 25-30% of oropharyngeal carcinomas. Some of the mechanistic aspects are discussed in this review.
KEY WORDS: papillomaviruses, cervical cancer, vaccinationDOI: 10.1134/S0006297908050027
Abbreviations: HPV) human papillomavirus; LSIL, HSIL) low and high grade squamous intraepithelial lesions, respectively; VLP) virus-like particles.
This manuscript is dedicated to Professor Gary I. Abelev for
his pioneering studies opening the field of cancer biomarkers
The identification of high risk human papillomavirus (HPV) types as causative agents of anogenital and oropharyngeal cancers paved the way for the development of preventive vaccines against these widespread sexually transmitted infections. Two vaccines are presently available, both directed against HPV 16 and 18 infections, one of them in addition against the genital wart virus types 6 and 11. Although these vaccines provide very effective protection in previously non-exposed women, they do not seem to possess a significant therapeutic effect in already infected persons. In addition, their price is presently prohibitive for their application in most resource-constrained countries. Thus, more affordable vaccines, preferentially not requiring cold chains and non-invasive application, are desirable in the future. In addition, group-specific immunogenicity, as observed for some antigenic epitopes of the L2 structural protein, will hopefully be further explored in third generation vaccines. The availability of immunogens protecting against HPV 16 and 18 infections marks the establishment of a first vaccine directly developed to protect against a major human cancer.
INFECTIONS AND CANCER--GLOBAL ASPECTS
We can presently estimate that between 20-21% of the global cancer incidence can be linked to infections [1]. This includes not only viral infections (e.g. Epstein-Barr virus, human herpes virus type 8, high risk HPV types, hepatitis B and C viruses, human T-lymphotropic retrovirus), but also bacterial (Helicobacter pylori) and parasitic infections (Schistosoma haematobium, Opisthorchis viverenni, Clonorchis sinensis). Among these infection-linked human cancers, in males H. pylori emerges as the major contributor, accounting for about 47%, mainly due to the global high frequency of gastric cancer. In females, the situation differs substantially, where H. pylori accounts only for 26% of the infection-linked carcinomas. Here HPV infections play by far the most dominant role, contributing to more than 51% of these cancer cases, whereas HPV in males is responsible for slightly more than 4% of infection-linked cancer cases.
There exist also major geographic differences in the incidence of infection-caused human cancers [2]: whereas in Sub-Saharan Africa and in some East Asian regions up to 40% of all cancers are linked to infectious events, in Europe and Northern America less than 10% can presently be linked to infections.
Immunosuppression induced by infections with human immunodeficiency viruses HIV 1 and 2 may result in activation of persisting tumor viruses, like Epstein-Barr virus or human herpes virus type 8. The high prevalence of HIV infections in Sub-Saharan Africa emerges as the trigger for the presently excessively high rate of Kaposi's sarcomas in this region, caused by activated human herpes virus type 8 [3].
PAPILLOMAVIRUSES AND CANCER: HISTORICAL DEVELOPMENTS
The infectious nature of human warts was initially described in 1907 by Ciuffo in Italy [4]. A first link between papillomatous lesions and cancer was reported for a rare hereditary disease, epidermodysplasia verruciformis, in 1922 [5]; the contribution of papillomavirus infections in the causation of these lesions was proven much later [6-8].
Since 1968, cervical cancer was suspected to be linked to human Herpes simplex virus (HSV) type 2 infections (reviewed in [1]). Our own negative attempts to find HSV-2 DNA in cervical cancer biopsies prompted considerations to search for other candidate agents involved in this cancer, whose epidemiology suggested the involvement of a sexually transmitted factor. Thus, we started in 1972 experimental studies to analyze the possible involvement of human papillomavirus in this malignancy. In view of some reports claiming the rare conversion of genital warts into malignant tumors, the virus supposedly present in genital warts appeared to us as the prime candidate. This was hypothesized in several publications [9-11]. In the 1970s, two groups almost simultaneously established the plurality of the HPV family [12, 13] containing by now far more than 100 well characterized members [14]. Between 1980 and 1982, we published the isolation and characterization of two HPV DNAs from genital warts and laryngeal papillomas [15-17]. Although we did not find this DNA in cervical malignancies, using these probes under conditions of low stringency hybridization permitted initial cloning and characterization of HPV 16 and 18 DNA directly from cervical cancer biopsies [18, 19] and from precursor lesions of anogenital cancer [20].
