
|
REVIEW: Small RNAs and CancerogenesisS. S. Ryazansky1 and V. A. Gvozdev1,2*1Institute of Molecular Genetics, Russian Academy of Sciences, 123182 Moscow, Russia; fax: (499) 196-0221; E-mail: ryazansky@img.ras.ru; gvozdev@img.ras.ru2Lomonosov Moscow State University, 119991 Moscow, Russia * To whom correspondence should be addressed. |
Received December 18, 2007; Revision received January 30, 2008
Disturbances of microRNA generation and functioning as inhibitors of gene expression at the translational level are considered as specific and diagnostic features of cancer. This review also highlights the role of short interfering RNA (siRNA) in modified epigenomic chromatin structure, which may cause cancer transformation. Future directions of cancer epigenomics are considered in the light of the involvement of siRNA in epigenomic modification of chromatin.
KEY WORDS: miRNA, siRNA, cancerogenesis, epigenomeDOI: 10.1134/S000629790050040
During recent years, new mechanisms of regulation of gene expression have been discovered. These mechanisms are based on complementary interaction of short RNA (of 20-30 nucleotides in length) with their mRNA targets or nascent transcripts within chromatin [1-3]. siRNA-mRNA recognition results in suppression of translation, and the interaction of siRNA with nascent transcript may be accompanied by heterochromatinization and chromatin silencing. Are impairments in these processes related to the appearance of tumors? The large experimental material summarized in recently published reviews demonstrates how impairments in formation and functioning of microRNA (miRNA) suppressing gene expression at the level of translation may cause malignization [4-6]. A possible role of short RNA in epigenomic modification of genes (and impairments in this regulation, which is now considered as one of the reasons underlying the development of cancer) remains less studied. The goal of numerous studies consists in investigation of epigenomic modifications that change gene expression and are inherited in subsequent cell generation, but are not accompanied by changes in DNA nucleotide sequences [7-9]. Taking into consideration results of studies on mammalian and other model objects, we consider here the possible effect of short RNA-dependent epigenomic modification on the development of cancer. Discussion of this topic may help to foresee future directions in studies of the epigenomics of cancer.
The discovery of the phenomenon known as RNA interference (RNAi) has demonstrated, that exogenously administered or artificially expressed double stranded RNAs (dsRNA) selectively inhibit expression of target genes with homologous nucleotide sequence [10]. This effect is associated with formation of siRNA (short interfering RNA) of 21-23 nucleotides in length. The role of miRNA (having the same size) in regulation of nematode development has been elucidated slight earlier; one type of miRNA has been shown to be highly conservative and functionally active in various eukaryotes [11]. The mechanisms of miRNA biogenesis employ the same biochemical reactions, which are also responsible for siRNA formation.
Results of these studies have been summarized in several reviews [12-14] and are shown at Fig. 1. The miRNA genes are transcribed by RNA-polymerase II followed by formation of primary transcript of ~1 kb. Subsequent excision of the miRNA precursor (pre-miRNA) of 60-90 nucleotides, representing imperfect hairpin with overhang dinucleotide at 3´ end involves endoribonuclease RNase III (Drosha) and RNA-binding protein DGCR8. Pre-miRNA is transported into the cytoplasm by a Exp5/Ran-GTP complex; in the cytoplasm RNase III Dicer functioning with other RNA-binding proteins cleaves a terminal loop. The resulting double-stranded intermediate with overhanging 3´_ends is then untwisted, and one of these strands (mature miRNA) becomes a component of the multiprotein effector RISC (RNA-induced silencing complex). The protein Argonaute is a key component of RISC; it recognizes short RNA and exhibits potential endonuclease activity. When miRNA and mRNA are fully complementary, the latter undergoes endonuclease cleavage; in the case of incomplete complementarity between miRNA and mRNA, translation process is blocked. The siRNA is formed from endogenous or artificially administered dsRNA, which undergoes processing by Dicer endonuclease. As in the case of miRNA, RISC contains only single stranded siRNA. Suppression of gene expression at the level of translation is determined by siRNA binding with complementary sequence in mRNA. The binding of siRNA to complementary sites of nascent transcript can also induce chromatin compactization (Fig. 1).
Similarity in processing of miRNA and siRNA suggest that functional pathways of these short RNAs may intercross because, in contrast to genomes of other organisms (Drosophila, plants), the human genome contains a single gene encoding Dicer. The viewpoints that delivery of miRNA and siRNA to their targets is strictly determined by one of the corresponding variants of the Argonaute protein have very recently been corrected. Studies on Drosophila have shown that these two classes of short RNA may compete for the binding at the same effector complex containing the same variant of the Argonaute protein; this suggests lack of “division of labor” between siRNA and miRNA [15]. Diversity of miRNA in mammals is determined by hundreds of corresponding genes, whereas the origin of endogenous dsRNA and siRNA in mammalian cells still requires detailed investigation. Probably sources for their formation may be associated not only with repeated elements of the genome (both strands may be templates during transcription) but also with other non-protein-coding transcripts, which overlap by nucleotide sequence and are complementary to each other [16]. Recently, another type of short RNA known as piRNA, differing from miRNA and siRNA by length (26-30 nucleotides) and methylation of 3´-terminal nucleotide at its 2´-OH-group, has been found in Drosophila and vertebrates (including mammals) [17]. The functioning of piRNA is associated with the development and differentiation of germinal tissues.Fig. 1. a) Mechanisms of formation and action of miRNAs and siRNAs (in mammals as an example). Ovals show effector complex containing the Argonaute protein. b) Nucleotide sequences of two human pre-miRNAs and mature miRNAs (marked in bold).
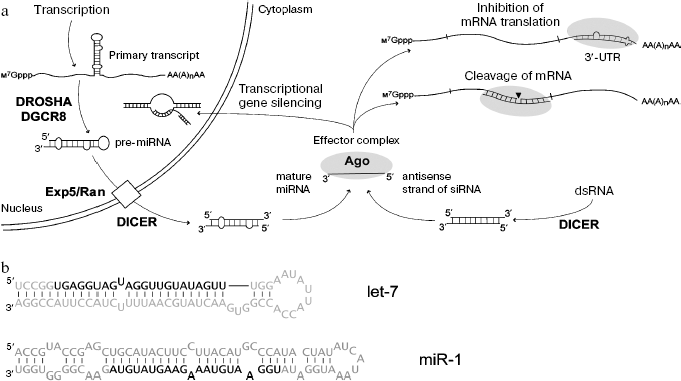
microRNAs
miRNAs are a large new class of regulatory RNAs, which are involved in control of expression of many genes. More than 5000 miRNAs are known in 58 species; these include miRNAs encoded by viral genomes (miRBase, Release 10.0, http://microrna.sanger.ac.uk). Obviously, mammalian miRNAs (~500 miRNAs in humans) regulate gene expression only at the translational level, but in plants miRNAs can induce gene repression at the chromatin level (this is accompanied by DNA methylation) [18]. Usually miRNA is complementary to 3´-untranslated region (UTR) of mRNA (Fig. 1). Each mRNA can be regulated by several miRNAs, and one miRNA can recognize several targets [11, 13, 14]. Rules have been developed for predicting mRNA-target recognition and so identification of mRNA targets employs computer programs that predict targets with high probability. However, prediction of target candidates requires experimental validation. It is believed that expression of 30% of human genes may be regulated by miRNA [19].
The mechanisms responsible for the effect of miRNA in animal cells still represent a subject for hot discussions [20]. Inhibition may occur at the stage of translation initiation [21-25], and this requires the presence of mRNA cap (m7Gppp-modification) [23-25]. However, suppression of translation may also occur during elongation [26-29]. Proteins of the Ago subfamily, the key components of the effector complex, contain an obligate cap-recognizing motif found in the initiation translation factor eIF4E [30]. In Drosophila, inhibition involves factor eIF6 blocking assembly of 80S ribosomes [21]. A marked fraction of repressed mRNA was found in the processing bodies (P-bodies) representing perinuclear cytoplasmic aggregates of RNA-protein complexes [31-36]. The mRNAs (including miRNA targets) accumulated in P-bodies either undergo degradation by means of decapping and deadenylating enzymes, and also RNA exonucleases or leave these bodies and are employed in translation [37-39].
It is possible that miRNAs form a regulatory network controlling many fundamental biological process: tissue differentiation [40-42], development [43, 44], metabolism [45], apoptosis [46], cell cycle [47-49]. Results of recent studies suggest involvement of miRNA in coordination of the gene expression program determining tumor metastases [50]. So it is not surprisingly that defects in miRNA expression result in appearance of cancer, and miRNA genes may act as oncogenes or tumor suppressors. Involvement of miRNA in cancerogenesis caused the development of the notion about oncomirs, i.e. miRNAs (miRs) whose impaired expression promotes oncoformation [6].
miRNAs AS KEY PLAYERS IN CANCEROGENESIS
Mammalian miRNAs. The expression profile of miRNA is highly specific for a particular type of tissue and stage of cell differentiation [11, 13, 14]. Impaired miRNA functioning, which occurs during tumor transformation, can be evaluated as a consequence rather than the cause of loss of cell identity. However, detection of deletions, local amplifications, and chromosomal breakpoints in the regions of miRNA genes (causing impairments in miRNA expression during cancerogenesis) is a good demonstration of direct role of miRNA in these processes. More than half of 186 predicted or known (at that time) miRNA genes were located inside or near regions of tumor-associated chromosomal rearrangements [51]. There is a specific change in expression of only miRNAs encoded by genes located in the regions of known chromosomal breakpoints typical for a particular type of cancer [52, 53].