Subsequent studies performed in a number of laboratories demonstrated the DNA of these two types in approximately 70% of all biopsies tested. In addition, a large number of further types found later in some biopsies eventually resulted in the detection of HPV DNA in virtually all cervical cancer biopsies carefully analyzed. Mainly HPV 16, but also HPV 18 and 31, or 33 DNA were also found in other anogenital cancers and in 25-30% of oropharyngeal carcinomas (reviewed in [1]).
MECHANISTIC ASPECTS OF MALIGNANT CONVERSION OF CELLS INFECTED BY
HIGH RISK HPV TYPES
Several infectious agents act as indirect carcinogens either by inducing immunosuppression resulting in the activation of other persisting tumorviruses or by prevention of apoptosis, permitting damaged cells the continuation of proliferation [1]. High risk papillomaviruses represent typical direct carcinogenic factors. Two viral early genes E6 and E7 are not only required for the development of precursor lesions of cervical cancer, but also necessary for maintaining the malignant phenotype of cervical cancer cells (reviewed in [1]). Their silencing in cervical carcinoma cells commonly results in apoptosis or senescence. In the stepwise progression from early infection to invasive cancer, regularly spanning a period of 15-25 years, the individual steps are characterized by an increase in E6/E7 gene activity within infected proliferating cells. E6/E7 expression is most pronounced in carcinomata-in-situ and in invasive cancer.
Progression is accompanied by modifications in genes involved in host cell signaling cascades. The inability to present viral antigenic epitopes at the cell surface is mediated by alterations within the HLA class I pathway and seems to account for the persistence of HPV infections over periods for more than 2 years in close to 10% of infected women, whereas the remaining infections are cleared by immunological interferences within this time period (reviewed in [21]). Two additional mutational events apparently contribute to further steps in the progression: one involves modifications in the regulation of the cyclin-dependent kinase inhibitors p16INK4 [22] and p14ARF [23]. These proteins exert a functional control of viral oncoproteins in normal proliferating cells. The inhibitory effect of p16INK4 for cell proliferation is blocked by E6. This is circumvented by E7 functions, which stimulate directly cyclins E and A. p14ARF negatively interferes with E7 via p53 activation [24]. This is blocked by E6 expression and the resulting degradation of p53. Thus, both viral oncogenes interact synergistically with each other. The loss of this second regulatory cascade by the modification of specific cellular genes seems to correspond clinically to the development of low grade squamous intraepithelial lesions (LSIL) and an “immortalized” growth of such cells under tissue culture conditions.
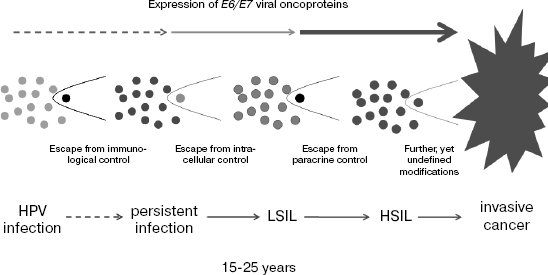
A third transcriptional control is exerted by a paracrine mechanism and obviously results from the excretion of specific cytokines, mainly tumor necrosis factor alpha, from activated macrophages [25, 26]. In HPV infected cells, which are still able to proliferate, this interference mechanism suppresses the majority of viral transcripts. In later stages of progression, the loss of this control is accompanied by a high expression level of viral oncoproteins. Clinically this corresponds to the development of high grade squamous intraepithelial lesions (HSIL), carcinoma in situ, and subsequently, probably by additional genomic modifications, to invasive carcinoma with metastasis formation. A scheme of this mode of progression is shown in Fig. 1. The individual steps are schematically outlined in Fig. 2.
Fig. 1. The development of cervical cancer after primary infection commonly takes between 15 and 25 years. The events are schematically outlined in this figure. After primary infection, individual clones develop escaping from existing extra- and intracellular control mechanisms acting against uninhibited expression of viral oncogenes within proliferating cells. In the course of these developments E6/E7 oncogene expression increases substantially. The viral oncoproteins contribute effectively to chromosomal instability and aneuploidy.