Defects in miRNA expression cause the development of cancer associated with impaired formation of oncoproteins or tumor suppressors regulated by these miRNAs (Fig. 2). For example, miR-15a and miR-16-11 (the first miRNAs with documented role in cancerogenesis) suppressed expression of the inhibitor of apoptosis, Bcl-2 [54-56]. Deletion of the region 13q14.1 encoding these miRNAs was partially associated with B-cell chronic lymphocytic leukemia (B-CLL), brain cortex cell lymphoma, and multiple myeloma [5]. Expression of miR-15a and miR-16-1 decreased in 68% of B-CLL cases [56]; this was accompanied by Bcl-2 overexpression [57].
[1Footnote: The name of miRNA contains an index number reflecting the order of its discovery and number or letter suffix, which usually discriminates miRNAs from the same family, which have the same index number; the latter consists of miRNAs with identical or very similar nucleotide sequences [54]. Some miRNAs (let-7, etc.) still have trivial names.]
One of the first identified miRNAs, let-7 found in the nematode Caenorhabditis elegans as well as in humans, is also a tumor suppressor decreasing formation of the oncoproteins Ras and HMGA2. In patients with lung cancer, there was inverse correlation between expression of let-7 and Ras/HMGA2 [58, 59]. Overexpression of let-7 suppressed growth of lung cancer cell culture [59]. Involvement of conservative miRNA let-7 in regulation of ras expression was also found in nematodes [58, 60, 61]. Human lung cancer is often associated with translocation of the first three exons of hmga2 followed by subsequent fusion with another gene [62]. It is suggested that during translocation, the hmga2 gene loses 3´-UTR with let-7 binding sites; this results in overexpression of oncoprotein and tumor transformation [63]. It is possible that HMGA2 and Ras proteins regulated by let-7 cooperate during tumor transformation of lung cells.Fig. 2. Participation of miRNAs in regulation of some key components of the cell cycle and apoptosis. Putative interactions are shown by dashed lines.
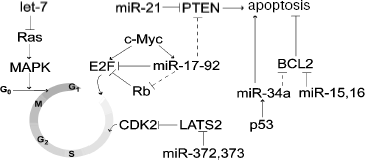
Under conditions of stress, known tumor suppressor p53 activated transcription of several miRNAs and gene miR-34a exhibited the highest activation [64-68]. Overexpression of miR-34a was characterized by arrest of the cell cycle and apoptosis accompanied by the decrease in expression of many genes responsible for cell proliferation and angiogenesis. Most of these genes were predicted as targets of miR-34a. These included the apoptosis inhibitor Bcl-2 [65, 67, 69] (Fig. 2). Inhibition of miR-34a by antisense LNA (locked nucleic acid) oligonucleotides strongly attenuated p53-dependent apoptosis during DNA damage [67]. The region 1p36 of chromosome 1 containing gene mir-34a is often deleted in many types of cancer, for example, in neuroblastoma [70]. These observations demonstrate that miR-34 and possibly other miRNAs are involved in p53-dependent cell response to stress. Thus, miR-34a may be considered as a tumor suppressor, which plays (together with p53) an important role in regulation of cell proliferation and apoptosis.
miR-155 is an example of miRNA exhibiting oncogenic properties; this miRNA is required for functioning of B- and T-cells [71-73]. Increased expression of miR-155 is observed in pediatric Burkitt's lymphoma, classic Hodgkin's disease, diffuse large cell lymphoma, B-CLL, and also in lung, breast, and pancreatic cancers [74-80]. Overexpression of miR-155 observed in B-CLL obviously occurs due to impairments in regulation of expression, and it is not associated with amplification of a corresponding genome locus [80]. Bioinformatic analysis predicts that cytokines, chemokines, and transcription factors are possible miR-155 targets [72]. In pancreatic cancer cells, this miRNA suppresses the activator of apoptosis TP53INP1 [79]. Immature B-cells from transgenic mice overexpressing miR-155 are characterized by induction of early polyclonal malignization [81].
Artificial expression of miR-372 and miR-373 induced proliferation and malignization of human primary cell culture overexpressing ras oncogene [82]. The target for both miRNAs is LATS2, a cyclin-dependent kinase (CDK2) [83]. Suppression of LATS2 by miR-372 and miR-373 activated CDK2 and the cell cycle (Fig. 2) [82].
We have considered possible functioning of various miRNA genes as tumor suppressors and also as oncogenes. Genes encoding miRNAs with oncogenic and suppressor properties may be located within one cluster. The cluster of miRNA genes mir-17-92, located in the region 13q31-32, includes seven miRNA genes (mir-17-5p, -17-3p, -18, -19a, -20a, -19b-1, and -92-1). It is transcribed with formation of a single transcript, which is a precursor of all encoded miRNAs. This cluster generally exhibits oncogenic properties. Frequent amplification of the region 13q31-32 has been recognized in B-cell lymphomas, follicular lymphomas, brain cortex lymphomas, and some other tumors [84]. Induction of transformation of mouse hemopoietic stem cells into B-cell lymphoma caused by human transgenic oncogene c-Myc accelerated by additional transfection of overexpressing truncated cluster fragment lacking the mir-92-1 gene [85]. Expression of this cluster also increased the rate of proliferation of lung cancer cell culture [86]. The predicted targets for some cluster miRNA include tumor suppressors: PTEN inducing apoptosis [87] and one protein of the Rb family (Rb12) inhibiting E2F [88] (Fig. 2). Interestingly, proto-oncogene c-Myc activating transcription of E2F1 can also activate this cluster (Fig. 2) [89]. However, some miRNA can act as tumor suppressors. miR-17-5p and miR-20a of this cluster can suppress translation of E2F1 mRNA [89]. In the same time, it is known that high level of E2F1 expression results in apoptosis [90]. In this case, negative regulation by miR-17-5p and miR-20a will exhibit oncogenic effect. These observations demonstrate complex regulatory effects of the miRNA cluster; they combine both oncogenic effects (determined by suppression of tumor suppressors) and some anti-oncogenic effects determined by complex modulation of E2F1 expression.
In some tumor cells miR-21 and miR-24 act as oncogenes, and in others they act as tumor suppressors. In HeLa cells, inhibition of miR-21 or miR-24 activity by modified anti-miR oligonucleotides accelerated proliferation [47]. Inhibition of miR-24 in A549 cells caused potent inhibition of cell growth, whereas inhibition of miR-21 had no influence on cell growth. Some tumors (hepatomas, glioblastoma, pancreatic or breast cancer) are accompanied by high expression of mir-21 [91-93]. Suppression of miR-21 in glioblastoma cell culture activates caspases in apoptosis [93], whereas in hepatomas the effect of miR-21 is probably determined by suppression of tumor suppressor PTEN [92] (Fig. 2).
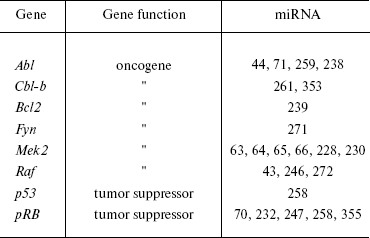
In general, analysis of bioinformatic predictions of putative targets suggests that proto- and anti-oncogenic activity is typical for many other miRNAs (Table 1). However, the proportion of false positive predictions of such targets is very high and may reach 30% [94]. Clearly, these predictions require experimental validation.
Table 1. Oncogenes and tumor suppressors as
potential targets for miRNA (modified from [202])
Defects in miRNA expression associated with carcinogens are possible not only during chromosomal rearrangements, but also due to impairments in the machinery responsible for miRNA formation and processing. For example, inhibition of expression of ribonucleases Dicer and Drosha by complementary siRNA caused acceleration of growth of lung adenocarcinoma cells of naked mice [95]. Expression of miR-143 and miR-145 strongly decreased in colon and breast cancers [96, 97], possibly due to blockade of processing of corresponding pre-miRNA [97].
Tumor transformation may be also determined by primary impairments in regulation of expression of a single miRNA, which is then accompanied by imbalance in the whole miRNA network. It is suggested that under normal conditions the regulatory network of miRNA suppresses expression of “unwanted” (for a particular tissue) genes and provides “canalization” for the development of particular cell types via a strictly determined pathway [98, 99]. Damage to this network induced, for example, by defect in expression of single but key miRNA may result in “de-canalization” of this cell followed by its de-differentiation and reduced resistance to various oncogenic stimuli.
Viral miRNAs. Genes encoding miRNAs have been found not only in eukaryotes, but also in DNA viruses preferentially related to herpesviruses. Functions of several miRNAs are known to date; these include suppression of expression of both viral and cell genes, which is required for optimal production of viral proteins [100]. Most of these viruses are associated with tumors [100, 101]. Epstein-Barr virus (EBV) encodes 23 miRNAs and can induce the development of B- and T-cell lymphomas and nasopharyngeal carcinomas. KSHV virus encoding 12 miRNAs is associated with Kaposi's sarcoma and some aggressive lymphomas. Polyoma virus SV40 (1 miRNA) induces tumors in newborn hamsters and mice with attenuated immunity. There is indirect evidence that cancerogenesis may be associated with viral miRNAs. For example, bioinformatic prediction has shown that proteins p53 and Bcl-2 are potential targets for two EBV miRNAs [101]. KSHV miR-K12-11 is the ortholog of cellular miR-155 [102] exhibiting oncogenic properties. Viruses also can alter cell growth by influencing formation or stability of host cell miRNA. For example, cells infected with HIV-1 markedly change the level of expression of ~40% of cell miRNAs [103]. More detailed discussion of the interrelationship between viruses, miRNA, and tumor cells can be found in [101].
miRNAs IN TUMOR DIAGNOSTICS
Impairments in miRNA functioning seen in cancerogenesis can be used for determination of miRNA expression for diagnostics of tumor origin. Each type of cancer is characterized by a certain profile of miRNA expression (Table 2). Analysis of expression of more than 200 miRNAs in more than 1000 samples (from three studies) of lymphomas and solid cancers resulted in successful classification of tumors into subtypes by their origin and stage of differentiation [91, 104, 105]. For example, cluster analysis of miRNA expression profiles in tumors accurately determines not only type of the tissue (e.g. epithelium or hemopoietic system), but also discriminates tumors within the same type of tissue; this may reflect mechanism of transformation [104]. The miRNA profile can be evaluated by means of microarrays [91]. It has been calculated that accuracy of tumor classification by means of miRNA profiles is higher than that employing mRNA profiles [104]. Obviously, evaluation of miRNA profiles can be used for prognosis of the development of tumors [59, 78, 106]. Such an approach for tumor diagnostics is very promising. However, it is not widely employed yet due to not-well developed technology, lack of standards, requirements of very high purity of RNA samples, and not always reproducible results.
Table 2. miRNA expression increased (↑) or
decreased (↓) in some of the most common tumors compared with normal
tissues (modified from [203])

EPIGENOMIC MODIFICATIONS AND THE CANCER EPIGENOME
The importance of epigenomic modifications of chromatin structure for the development of tumors has already been recognized [107-109]. We will cite here the results of studies of epigenomic modifications in cancer, which will be necessary for subsequent discussion of a possible role of short RNAs in elucidation of these modifications2. Heterogeneity of tumor cells is significantly influenced by variations in epigenomic structure of chromatin (epigenome) in tumor precursor cells. The epigenome undergoes significant rearrangements during the development of cancer [110]. Tumor progression is determined not only by genetic changes but also by impairments in epigenomic characteristics, which appear more often than true mutations [111]. Chromatin contains inherited epigenetic information determining important characteristics of the epigenome. The epigenomic modifications include: DNA methylation, post-transnational modification of histones, and substitution of canonic variants of histones for others differing in amino acid sequence.
[2Footnote: Most of the references are recent reviews rather than particular studies.]
Methylation of cytosine in DNA followed by formation of 5-methylcytosine occurs in the dinucleotide sequences CpG [8]; most of them are methylated, except those which constitute CpG islands, the CpG clusters located in most promoters of human genes. Methylation can cause suppression of transcription due to “attraction” of proteins recognizing methylated CpG. Such proteins cause gene repression via competition with other regulatory proteins [112] or due to binding of histone deacetylases removing acetyl groups from lysine residues, the typical markers of active chromatin [113]. Methylation of DNA may also change parameters of nucleosome structure of chromatin, which depends on functioning of various remodeling complexes [114-116]. The remodeling complexes can remove nucleosomes, change their location on DNA, and regulate the presence of histone variants differing by amino acid sequence in a nucleosome. Functions of the remodeling complexes can depend on the presence of proteins recognizing methylated DNA. Nucleosome DNA sites are either protected against interaction with regulatory complexes of non-histone regulatory proteins or may promote such interactions. Remodeling can result in activation of repression of gene activity, thus determining gene expression in the development and specificity of cell differentiation. Three processes (DNA methylation, histone modification, and remodeling of nucleosome structure of chromatin) are mutually coupled and so mutations in genes encoding proteins, direct participants of these processes, can be a reason for the development of various types of cancer.
DNA methylation in the presence of DNA methyltransferase Dnmt1 may be passively maintained during DNA replication for preservation of this type of epigenetic information in cell generations. However, methylation of the mammalian genome can be changed during development [117, 118]. After fertilization but before fusion of pronuclei, the father genome undergoes active demethylation. Subsequently, during cell divisions in the absence of active DNA methyltransferase, the maternal genome undergoes passive demethylation. Genome methylation de novo begins at the stage of the blastocyst. “Methylation waves” also occur during gastrulation; this promotes tissue specific methylation. The pattern of DNA methylation (“methylome”) is subjected to changes in cancer, and the mode of these changes is specific for various tumors [119]. Although there is general decrease of methylation, some CpG islands are characterized by local aberrant excessive methylation typical for the development of cancer and leading to gene repression [109, 120]. Such excessive methylation results in suppression of activity of tumor suppressor genes or may be accompanied by mutations in these genes, because 5-methylcytosine is a mutagenic base, which can be converted into thymine during deamination [109, 121]. Suppression of tumor suppressors caused by methylation is the most important epigenomic impairment leading to growth of tumor cells. One type of tumor can be characterized by hundreds of genes with aberrantly methylated promoters. Recently, excessive methylation of CpG islands of miRNA genes has been found in cancer cells [122, 123]. The authors attribute the effect of this methylation to suppression of tumor suppressor genes represented by miRNA genes. Some types of cancer are accompanied by methylation of extended parts of the genome (about 106 DNA base pairs) [124].
Besides gene specific methylation, which can also occur in the coding region of a gene, there is so-called global decrease of genome methylation, which occurs in tumor cells (especially in metastasizing cells) [125, 126]. This mainly reflects changes in methylation of CpG in non-coding sequences of the mammalian genome. Under normal conditions satellite DNA sequences located within the centromeric heterochromatin are strongly methylated. Global decrease of methylation involving these regions has been described for human glioblastoma [127]. Insufficient DNA methylation in the pericentromeric region of human chromosomes can induce their instability due to decondensation of heterochromatin and increased recombination in these regions [117]. Chromosome rearrangements caused by hypomethylation have been described in several studies [128, 129]. Impairments in the structure of centromere regions cause chromosome nondisjunction and aneuploidy typical for tumor cells. These processes have been shown to correlate with the development of urinary bladder carcinomas (e.g. by inducing loss of chromosome 9) [130].
Processes of DNA methylation may be closely associated with modification of chromatin histones. Several recent reviews highlight the role of histone modification in gene expression [131, 132]. The mechanism underlying dependence of histone modification on DNA methylation is based on complex formation between proteins forming epigenome characteristics: DNA methyltransferases; proteins recognizing methylated sites; histone acetylases and histone methyltransferases [107, 110, 133, 134]. DNA methyltransferase can bind not only histone methyltransferase, but also histone deacetylase and the heterochromatin protein HP1. Histone methyltransferase recruits DNA methyltransferase into the complex of Polycomb (PcG) proteins involved in hereditary repression of genes responsible for differentiation. Formation of such complexes may represent a basis for the reverse process--the dependence of DNA methylation on histone modification; this demonstrates the possibility of tight interactions between these two systems of epigenomic silencing.
Active chromatin is usually characterized by acetylation of lysine residues (K) in histones and trimethylation of lysine residue at position 4 of the histone H3 (H3K4me3), whereas modification H3K9me3 and H3K27me3 are typical for repressed heterochromatin and inactive euchromatin, respectively [131]. Gene silencing determined by characteristic modification of H3K27me3 and H3K9me3 can be accompanied by histone deacetylation and DNA methylation. The H3K9me3 modification attracts structural protein HP1, which plays an important role in gene silencing and formation of heterochromatin structures [133]. Loss of H3K9me3 causes removal of HP1 from the pericentromeric heterochromatin, aneuploidy, and cancerogenesis [135]. Appearance and metastasizing of breast cancer is associated with decreased level of HP1 expression [136]. It should be noted that separate modification of histones has to be considered in context with other modifications: a single modification cannot be unequivocally interpreted as positive or negative without analysis of other modifications; a common pattern of localization of such modifications on histone nucleosomes will determine specificity of interaction with chromatin of activating or repressing protein complexes [137].
Impairments of histone modifications in tumor cells have been found in many studies; we just mention here that the typical change in histone modification in human cancer includes loss of K16 acetylation and K20 methylation in histone H4 [120, 138]. There is a correlation between the decrease of histone acetylase activity with respect of H3K27 and H3K9 sites and impairments in Rb protein functioning and also with abnormalities in regulation of the cell cycle observed in tumor cells [133]. Some types of cancers are characterized by a sharp increase in expression of proteins of the PcG protein complex, which is responsible for modifications of H3K27me3 and H3K9me [107]. One of the PcG proteins is histone methyltransferase [139]. In embryonic mammalian stem cells, the PcG proteins are required for maintenance of cell pluripotency by transient (reversible) suppression of activity of genes, which are actively expressed during differentiation. In cancer cells, there is sharp methylation of promoters of the genes that are targets for PcG proteins. This results in reliable gene silencing, which prevents differentiation and maintains stem cell self-renovation, predisposing to malignization [140].
RNA-MEDIATED EPIGENOMIC SILENCING
In mammals RNA not coding proteins (further defined as non-coding RNA) determines genomic imprinting--selective expression of only one of two alleles inherited from the parents [141, 142], inactivation of one of two X-chromosomes in mammalian cells [143], and also PcG protein associated regulation of HOX cluster genes responsible for realization of the plan of the body formation [144]. These cases of RNA silencing involve large non-coding RNA molecules, which consist of tens and even hundreds of thousands of nucleotides [145, 146]. Impairments in genomic imprinting may cause expression of both alleles and overproduction of potential growth factors leading to tumor formation [107]. Gene silencing during genomic imprinting and inactivation of X-chromosome are accompanied not only by various modifications of histone molecules but also by DNA methylation. Recently it has been reported that overexpression of non-coding long RNA (>2 kb) causes an antiapoptotic effect in human cancer cells [147].
Processes of epigenomic silencing may involve another type of non-coding RNAs, short siRNAs. This demonstrates the existence of nuclear RNA-interference, which is based (in contrast to RNAi) on suppression of mRNA translation in cytoplasm. Involvement of siRNAs in gene silencing at the level of chromatin was originally found in model objects. There is still no convincing evidence that this type of silencing is a cause for tumor development. However, some experimental data suggest that siRNA plays a certain role in gene silencing at the level of chromatin in mammalian cells (including human cancer cells). Short RNAs play a certain role in stabilization of non-coding pericentromeric regions of mammalian chromosomes (it has already been mentioned that impairments in chromatin structure of these regions are associated with appearance of tumors). Subsequent studies of mechanisms responsible for cancer epigenome formation would clarify the role of these short RNAs in these processes. We consider examples of involvement of short RNAs in epigenomic silencing, which is coupled to DNA methylation, histone modification of target genes, and attraction of the heterochromatin HP1 protein to them. All these chromatin modifications are typical for the cancer epigenome.