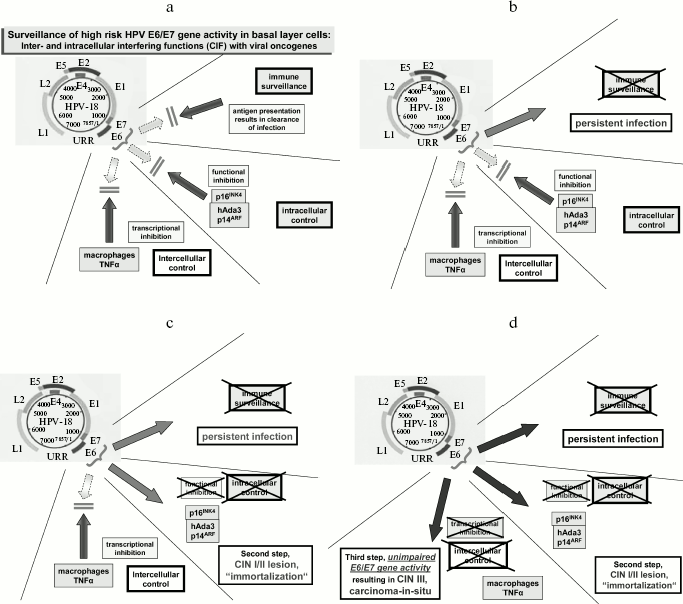
Two key observations in understanding the function of E6 and E7 oncoproteins were made in 1989 [27] and 1990 [28, 29]. The discoveries by Dyson et al. in 1989 of an interaction of E7 with pRb and by Werness et al. and Scheffner et al. in 1990 of E6 interacting and degrading p53 permitted molecular approaches to study functions of viral oncoproteins and to contribute to basic understanding of viral carcinogenesis. In both instances, these interactions result in the degradation of the respective cellular proteins (reviewed in [1]). In particular, the formation of complex between E6 and the E6/E3 ubiquitin ligase has consequences for a number of intracellular pathways: it results in the activation of telomerase, the degradation of several PDZ proteins regulating intracellular pathways, and the activation of cyclin D/cdk4/6 complexes. The latter is counteracted by the high induction of p16INK4 as a consequence of E7/pRb interaction followed by the degradation of pRb. A number of further interactions of viral oncoproteins with host cell components have been described in recent years, which will not be reviewed here.Fig. 2. Schematic representation of control mechanisms blocking viral oncoproteins of viral RNA transcription (a). Viral DNA persistence seems to occur after mutational events within the HLA class I pathway (b). The interference with the pathway blocking the function of viral oncoproteins results in early lesions (c) and seems to correspond in tissue culture to the state of immortalization. The eventual disappearance of a paracrine transcriptional control is correlated by a high rate of E6/E7 oncoprotein expression and the development of high grade lesions (carcinoma in situ) (d). Further molecular events are probably required to result in invasive growth of the lesions.
VACCINATION AGAINST HPV TYPES
Accumulating data on the immunological control of persisting high risk HPV infections raised early hopes for the prevention of cervical cancer by vaccination [30]. The interest of pharmaceutical companies in the production of an HPV vaccine arose only when epidemiological studies were published supporting the existing experimental evidence [31].
Presently available vaccines are based on virus-like particles [32, 33], initially produced by inserting the L1 and L2 open reading frames of HPV 16 into vaccinia virus vector systems, subsequently by exclusively expressing L1 in yeast cells. The expression of the L1 open reading frame, coding for the major capsid protein, results in the formation of empty capsid structures, virus-like particles (VLP). After purification and addition of specific adjuvants, these VLPs are being used for vaccination. Two vaccines are presently available and in clinical use: one contains VLPs of HPV 6, 11, 16, and 18, the other only HPV 16 and 18. Since HPV 16 covers slightly more than 50% of all cervical cancers and HPV 18 close to 20%, it is anticipated that these vaccines should protect against at least 70% of cervical carcinomas and their precursor lesions. Recent data also indicate that there exists a certain cross-protection against the HPV 16-related type HPV 31, and the HPV 18-related type HPV 45, which may bring up the rate of protection to close to 80%.