Short RNAs and epigenomic silencing. Studies on the role of siRNA and the Dicer protein in suppression of gene expression at the chromatin level originally employed model objects, plants (Arabidopsis thaliana) and dividing yeasts (Schizosaccharomyces pombe) [148, 149], and then they were continued using Drosophila. In plants, siRNA-mediated RNA silencing was accompanied by repressor modification of histones and DNA methylation. It was induced by endogenous dsRNA; its nucleotide sequence was identical to gene promoters [149]. The dsRNAs were formed during transcription of self-complementary inverted repeats or RNA-dependent RNA synthesis. In yeasts, siRNAs are involved in heterochromatinization of pericentromeric regions of chromosomes. In this case, dsRNAs are formed due to transcription of both strands of DNA repeats of these regions or (as in plants) using RNA-dependent RNA polymerase. Processing of dsRNA involves the Dicer protein; the resulting siRNAs bind to the Argonaute (AGO) protein required for their interactions with nascent transcripts (Fig. 1). The forming complex attracts histone methyltransferase responsible for the repressor modification H3K9me and association of the heterochromatin protein, yeast homolog of HP1. This results in heterochromatinization and silencing of centromeres required for chromatid cohesion and their normal disjunction during cell division [150, 151]. Yeasts lack DNA methylation. Consequently, it is reasonable to propose the rather paradoxical (at first glance) but experimentally confirmable suggestion that formation of compact inactive chromatin requires chromatin transcription, because nascent transcripts are a platform for assembly of complexes responsible for silencing. In plants, heterochromatin formation requires transcription of corresponding regions of the chromosome, which involves special RNA polymerase IV [9, 149]. Thus, heterochromatinization is determined by transcription, and notions that heterochromatin is not transcribed should therefore be essentially corrected.
In mammalian cells lacking RNA-dependent RNA polymerase, siRNA can originate from dsRNA via bi-directional transcription of the repeated elements of the genome (heterochromatin repeats and transposable elements). Non-coding intergenic regions of the mammalian genome are also transcribed, and the forming transcripts can be oppositely oriented and complementary to each other [16, 152, 153]. Finally, formation of a special type of short RNAs from single stranded transcripts is also possible.
Studies on human cell cultures (mainly of tumor origin) have shown that siRNA administered into cells or endogenously expressed siRNA can be involved in transcription silencing provided that complementary nucleotide sequence of gene promoter is a target [154-159]. Silencing requires transcription of a promoter region [156, 157] as previously shown using model objects [148, 149]. However, it is also possible that promoter DNA can be also a target. Transcription silencing is characterized by appearance of repressor modification (H3K27me3 and H3K9me2) of histones; it was accompanied [155] or not accompanied [154, 157] by methylation of the target gene. In HeLa cells expression of siRNA complementary to the promoter region and CpG islands in recently found tumor suppressor RASSF1A (Ras association domain family 1 isoform A) resulted in gene silencing due to DNA methylation without any signs of repressor modification of histones [159]. However, results of another study demonstrated that siRNA-dependent transcription silencing of rassf1a gene was accompanied by insertion of the H3K9me2 modification into histone promoter and association of AGO1 protein with PcG (attracting DNA methyltransferase) on the promoter [160]. Using siRNAs complementary to promoter region of progesterone receptor gene, it was possible to suppress its expression in breast cancer cells, but this, however, was not accompanied by changes in histone modification [158]. In this case, inhibition of expression of the Argonaute family proteins (AGO1 and AGO2) abolished both silencing induced by siRNA complementary to the promoter and siRNA acting at cytoplasmic mRNA as the target. These data indicate cross-talk between pathways of transcription silencing and mechanisms of RNAi realized at the translation level. Results of these studies demonstrate similarity of siRNA-dependent mechanisms of sequence-specific transcription silencing in humans and model objects. The examples of transcription silencing found in tumor cell cultures reflect interest of researchers in the development of new approaches to “epigenomic therapy” of cancerogenesis, which may be highly specific. However, paradoxical activation of progesterone receptor gene expression induced by siRNA complementary to the promoter sequence accompanied by active histone modification in the target has also been reported [161].
The role of RNAi is recognized not only in silencing of tumor suppressors or proto-oncogenes, but also in maintenance of heterochromatin structure of centromeric regions in mammalian cells [162, 163]. Transcription of both strands of centromeric DNA regions can result in formation of dsRNA, which undergoes Dicer endonuclease dependent processing followed siRNA formation [164]. Knockout of the Dicer gene in mouse embryonic stem cells resulted in impairments of epigenomic silencing of centromeric repeats; these impairments included the decrease in DNA methylation and disappearance of characteristic histone modifications [163]. In cell cultures possessing a hybrid genome (human chromosome 21 in chicken genome), deletion of the Dicer gene caused accumulation of abnormal mitoses demonstrating premature separation of sister chromatids [162]. Studies on Drosophila demonstrate that impairments in expression of the Argonaute family protein (AGO2) may result in defects of compact centromeric heterochromatin and aberrations in the division cycles [165], whereas mutations in the Dicer gene are accompanied by disorganization of centromeric heterochromatin [166]. All these results reasonably suggest involvement of short RNAs in maintenance of heterochromatin structures in human cells; defects in these structures are typical for tumor cells.
TRANSPOSABLE ELEMENTS AND SHORT RNAs
The role of transposable genome elements in appearance of cancer requires separate consideration. Let us consider results of studies demonstrating association of cancerogenesis with derepression of transposable elements and mechanisms of their silencing by means of short RNAs. This usually includes consideration of potential effect on tumor progression of L1 retrotransposons lacking long terminal repeats (LINE elements). The LINE elements represent 17% of the human genome and about a hundred of their copies, which are capable of autonomous transposition [167, 168]. Reverse transcriptase and endonuclease encoded by L1 elements can provide transposition of nonautonomous Alu retroelements (~106 copies in humans) and SVA (~5000 copies) [169]. The presence of reverse transcriptase encoded by these elements and also by endogenous retroviruses is considered as functional characteristics of tumorigenic cells [170]. In two sublines of melanoma cells obtained from a common parent line, proliferative potential and tumorigenicity positively correlated with the expression level of L1 elements [171]. Activation of L1 transcription can somehow stimulate initiation and tumor progression [172]. Suppression of reverse transcription by means of an RNAi mechanism decreased proliferation and promoted differentiation of some cancer cell lines [173, 174]. L1 transposition makes a marked contribution to insertion mutagenesis accompanied by diseases in humans [175]. Although L1 does not play a leading role in cancerogenesis, nevertheless L1 insertion into proto-oncogene or suppressor gene has been shown to be associated with tumor formation [176, 177]. It also should be noted that L1 transposition (especially defects of these transpositions) can result in double strand breaks of DNA [178] accompanied by genetic instability typical for cancer cells [179].
The possibility of suppression of L1 expression by siRNA is discussed. It has been shown that the antisense promoter L1 is required for dsRNA formation; in vitro the latter may be processed to siRNA by means of Dicer. These siRNAs suppress expression of artificial constructs with transcribed L1 sequences [180]. It has also been found that RNA binding protein encoded by L1 and required for element transposition binds to proteins AGO2 and FMRP (fragile mental retardation protein) of the effector complex RISC [181]. This suggests the role of RNAi in regulation of LINE element transposition in humans.
In eukaryotic genomes, expression of transposable elements can be regulated by means of epigenomic modifications. In mammals silencing of retrotransposons including L1 elements occurs due to their methylation of CpG sites [182]. Impairments of methylation of transposable elements trigger their expression [183]. Tumor progression is accompanied by global decrease of genome methylation and correlates with increased transcription of retrotransposons in cancer cells [184, 185]. It is suggested that the decrease of L1 methylation can be associated with urinary bladder carcinoma [186]. L1 methylation in tissues of mouse embryonic germline tissues is determined by functioning of special silencing system, employing a recently discovered type of short RNAs (average size of 26-30 nucleotides) [187]. In contrast to siRNA and miRNA, this type of short RNAs is defined as piRNAs because their functioning is determined by PIWI subfamily proteins, which represent structural variants of a large family of Argonaute proteins [188, 189]. Mutations in genes encoding mouse PIWI proteins result in derepression and demethylation not only of L1, but also other transposons possessing long terminal repeats (as retroviruses). This is accompanied by double strand breaks followed by formation of intranuclear repair compartments, the indicators of double strand breaks [190]. The role of short RNAs in suppression of transposable element expression in embryonic germline tissues including the role of short RNAs in chromatin silencing has been better studied in Drosophila [191-194]. Results of recent studies indicate that piRNAs are obviously formed from long single stranded transcripts, and this does not involve the Dicer protein [194, 195]. In vertebrates as well as in Drosophila, functioning of PIWI family proteins is associated with germline tissues, where transposable element transpositions, the sources of genomic instability, are considered as especially dangerous. High level of L1 expression is detected in tumors originating from human germinal tissues [126], and the decrease of their methylation has been demonstrated for various types of human tumors [196]. It is possible that piRNAs play an important role in the epigenomic regulation of retrotransposon expression in germinal tissues.
Besides consideration of the role of impairments of miRNA functions for cancerogenesis, we have discussed the role of other types of short RNAs (siRNA, piRNA) in regulation of epigenomic modifications and their impairments typical for cancer cells. This problem still requires further investigation, and studies in this direction may reveal new approaches for analysis of cancer epigenomics. It is possible that siRNA exhibits not only the cis-effect on repression of genes adjacent to the promoter, but it is also involved in long range, possibly trans-acting mechanisms of epigenomic regulation of gene expression. This is quite expectable because proteins involved in both siRNA-dependent (Dicer, Argonaute) and piRNA-dependent (PIWI) silencing have been found in intranuclear bodies, which include large evolutionarily conservative multiprotein complexes PcG; their impaired functioning is typical for cancer cells. The interrelationship between functioning of PcG complexes and the RNAi system has been found only for Drosophila [197], where these complexes are responsible for distant interactions, providing the repressor effect of regulatory elements positioned from the target genes at the distance of >=10 kb or located on other chromosomes [198].
Tumor therapy represents an important problem. New principles of tumor therapy can be based on our knowledge of the role of short RNAs in cancerogenesis. We have already considered examples of artificial overexpression or knockdown of miRNA in cell cultures resulted in suppression of their growth [47, 82, 93]. Deletion of a miRNA gene in cancer cells may be compensated by its delivery by means of viral vectors. In the case of overexpression of oncogenic miRNAs it is possible to use anti-miR oligonucleotides (antagomirs) complementary to miRNA and modified for their lifetime elongation for miRNA knockdown. Oligonucleotides carrying 2´-O-methyl [47, 93, 199] and 2´-O-methoxyethyl [200] groups and also LNA-oligonucleotides in which ribose oxygen atoms at C-2´ and C-4´ are linked together with a methylene bridge [93, 201] have already been employed for studies of miRNA functions. Results of basic studies of epigenomic RNA silencing may be used for the development of a new direction in epigenomic therapy, where the action of inhibitor onto a particular target will be based on its complementary interaction with RNA. This provides high specificity of interactions, which are not usually achieved in the case of use of other antitumor “epigenomic” agents, inhibitors of DNA methylation or histone-modifying proteins.
This work was financially supported by grants from the Russian Foundation for Basic Research (No. 08-04-00087a), Leading Scientific Schools, and Program of Russian Academy of Sciences on Molecular and Cell Biology and by State contract No. 02.522.11.2005.
REFERENCES
1.Bernstein, E., and Allis, C. D. (2005) Genes
Dev., 19, 1635-1655.
2.Cerutti, H., and Casas-Mollano, J. A. (2006)
Curr. Genet., 50, 81-99.
3.Sontheimer, E. J. (2005) Nat. Rev. Mol. Cell
Biol., 6, 127-138.
4.Calin, G. A., and Croce, C. M. (2006) Nat. Rev.
Cancer, 6, 857-866.
5.Calin, G. A., and Croce, C. M. (2006)
Oncogene, 25, 6202-6210.
6.Esquela-Kerscher, A., and Slack, F. J. (2006)
Nat. Rev. Cancer, 6, 259-269.
7.Allis, C. D., Junewein, T., Reinberg, D., and
Capparos, M. L. (eds.) (2006) Epigenetics, CSHL Press, N. Y.
8.Bird, A. (2007) Nature, 447,
396-398.
9.Klenov, M. S., and Gvozdev, V. A. (2005)
Biochemistry (Moscow), 70, 1187-1198.
10.Mello, C. C., and Conte, D., Jr. (2004)
Nature, 431, 338-342.
11.Bartel, D. P. (2004) Cell, 116,
281-297.
12.Kotel'nikov, R. N., Shpiz, S. G., Kalmykova, A.
I., and Gvozdev, V. A. (2006) Mol. Biol. (Moscow), 40,
1-14.
13.Zeng, Y. (2006) Oncogene, 25,
6156-6162.
14.Kim, V. N. (2005) Nat. Rev. Mol. Cell
Biol., 6, 376-385.
15.Bellare, P., and Sontheimer, E. J. (2007) Nat.
Struct. Mol. Biol., 14, 684-686.
16.Willingham, A. T., and Gingeras, T. R. (2006)
Cell, 125, 1215-1220.
17.Hartig, J. V., Tomari, Y., and Forstemann, K.
(2007) Genes Dev., 21, 1707-1713.
18.Bao, N., Lye, K. W., and Barton, M. K. (2004)
Dev. Cell, 7, 653-662.
19.Lewis, B. P., Burge, C. B., and Bartel, D. P.
(2005) Cell, 120, 15-20.
20.Eulalio, A., Huntzinger, E., and Izaurralde, E.
(2008) Cell, 132, 9-14.
21.Chendrimada, T. P., Finn, K. J., Ji, X., Baillat,
D., Gregory, R. I., Liebhaber, S. A., Pasquinelli, A. E., and
Shiekhattar, R. (2007) Nature, 447, 823-828.
22.Pillai, R. S., Bhattacharyya, S. N., Artus, C.
G., Zoller, T., Cougot, N., Basyuk, E., Bertrand, E., and Filipowicz,
W. (2005) Science, 309, 1573-1576.
23.Thermann, R., and Hentze, M. W. (2007)
Nature, 447, 875-878.
24.Wakiyama, M., Takimoto, K., Ohara, O., and
Yokoyama, S. (2007) Genes Dev., 21, 1857-1862.
25.Wang, B., Love, T. M., Call, M. E., Doench, J.
G., and Novina, C. D. (2006) Mol. Cell, 22, 553-560.
26.Nottrott, S., Simard, M. J., and Richter, J. D.
(2006) Nat. Struct. Mol. Biol., 13, 1108-1114.
27.Olsen, P. H., and Ambros, V. (1999) Dev.
Biol., 216, 671-680.
28.Petersen, C. P., Bordeleau, M. E., Pelletier, J.,
and Sharp, P. A. (2006) Mol. Cell, 21, 533-542.
29.Seggerson, K., Tang, L., and Moss, E. G. (2002)
Dev. Biol., 243, 215-225.
30.Kiriakidou, M., Tan, G. S., Lamprinaki, S., de
Planell-Saguer, M., Nelson, P. T., and Mourelatos, Z. (2007)
Cell, 129, 1141-1151.
31.Sheth, U., and Parker, R. (2003) Science,
300, 805-808.
32.Rehwinkel, J., Behm-Ansmant, I., Gatfield, D.,
and Izaurralde, E. (2005) RNA, 11, 1640-1647.
33.Meister, G., Landthaler, M., Peters, L., Chen, P.
Y., Urlaub, H., Luhrmann, R., and Tuschl, T. (2005) Curr. Biol.,
15, 2149-2155.
34.Liu, J., Rivas, F. V., Wohlschlegel, J., Yates,
J. R., III, Parker, R., and Hannon, G. J. (2005) Nat. Cell
Biol., 7, 1261-1266.
35.Liu, J., Valencia-Sanchez, M. A., Hannon, G. J.,
and Parker, R. (2005) Nat. Cell Biol., 7, 719-723.
36.Ding, L., Spencer, A., Morita, K., and Han, M.
(2005) Mol. Cell, 19, 437-447.
37.Parker, R., and Sheth, U. (2007) Mol.
Cell, 25, 635-646.
38.Chan, S. P., and Slack, F. J. (2006) RNA
Biol., 3, 97-100.
39.Standart, N., and Jackson, R. J. (2007) Genes
Dev., 21, 1975-1982.
40.Chen, C. Z., Li, L., Lodish, H. F., and Bartel,
D. P. (2004) Science, 303, 83-86.
41.Neilson, J. R., Zheng, G. X., Burge, C. B., and
Sharp, P. A. (2007) Genes Dev., 21, 578-589.
42.Zhao, Y., Ransom, J. F., Li, A., von Vedantham,
V. D. M., Muth, A. N., Tsuchihashi, T., McManus, M. T., Schwartz, R.
J., and Srivastava, D. (2007) Cell, 129, 303-317.
43.Lim, L. P., Lau, N. C., Garrett-Engele, P.,
Grimson, A., Schelter, J. M., Castle, J., Bartel, D. P., Linsley, P.
S., and Johnson, J. M. (2005) Nature, 433, 769-773.
44.Karp, X., and Ambros, V. (2005) Science,
310, 1288-1289.
45.Xu, P., Vernooy, S. Y., Guo, M., and Hay, B. A.
(2003) Curr. Biol., 13, 790-795.
46.Xu, P., Guo, M., and Hay, B. A. (2004) Trends
Genet., 20, 617-624.
47.Cheng, A. M., Byrom, M. W., Shelton, J., and
Ford, L. (2005) Nucleic Acids Res., 33, 1290-1297.
48.Hatfield, S. D., Shcherbata, H. R., Fischer, K.
A., Nakahara, K., Carthew, R. W., and Ruohola-Baker, H. (2005)
Nature, 435, 974-978.
49.Linsley, P. S., Schelter, J., Burchard, J.,
Kibukawa, M., Martin, M. M., Bartz, S. R., Johnson, J. M., Cummins, J.
M., Raymond, C. K., Dai, H., Chau, N., Cleary, M., Jackson, A. L.,
Carleton, M., and Lim, L. (2007) Mol. Cell Biol., 27,
2240-2252.
50.Ma, L., Teruya-Feldstein, J., and Weinberg, R. A.
(2007) Nature, 449, 682-688.
51.Calin, G. A., Sevignani, C., Dumitru, C. D.,
Hyslop, T., Noch, E., Yendamuri, S., Shimizu, M., Rattan, S., Bullrich,
F., Negrini, M., and Croce, C. M. (2004) Proc. Natl. Acad. Sci.
USA, 101, 2999-3004.
52.Calin, G. A., Liu, C. G., Sevignani, C.,
Ferracin, M., Felli, N., Dumitru, C. D., Shimizu, M., Cimmino, A.,
Zupo, S., Dono, M., Dell'Aquila, M. L., Alder, H., Rassenti, L., Kipps,
T. J., Bullrich, F., Negrini, M., and Croce, C. M. (2004) Proc.
Natl. Acad. Sci. USA, 101, 11755-11760.
53.Zhang, L., Huang, J., Yang, N., Greshock, J.,
Megraw, M. S., Giannakakis, A., Liang, S., Naylor, T. L., Barchetti,
A., Ward, M. R., Yao, G., Medina, A., O'Brien-Jenkins, A., Katsaros,
D., Hatzigeorgiou, A., Gimotty, P. A., Weber, B. L., and Coukos, G.