The available data covering by now more than five years of follow-up are remarkably impressive. Both vaccines appear to be highly efficient in preventing infections by the respective HPV types in previously non-exposed women [34, 35]. The vaccines do not exhibit a significant protective effect in already infected women, thus they are recommended to be applied to girls and young women in age groups between 9 and 25 years.
The vaccines are proposed to be intramuscularly injected in three shots at months 0, 1 or 2, and 6. The antibody conversion occurred in approximately 100% of vaccinated persons. Antibodies usually persisted at high titers for up to 6 years, whereas the titers were commonly low or negative in placebo controls.
Approximately 83% of all cervical cancers occur in resource-constrained countries in Sub-Saharan Africa, Central and South America, and South East Asia. The present high costs of the vaccines, ranging from US $360 in the United States to about 500 Euro in European countries are prohibitive for a global application specifically in those countries that most badly need it.
A number of additional questions still require further exploration. This concerns in particular the age groups, which should be vaccinated. The recommendations vary between individual countries, usually in the range of 9-25 years. It is likely, however, that even persons infected with one or two of the virus types present in the vaccine will profit from the vaccination by acquiring protection against additional types present in the vaccine. In view of the ambiguities of negative PCR or antibody results, it is difficult to come up with a uniform proposal. Clearly, in counties without sufficient screening and gynecological control of women, vaccination should start early prior to the onset of sexual activities.
Two other aspects deserve consideration: should the vaccine also be applied to boys and young adult males, and does vaccination result in a prolongation of intervals for cervical cancer screening?
Vaccination of boys and young adult males is often not considered to be desirable. There exist, however, several reasons to consider this group also in vaccination strategies. Two types of high risk HPV-linked cancers occur even more frequently in males than in females. This accounts for anal and perianal cancers and for 25-30% of oropharyngeal cancers. In addition, genital warts occur at relatively high frequency in both sexes. At least one of the vaccines protects against approximately 90% of these infections. A further aspect is the consideration of gender solidarity, since high risk HPV are transmitted from males to females and vice versa. Although controlled clinical trials after vaccination of boys have not yet been published, it is highly likely that their reactivity will not significantly differ from that of girls.
Successful application of the presently available vaccines should not result in diminished screening of women. The present vaccines do not protect against other high risk types which may still account for 20-30% of cervical cancers. Thus, a follow-up of early cervical lesions and their removal in case of progression will remain mandatory.
Besides these general aspects, some global perspectives of HPV vaccination should be considered. The remaining major problem is the question how can we achieve a global application of the vaccine? This is of course very closely linked to a drastic reduction in the price for the vaccine. But it also involves the mobilization of the political will, necessary regulatory actions, effective delivery, and sufficient financial support. In addition, careful studies are still missing, indicating a possible protective effect of only two injections. There exists the problem of creating a global awareness for the importance of cervical cancer and for sustained information of local physicians, health workers, politicians, school teachers, and company employees. How can we develop suitable management structures for a global application of this vaccine? The involvement of the World Health Organization, the United Nations, and of a large number of NGOs seems to be mandatory. Thus, we are only in the beginning of hopefully solving an important global cancer problem. Cervical cancer still ranks number two in the global cancer incidence of women with close to 500,000 women acquiring this disease annually and approximately one half of them succumbing to this cancer.
FUTURE PERSPECTIVES
Several additional alternatives exist for future generations of vaccines. The foremost question will be the provision of affordable vaccines at a price permitting their application in regions of the developing world. This will require new concepts in vaccine production. Some of them are already presently being explored. This concerns mainly the production of vaccines in bacteria. This commonly does not seem to lead to VLP production, but only in capsomer assembly. Yet, these capsomers are immunogenic and could replace VLP vaccines, albeit probably at higher protein concentrations.
A number of alternative concepts are also under consideration. The N-terminal part of the L2 protein induces a broad range of group-specific immunity, resulting in neutralizing antibodies for a large number of HPV types. This is the basis for anticipating the future availability of a vaccine covering a large spectrum of papillomavirus infections. The present problem for an application exists in the relatively low immunogenicity of the respective L2 antigenic epitopes. Other concepts involve the application of modified “naked” viral DNA, viral RNA vector systems that may turn out to be useful for prophylactic and therapeutic vaccinations, or intranasal application of genetically modified attenuated Salmonella strains expressing HPV proteins. These opportunities have not yet been tested extensively.