(2006) Proc. Natl. Acad. Sci. USA, 103, 9136-9141.
54.Ambros, V., Bartel, B., Bartel, D. P., Burge, C.
B., Carrington, J. C., Chen, X., Dreyfuss, G., Eddy, S. R.,
Griffiths-Jones, S., Marshall, M., Matzke, M., Ruvkun, G., and Tuschl,
T. (2003) RNA, 9, 277-279.
55.Cimmino, A., Calin, G. A., Fabbri, M., Iorio, M.
V., Ferracin, M., Shimizu, M., Wojcik, S. E., Aqeilan, R. I., Zupo, S.,
Dono, M., Rassenti, L., Alder, H., Volinia, S., Liu, C. G., Kipps, T.
J., Negrini, M., and Croce, C. M. (2005) Proc. Natl. Acad. Sci.
USA, 102, 13944-13949.
56.Calin, G. A., Dumitru, C. D., Shimizu, M., Bichi,
R., Zupo, S., Noch, E., Aldler, H., Rattan, S., Keating, M., Rai, K.,
Rassenti, L., Kipps, T., Negrini, M., Bullrich, F., and Croce, C. M.
(2002) Proc. Natl. Acad. Sci. USA, 99, 15524-15529.
57.Sanchez-Beato, M., Sanchez-Aguilera, A., and
Piris, M. A. (2003) Blood, 101, 1220-1235.
58.Johnson, S. M., Grosshans, H., Shingara, J.,
Byrom, M., Jarvis, R., Cheng, A., Labourier, E., Reinert, K. L., Brown,
D., and Slack, F. J. (2005) Cell, 120, 635-647.
59.Takamizawa, J., Konishi, H., Yanagisawa, K.,
Tomida, S., Osada, H., Endoh, H., Harano, T., Yatabe, Y., Nagino, M.,
Nimura, Y., Mitsudomi, T., and Takahashi, T. (2004) Cancer Res.,
64, 3753-3756.
60.Reinhart, B. J., Slack, F. J., Basson, M.,
Pasquinelli, A. E., Bettinger, J. C., Rougvie, A. E., Horvitz, H. R.,
and Ruvkun, G. (2000) Nature, 403, 901-906.
61.Grosshans, H., Johnson, T., Reinert, K. L.,
Gerstein, M., and Slack, F. J. (2005) Dev. Cell, 8,
321-330.
62.Goodwin, G. (1998) Int. J. Biochem. Cell
Biol., 30, 761-766.
63.Young, A. R., and Narita, M. (2007) Genes
Dev., 21, 1005-1009.
64.Bommer, G. T., Gerin, I., Feng, Y., Kaczorowski,
A. J., Kuick, R., Love, R. E., Zhai, Y., Giordano, T. J., Qin, Z. S.,
Moore, B. B., Macdougald, O. A., Cho, K. R., and Fearon, E. R. (2007)
Curr. Biol., 17, 1298-1307.
65.Chang, T. C., Wentzel, E. A., Kent, O. A.,
Ramachandran, K., Mullendore, M., Lee, K. H., Feldmann, G., Yamakuchi,
M., Ferlito, M., Lowenstein, C. J., Arking, D. E., Beer, M. A., Maitra,
A., and Mendell, J. T. (2007) Mol. Cell, 26, 745-752.
66.He, L., He, X., Lim, L. P., De, S. E., Xuan, Z.,
Liang, Y., Xue, W., Zender, L., Magnus, J., Ridzon, D., Jackson, A. L.,
Linsley, P. S., Chen, C., Lowe, S. W., Cleary, M. A., and Hannon, G. J.
(2007) Nature, 447, 1130-1134.
67.Raver-Shapira, N., Marciano, E., Meiri, E.,
Spector, Y., Rosenfeld, N., Moskovits, N., Bentwich, Z., and Oren, M.
(2007) Mol. Cell, 26, 731-743.
68.Tarasov, V., Jung, P., Verdoodt, B., Lodygin, D.,
Epanchintsev, A., Menssen, A., Meister, G., and Hermeking, H. (2007)
Cell Cycle, 6, 1586-1593.
69.Raver-Shapira, N., and Oren, M. (2007) Cell
Cycle, 6, 2656-2661.
70.Welch, C., Chen, Y., and Stallings, R. L. (2007)
Oncogene, 26, 5017-5022.
71.O'Connell, R. M., Taganov, K. D., Boldin, M. P.,
Cheng, G., and Baltimore, D. (2007) Proc. Natl. Acad. Sci. USA,
104, 1604-1609.
72.Rodriguez, A., Vigorito, E., Clare, S., Warren,
M. V., Couttet, P., van Soond, D. R. D. S., Grocock, R. J., Das, P. P.,
Miska, E. A., Vetrie, D., Okkenhaug, K., Enright, A. J., Dougan, G.,
Turner, M., and Bradley, A. (2007) Science, 316,
608-611.
73.Thai, T. H., Calado, D. P., Casola, S., Ansel, K.
M., Xiao, C., Xue, Y., Murphy, A., Frendewey, D., Valenzuela, D.,
Kutok, J. L., Schmidt-Supprian, M., Rajewsky, N., Yancopoulos, G., Rao,
A., and Rajewsky, K. (2007) Science, 316, 604-608.
74.Kluiver, J., de Haralambieva, E. J. D., Blokzijl,
T., Jacobs, S., Kroesen, B. J., Poppema, S., and van den Berg, A.
(2006) Genes Chromosomes. Cancer, 45, 147-153.
75.Kluiver, J., de Poppema, S. J. D., Blokzijl, T.,
Harms, G., Jacobs, S., Kroesen, B. J., and van den Berg, A. (2005)
J. Pathol., 207, 243-249.
76.Eis, P. S., Tam, W., Sun, L., Chadburn, A., Li,
Z., Gomez, M. F., Lund, E., and Dahlberg, J. E. (2005) Proc. Natl.
Acad. Sci. USA, 102, 3627-3632.
77.Metzler, M., Wilda, M., Busch, K., Viehmann, S.,
and Borkhardt, A. (2004) Genes Chromosomes. Cancer, 39,
167-169.
78.Yanaihara, N., Caplen, N., Bowman, E., Seike, M.,
Kumamoto, K., Yi, M., Stephens, R. M., Okamoto, A., Yokota, J., Tanaka,
T., Calin, G. A., Liu, C. G., Croce, C. M., and Harris, C. C. (2006)
Cancer Cell, 9, 189-198.
79.Gironella, M., Seux, M., Xie, M. J., Cano, C.,
Tomasini, R., Gommeaux, J., Garcia, S., Nowak, J., Yeung, M. L., Jeang,
K. T., Chaix, A., Fazli, L., Motoo, Y., Wang, Q., Rocchi, P., Russo,
A., Gleave, M., Dagorn, J. C., Iovanna, J. L., Carrier, A., Pebusque,
M. J., and Dusetti, N. J. (2007) Proc. Natl. Acad. Sci. USA,
104, 16170-16175.
80.Fulci, V., Chiaretti, S., Goldoni, M., Azzalin,
G., Carucci, N., Tavolaro, S., Castellano, L., Magrelli, A., Citarella,
F., Messina, M., Maggio, R., Peragine, N., Santangelo, S., Mauro, F.
R., Landgraf, P., Tuschl, T., Weir, D. B., Chien, M., Russo, J. J., Ju,
J., Sheridan, R., Sander, C., Zavolan, M., Guarini, A., Foa, R., and
Macino, G. (2007) Blood, 109, 4944-4951.
81.Costinean, S., Zanesi, N., Pekarsky, Y., Tili,
E., Volinia, S., Heerema, N., and Croce, C. M. (2006) Proc. Natl.
Acad. Sci. USA, 103, 7024-7029.
82.Voorhoeve, P. M., Le, S. C., Schrier, M., Gillis,
A. J., Stoop, H., Nagel, R., Van, D. J., Liu, Y. P., Drost, J.,
Griekspoor, A., Zlotorynski, E., Yabuta, N., De, V. G., Nojima, H.,
Looijenga, L. H., and Agami, R. (2006) Cell, 124,
1169-1181.
83.Aylon, Y., Michael, D., Shmueli, A., Yabuta, N.,
Nojima, H., and Oren, M. (2006) Genes Dev., 20,
2687-2700.
84.Ota, A., Tagawa, H., Karnan, S., Tsuzuki, S.,
Karpas, A., Kira, S., Yoshida, Y., and Seto, M. (2004) Cancer
Res., 64, 3087-3095.
85.He, L., Thomson, J. M., Hemann, M. T.,
Hernando-Monge, E., Mu, D., Goodson, S., Powers, S., Cordon-Cardo, C.,
Lowe, S. W., Hannon, G. J., and Hammond, S. M. (2005) Nature,
435, 828-833.
86.Hayashita, Y., Osada, H., Tatematsu, Y., Yamada,
H., Yanagisawa, K., Tomida, S., Yatabe, Y., Kawahara, K., Sekido, Y.,
and Takahashi, T. (2005) Cancer Res., 65, 9628-9632.
87.Lewis, B. P., Shih, I. H., Jones-Rhoades, M. W.,
Bartel, D. P., and Burge, C. B. (2003) Cell, 115,
787-798.
88.Lu, Y., Thomson, J. M., Wong, H. Y., Hammond, S.
M., and Hogan, B. L. (2007) Dev. Biol., 310, 442-453.
89.O'Donnell, K. A., Wentzel, E. A., Zeller, K. I.,
Dang, C. V., and Mendell, J. T. (2005) Nature, 435,
839-843.
90.Trimarchi, J. M., and Lees, J. A. (2002) Nat.
Rev. Mol. Cell Biol., 3, 11-20.