One of the more promising concepts is the application of adeno-associated virus (AAV) vector systems carrying the gene for HPV L1 proteins. This system has the advantage of being relatively heat-resistant (AAV tolerate temperatures of up to 60°C), thus it does not require a cold chain. In addition, these preparations may be applied as intranasal sprays, avoiding injections. A first preclinical test revealed the immunogenicity and efficacy of this vaccination after intranasal application in mice [36].
In spite of the very remarkable progress made in the prevention of specific high risk HPV infections, several problems remain to be resolved: they concern the inclusion of other high risk types into the vaccine preparations and hopefully the eradication of all types engaged in cancer of the cervix. Of course, the most important problem to be resolved remains the production of an affordable vaccine for developing parts of the world.
Other problems requiring urgent attention are the immunotherapeutic or chemotherapeutic interference with persisting infections, early and late cervical intraepithelial neoplasias, carcinomas in situ, and invasive cancer. At this moment, it seems that immunotherapy may have a chance in interfering with viral persistence and very early lesions. This remains, however, to be seen in future studies. Targeted chemotherapy is a promising area of research. The three dimensional structure of various viral oncoproteins is now available. This may provide hope for successful future interventions.
There still exist other modes of intervention by small interfering RNAs, where at least in tissue culture experiments shut-off of E7 functions in cervical cancer cells results in apoptosis [37]. In addition, other non-coding RNAs, as exemplified by viral and cellular microRNAs, may fulfill important functions in the positive or negative regulation of viral oncogenes. Some of those may also turn out to be useful for therapeutic interferences. A major question still to be resolved remains the mode of application of RNA-based pharmaceuticals. Hopefully, new concepts will arise along those lines in forthcoming years.
REFERENCES
1.Zur Hausen, H. (2006) Infections Causing Human
Cancer, Wiley-VCH, Weinheim-New York, pp. 1-517.
2.Munoz, N., Bosch, F. X., Castellsague, X., Diaz,
M., de Sanjose, S., Hammouda, D., Shah, K. V., and Meijer, C. J. (2004)
Int. J. Cancer, 111, 278.
3.Parkin, D. M. (2006) Int. J. Cancer,
118, 3030.
4.Ciuffo, G. (1907) Giorn. Ital. Mal.
Venereol., 48, 12.
5.Lewandowsky, F., and Lutz, W. (1922) Arch.
Dermatol. Syph. (Berlin), 141, 193-203.
6.Jablonska, S., and Milewski, B. (1957)
Dermatologica, 115, 1-22.
7.Jablonska, S., Dabrowski, J., and Jakubowicz, K.
(1972) Cancer Res., 32, 583-589.
8.Orth, G., Jablonska, S., Favre, M.,
Jarzabek-Chorzelska, M., and Rzesa, G. (1978) Proc. Natl. Acad. Sci.
USA, 75, 1537-1541.
9.Zur Hausen, H., Meinhof, W., Scheiber, W., and
Bornkamm, G. W. (1974) Int. J. Cancer, 13, 650-656.
10.Zur Hausen, H. (1976) Cancer Res.,
36, 794.
11.Zur Hausen, H. (1977) Curr. Top. Microbiol.
Immunol., 78, 1-30.
12.Gissmann, L., Pfister, H., and zur Hausen, H.
(1977) Virology, 76, 569-580.
13.Orth, G., Favre, M., and Croissant, O. (1977)
J. Virol., 24, 108-120.
14.De Villiers, E.-M., Fauquet, C., Broker, T. R.,
Bernard, H.-U., and zur Hausen, H. (2004) Virology, 324,
17-27.
15.Gissmann, L., and zur Hausen, H. (1980) Int.
J. Cancer, 25, 605-609.
16.De Villiers, E.-M., Gissmann, L., and zur Hausen,
H. (1981) J. Virol., 40, 932-935.
17.Gissmann, L., Diehl, V., Schultz-Coulon, H. J.,
and zur Hausen, H. (1982) J. Virol., 44, 393-400.
18.Durst, M., Gissmann, L., Ikenberg, H., and zur
Hausen, H. (1983) Proc. Natl. Acad. Sci. USA, 80,
3812-3815.