91.Volinia, S., Calin, G. A., Liu, C. G., Ambs, S.,
Cimmino, A., Petrocca, F., Visone, R., Iorio, M., Roldo, C., Ferracin,
M., Prueitt, R. L., Yanaihara, N., Lanza, G., Scarpa, A., Vecchione,
A., Negrini, M., Harris, C. C., and Croce, C. M. (2006) Proc. Natl.
Acad. Sci. USA, 103, 2257-2261.
92.Meng, F., Henson, R., Wehbe-Janek, H., Ghoshal,
K., Jacob, S. T., and Patel, T. (2007) Gastroenterology,
133, 647-658.
93.Chan, J. A., Krichevsky, A. M., and Kosik, K. S.
(2005) Cancer Res., 65, 6029-6033.
94.Enright, A. J., John, B., Gaul, U., Tuschl, T.,
Sander, C., and Marks, D. S. (2003) Genome Biol., 5,
R1.
95.Kumar, M. S., Lu, J., Mercer, K. L., Golub, T.
R., and Jacks, T. (2007) Nat. Genet., 39, 673-677.
96.Iorio, M. V., Ferracin, M., Liu, C. G., Veronese,
A., Spizzo, R., Sabbioni, S., Magri, E., Pedriali, M., Fabbri, M.,
Campiglio, M., Menard, S., Palazzo, J. P., Rosenberg, A., Musiani, P.,
Volinia, S., Nenci, I., Calin, G. A., Querzoli, P., Negrini, M., and
Croce, C. M. (2005) Cancer Res., 65, 7065-7070.
97.Michael, M. Z., O' Connor, S. M., van Holst
Pellekaan, N. G., Young, G. P., and James, R. J. (2003) Mol. Cancer
Res., 1, 882-891.
98.Hornstein, E., and Shomron, N. (2006) Nat.
Genet., 38 (Suppl.), S20-S24.
99.Cohen, S. M., Brennecke, J., and Stark, A. (2006)
Genes Dev., 20, 2769-2772.
100.Cullen, B. R. (2006) Nat. Genet., 38
(Suppl.), S25-S30.
101.Pfeffer, S., and Voinnet, O. (2006)
Oncogene, 25, 6211-6219.
102.Skalsky, R. L., Samols, M. A., Plaisance, K.
B., Boss, I. W., Riva, A., Lopez, M. C., Baker, H. V., and Renne, R.
(2007) J. Virol., 81, 12836-12845.
103.Yeung, M. L., Bennasser, Y., Myers, T. G.,
Jiang, G., Benkirane, M., and Jeang, K. T. (2005) Retrovirology,
2, 81.
104.Lu, J., Getz, G., Miska, E. A., Varez-Saavedra,
E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B. L., Mak, R. H.,
Ferrando, A. A., Downing, J. R., Jacks, T., Horvitz, H. R., and Golub,
T. R. (2005) Nature, 435, 834-838.
105.Blenkiron, C., Goldstein, L. D., Thorne, N. P.,
Spiteri, I., Chin, S. F., Dunning, M. J., Barbosa-Morais, N. L.,
Teschendorff, A. E., Green, A. R., Ellis, I. O., Tavare, S., Caldas,
C., and Miska, E. A. (2007) Genome Biol., 8, R214.
106.Calin, G. A., Ferracin, M., Cimmino, A., Di, L.
G., Shimizu, M., Wojcik, S. E., Iorio, M. V., Visone, R., Sever, N. I.,
Fabbri, M., Iuliano, R., Palumbo, T., Pichiorri, F., Roldo, C., Garzon,
R., Sevignani, C., Rassenti, L., Alder, H., Volinia, S., Liu, C. G.,
Kipps, T. J., Negrini, M., and Croce, C. M. (2005) N. Engl. J.
Med., 353, 1793-1801.
107.Ting, A. H., McGarvey, K. M., and Baylin, S. B.
(2006) Genes Dev., 20, 3215-3231.
108.Bernstein, B. E., Meissner, A., and Lander, E.
S. (2007) Cell, 128, 669-681.
109.Jones, P. A., and Baylin, S. B. (2007)
Cell, 128, 683-692.
110.Lund, A. H., and van Lohuizen, M. (2004)
Genes Dev., 18, 2315-2335.
111.Feinberg, A. P., Ohlsson, R., and Henikoff, S.
(2006) Nat. Rev. Genet., 7, 21-33.
112.Hark, A. T., Schoenherr, C. J., Katz, D. J.,
Ingram, R. S., Levorse, J. M., and Tilghman, S. M. (2000)
Nature, 405, 486-489.
113.Bird, A. (2002) Genes Dev., 16,
6-21.
114.Saha, A., Wittmeyer, J., and Cairns, B. R.
(2006) Results Probl. Cell Differ., 41, 127-148.
115.Cairns, B. R. (2007) Nat. Struct. Mol.
Biol., 14, 989-996.
116.Smith, C. L., and Peterson, C. L. (2005)
Curr. Top. Dev. Biol., 65, 115-148.
117.Jaenisch, R., and Bird, A. (2003) Nat.
Genet., 33 (Suppl.), 245-254.
118.Rougier, N., Bourc'his, D., Gomes, D. M.,
Niveleau, A., Plachot, M., Paldi, A., and Viegas-Pequignot, E. (1998)
Genes Dev., 12, 2108-2113.
119.Keshet, I., Schlesinger, Y., Farkash, S., Rand,
E., Hecht, M., Segal, E., Pikarski, E., Young, R. A., Niveleau, A.,
Cedar, H., and Simon, I. (2006) Nat. Genet., 38,
149-153.
120.Esteller, M. (2007) Nat. Rev. Genet.,
8, 286-298.
121.Rideout, W. M., III, Coetzee, G. A., Olumi, A.
F., and Jones, P. A. (1990) Science, 249, 1288-1290.
122.Lujambio, A., Ropero, S., Ballestar, E., Fraga,
M. F., Cerrato, C., Setien, F., Casado, S., Suarez-Gauthier, A.,
Sanchez-Cespedes, M., Git, A., Spiteri, I., Das, P. P., Caldas, C.,
Miska, E., and Esteller, M. (2007) Cancer Res., 67,
1424-1429.
123.Saito, Y., Liang, G., Egger, G., Friedman, J.
M., Chuang, J. C., Coetzee, G. A., and Jones, P. A. (2006) Cancer
Cell, 9, 435-443.
124.Frigola, J., Song, J., Stirzaker, C.,
Hinshelwood, R. A., Peinado, M. A., and Clark, S. J. (2006) Nat.
Genet., 38, 540-549.
125.Feinberg, A. P., and Vogelstein, B. (1983)
Nature, 301, 89-92.
126.Hoffmann, M. J., and Schulz, W. A. (2005)
Biochem. Cell Biol., 83, 296-321.
127.Cadieux, B., Ching, T. T., van den Berg, S. R.,
and Costello, J. F. (2006) Cancer Res., 66,
8469-8476.
128.Eden, A., Gaudet, F., Waghmare, A., and
Jaenisch, R. (2003) Science, 300, 455.
129.Dodge, J. E., Okano, M., Dick, F., Tsujimoto,
N., Chen, T., Wang, S., Ueda, Y., Dyson, N., and Li, E. (2005) J.
Biol. Chem., 280, 17986-17991.
130.Nakagawa, T., Kanai, Y., Ushijima, S.,
Kitamura, T., Kakizoe, T., and Hirohashi, S. (2005) J. Urol.,
173, 243-246.
131.Taverna, S. D., Li, H., Ruthenburg, A. J.,
Allis, C. D., and Patel, D. J. (2007) Nat. Struct. Mol. Biol.,
14, 1025-1040.
132.Kouzarides, T. (2007) Cell, 128,
693-705.
133.Moss, T. J., and Wallrath, L. L. (2007)
Mutat. Res., 618, 163-174.
134.Jones, P. L., Veenstra, G. J., Wade, P. A.,
Vermaak, D., Kass, S. U., Landsberger, N., Strouboulis, J., and Wolffe,
A. P. (1998) Nat. Genet., 19, 187-191.
135.Peters, A. H., O'Carroll, D., Scherthan, H.,
Mechtler, K., Sauer, S., Schofer, C., Weipoltshammer, K., Pagani, M.,
Lachner, M., Kohlmaier, A., Opravil, S., Doyle, M., Sibilia, M., and
Jenuwein, T. (2001) Cell, 107, 323-337.
136.Kirschmann, D. A., Lininger, R. A., Gardner, L.
M., Seftor, E. A., Odero, V. A., Ainsztein, A. M., Earnshaw, W. C.,
Wallrath, L. L., and Hendrix, M. J. (2000) Cancer Res.,
60, 3359-3363.
137.Berger, S. L. (2007) Nature, 447,
407-412.
138.Fraga, M. F., Ballestar, E., Villar-Garea, A.,
Boix-Chornet, M., Espada, J., Schotta, G., Bonaldi, T., Haydon, C.,
Ropero, S., Petrie, K., Iyer, N. G., Perez-Rosado, A., Calvo, E.,
Lopez, J. A., Cano, A., Calasanz, M. J., Colomer, D., Piris, M. A.,
Ahn, N., Imhof, A., Caldas, C., Jenuwein, T., and Esteller, M. (2005)
Nat. Genet., 37, 391-400.
139.Vire, E., Brenner, C., Deplus, R., Blanchon,
L., Fraga, M., Didelot, C., Morey, L., Van, E. A., Bernard, D.,
Vanderwinden, J. M., Bollen, M., Esteller, M., Di, C. L., de Launoit,
Y., and Fuks, F. (2006) Nature, 439, 871-874.
140.Widschwendter, M., Fiegl, H., Egle, D.,
Mueller-Holzner, E., Spizzo, G., Marth, C., Weisenberger, D. J.,
Campan, M., Young, J., Jacobs, I., and Laird, P. W. (2007) Nat.