19.Boshart, M., Gissmann, L., Ikenberg, H.,
Kleinheinz, A., Scheurlen, W., and zur Hausen, H. (1984) EMBO
J., 3, 1151-1157.
20.Ikenberg, H., Gissmann, L., Gross, G.,
Grussendorf-Conen, E.-I., and zur Hausen, H. (1983) Int. J.
Cancer, 32, 563-565.
21.Zur Hausen, H. (2002) Nature Rev. Cancer,
2, 342-350.
22.Reznikoff, C. A., Yeager, T. R., Belair, C. D.,
Savelieva, E., Puthenveettil, J. A., and Stadler, W. M. (1996)
Cancer Res., 56, 2886-2890.
23.Sekaric, P., Shamanin, V. A., Luo, J., and
Androphy, E. J. (2007) Oncogene, 26, 6261.
24.Pan, W., Datta, A., Adami, G. R., Raychaudhuri,
P., and Bagchi, S. (2003) Oncogene, 22, 5496-5503.
25.Soto, U., Das, B. C., Lengert, M., Finzer, P.,
zur Hausen, H., and Rosl, F. (1999) Oncogene, 18,
3187-3198.
26.Soto, U., Denk, C., Lengert, M., Finzer, P.,
Hutter, K.-J., zur Hausen, H., and Rosl, F. (2000) Int. J.
Cancer, 86, 811-817.
27.Dyson, N., Howley, P. M., Munger, K., and Harlow,
E. (1989) Science, 243, 934-937.
28.Werness, B. A., Levine, A. J., and Howley, P. M.
(1990) Science, 248, 76-79.
29.Scheffner, M., Werness, B. A., Huibregtse, J. M.,
Levine, A. J., and Howley, P. M. (1990) Cell, 63,
1129-1136.
30.Zur Hausen, H. (1986) in Vaccine Intervention
against Virus-Induced Tumours (Goldman, J. M., and Epstein, M. A.,
eds.) MacMillan Press, pp. 63-80.
31.Munoz, N., Bosch, F. X., de Sanjose, S., Tafur,
L., Izarzugaza, I., Gili, M., Viladiu, P., Navarro, C., Martos, C.,
Asunce, N., et al. (1992) Int. J. Cancer, 52,
743-749.
32.Zhou, J., Sun, X. Y., Stenzel, D. J., and Frazer,
I. H. (1991) Virology, 185, 251-257.
33.Kirnbauer, R., Booy, F., Cheng, N., Lowy, D. R.,
and Schiller, J. T. (1992) Proc. Natl. Acad. Sci. USA,
89, 12180-12184.
34.Harper, D. M., Franco, E. L., Wheeler, C. M.,
Ferris, D. G., Jenkins, D., Schuind, A., Zahaf, T., Innis, B., Naud,
P., de Carvalho, N. S., Roteli-Martins, C. M., Teixeira, J., Blatter,
M. M., Korn, A. P., Quint, W., and Dubin, G. (2004) Lancet,
364, 1757-1765.
35.Villa, L. L., Costa, R. L., Petta, C. A.,
Andrade, R. P., Ault, K. A., Giuliano, A. R., Wheeler, C. M., Koutsky,
L. A., Malm, C., Lehtinen, M., Skjeldestad, F. E., Olsson, S. E.,
Steinwall, M., Brown, D. R., Kurman, R. J., Ronnett, B. M., Stoler, M.
H., Ferenczy, A., Harper, D. M., Tamms, G. M., Yu. J., Lupinacci, L.,
Railkar, R., Taddeo, F. J., Jansen, K. U., Esser, M. T., Sings, H. L.,
Saah, A. J., and Barr, E. (2005) Lancet Oncol., 6,
271-278.
36.Kuck, D., Lau, T., Leuchs, B., Kern, A., Muller,
M., Gissmann, L., and Kleinschmidt, J. A. (2006) J. Virol.,
80, 2621-2630.
37.Sima, N., Wang, W., Kong, D., Deng, D., Xu, Q.,
Zhou, J., Xu, G., Meng, L., Lu, Y., Wang, S., and Ma, D. (2007)
Apoptosis, 13, 273-281.