Genet., 39, 157-158.
141.Yang, P. K., and Kuroda, M. I. (2007)
Cell, 128, 777-786.
142.Barlow, D. P., and Bartolomei, M. S. (2006) in
Epigenetics (Allis, C. D., Junewein, T., Reinberg, D., and
Capparos, M. L., eds.) CSHL Press, N. Y., pp. 357-375.
143.Brockdorf, N., and Turner, B. M. (2006) in
Epigenetics (Allis, C. D., Junewein, T., Reinberg, D., and
Capparos, M. L., eds.) CSHL Press, N. Y., pp. 321-340.
144.Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo,
S. L., Xu, X., Brugmann, S. A., Goodnough, L. H., Helms, J. A.,
Farnham, P. J., Segal, E., and Chang, H. Y. (2007) Cell,
129, 1311-1323.
145.Gilfillan, G. D., Dahlsveen, I. K., and Becker,
P. B. (2004) FEBS Lett., 567, 8-14.
146.Wutz, A., Rasmussen, T. P., and Jaenisch, R.
(2002) Nat. Genet., 30, 167-174.
147.Tsang, W. P., Wong, T. W., Cheung, A. H., Co,
C. N., and Kwok, T. T. (2007) RNA, 13, 890-898.
148.Matzke, M. A., and Birchler, J. A. (2005)
Nat. Rev. Genet., 6, 24-35.
149.Wassenegger, M. (2005) Cell, 122,
13-16.
150.Zaratiegui, M., Irvine, D. V., and Martienssen,
R. A. (2007) Cell, 128, 763-776.
151.Buhler, M., and Moazed, D. (2007) Nat.
Struct. Mol. Biol., 14, 1041-1048.
152.Ravasi, T., Suzuki, H., Pang, K. C., Katayama,
S., Furuno, M., Okunishi, R., Fukuda, S., Ru, K., Frith, M. C.,
Gongora, M. M., Grimmond, S. M., Hume, D. A., Hayashizaki, Y., and
Mattick, J. S. (2006) Genome Res., 16, 11-19.
153.Chen, J., Sun, M., Kent, W. J., Huang, X., Xie,
H., Wang, W., Zhou, G., Shi, R. Z., and Rowley, J. D. (2004) Nucleic
Acids Res., 32, 4812-4820.
154.Ting, A. H., Schuebel, K. E., Herman, J. G.,
and Baylin, S. B. (2005) Nat. Genet., 37, 906-910.
155.Morris, K. V., Chan, S. W., Jacobsen, S. E.,
and Looney, D. J. (2004) Science, 305, 1289-1292.
156.Han, J., Kim, D., and Morris, K. V. (2007)
Proc. Natl. Acad. Sci. USA, 104, 12422-12427.
157.Weinberg, M. S., Villeneuve, L. M., Ehsani, A.,
Amarzguioui, M., Aagaard, L., Chen, Z. X., Riggs, A. D., Rossi, J. J.,
and Morris, K. V. (2006) RNA, 12, 256-262.
158.Janowski, B. A., Huffman, K. E., Schwartz, J.
C., Ram, R., Nordsell, R., Shames, D. S., Minna, J. D., and Corey, D.
R. (2006) Nat. Struct. Mol. Biol., 13, 787-792.
159.Castanotto, D., Tommasi, S., Li, M., Li, H.,
Yanow, S., Pfeifer, G. P., and Rossi, J. J. (2005) Mol. Ther.,
12, 179-183.
160.Kim, D. H., Villeneuve, L. M., Morris, K. V.,
and Rossi, J. J. (2006) Nat. Struct. Mol. Biol., 13,
793-797.
161.Janowski, B. A., Younger, S. T., Hardy, D. B.,
Ram, R., Huffman, K. E., and Corey, D. R. (2007) Nat. Chem.
Biol., 3, 166-173.
162.Fukagawa, T., Nogami, M., Yoshikawa, M., Ikeno,
M., Okazaki, T., Takami, Y., Nakayama, T., and Oshimura, M. (2004)
Nat. Cell Biol., 6, 784-791.
163.Kanellopoulou, C., Muljo, S. A., Kung, A. L.,
Ganesan, S., Drapkin, R., Jenuwein, T., Livingston, D. M., and
Rajewsky, K. (2005) Genes Dev., 19, 489-501.
164.Hutvagner, G., and Zamore, P. D. (2002)
Curr. Opin. Genet. Dev., 12, 225-232.
165.Deshpande, G., Calhoun, G., and Schedl, P.
(2005) Genes Dev., 19, 1680-1685.
166.Peng, J. C., and Karpen, G. H. (2007) Nat.
Cell Biol., 9, 25-35.
167.Kazazian, H. H., Jr., and Goodier, J. L. (2002)
Cell, 110, 277-280.
168.Brouha, B., Schustak, J., Badge, R. M.,
Lutz-Prigge, S., Farley, A. H., Moran, J. V., and Kazazian, H. H., Jr.
(2003) Proc. Natl. Acad. Sci. USA, 100, 5280-5285.
169.International Human Genome Sequencing
Consortium (2001) Nature, 409, 860-921.
170.Sinibaldi-Vallebona, P., Lavia, P., Garaci, E.,
and Spadafora, C. (2006) Genes Chromosomes. Cancer, 45,
1-10.
171.Oricchio, E., Sciamanna, I., Beraldi, R.,
Tolstonog, G. V., Schumann, G. G., and Spadafora, C. (2007)
Oncogene, 26, 4226-4233.
172.Collier, L. S., and Largaespada, D. A. (2006)
Curr. Opin. Genet. Dev., 16, 23-29.
173.Spadafora, C. (2004) Cytogenet. Genome
Res., 105, 346-350.
174.Sciamanna, I., Landriscina, M., Pittoggi, C.,
Quirino, M., Mearelli, C., Beraldi, R., Mattei, E., Serafino, A.,
Cassano, A., Sinibaldi-Vallebona, P., Garaci, E., Barone, C., and
Spadafora, C. (2005) Oncogene, 24, 3923-3931.
175.Chen, J. M., Stenson, P. D., Cooper, D. N., and
Ferec, C. (2005) Hum. Genet., 117, 411-427.
176.Wang, T., Lerer, I., Gueta, Z., Sagi, M.,
Kadouri, L., Peretz, T., and Abeliovich, D. (2001) Genes
Chromosomes. Cancer, 31, 91-95.
177.Miki, Y., Nishisho, I., Horii, A., Miyoshi, Y.,
Utsunomiya, J., Kinzler, K. W., Vogelstein, B., and Nakamura, Y. (1992)
Cancer Res., 52, 643-645.
178.Gasior, S. L., Wakeman, T. P., Xu, B., and
Deininger, P. L. (2006) J. Mol. Biol., 357,
1383-1393.
179.Symer, D. E., Connelly, C., Szak, S. T.,
Caputo, E. M., Cost, G. J., Parmigiani, G., and Boeke, J. D. (2002)
Cell, 110, 327-338.
180.Yang, N., and Kazazian, H. H., Jr. (2006)
Nat. Struct. Mol. Biol., 13, 763-771.
181.Goodier, J. L., Zhang, L., Vetter, M. R., and
Kazazian, H. H., Jr. (2007) Mol. Cell Biol., 27,
6469-6483.
182.Bourc'his, D., and Bestor, T. H. (2004)
Nature, 431, 96-99.
183.Carnell, A. N., and Goodman, J. I. (2003)
Toxicol. Sci., 75, 229-235.
184.Roman-Gomez, J., Jimenez-Velasco, A., Agirre,
X., Cervantes, F., Sanchez, J., Garate, L., Barrios, M., Castillejo, J.
A., Navarro, G., Colomer, D., Prosper, F., Heiniger, A., and Torres, A.
(2005) Oncogene, 24, 7213-7223.
185.Schulz, W. A. (2006) J. Biomed.
Biotechnol., 2006, 83672.
186.Jurgens, B., Schmitz-Drager, B. J., and Schulz,
W. A. (1996) Cancer Res., 56, 5698-5703.
187.Aravin, A. A., Sachidanandam, R., Girard, A.,
Fejes-Toth, K., and Hannon, G. J. (2007) Science, 316,
744-747.
188.O'Donnell, K. A., and Boeke, J. D. (2007)
Cell, 129, 37-44.
189.Hartig, J. V., Tomari, Y., and Forstemann, K.
(2007) Genes Dev., 21, 1707-1713.
190.Carmell, M. A., Girard, A., van de Kant, H. J.,
Bourc'his, D., Bestor, T. H., de Rooij, D. G., and Hannon, G. J. (2007)
Dev. Cell, 12, 503-514.
191.Klenov, M. C., Stolyarenko, A. D., Ryazansky,
S. S., Sokolova, O. A., Konstantinov, I. N., and Gvozdev, V. A. (2007)
Ontogenez, 38, 1-15.
192.Klenov, M. S., Lavrov, S. A., Stolyarenko, A.
D., Ryazansky, S. S., Aravin, A. A., Tuschl, T., and Gvozdev, V. A.
(2007) Nucleic Acids Res., 35, 5430-5438.
193.Kalmykova, A. I., Nurminsky, D. I., Ryzhov, D.
V., and Shevelyov, Y. Y. (2005) Nucleic Acids Res., 33,
1435-1444.
194.Vagin, V. V., Sigova, A., Li, C., Seitz, H.,
Gvozdev, V., and Zamore, P. D. (2006) Science, 313,
320-324.
195.Brennecke, J., Aravin, A. A., Stark, A., Dus,
M., Kellis, M., Sachidanandam, R., and Hannon, G. J. (2007)
Cell, 128, 1089-1103.
196.Chalitchagorn, K., Shuangshoti, S., Hourpai,
N., Kongruttanachok, N., Tangkijvanich, P., Thong-ngam, D., Voravud,
N., Sriuranpong, V., and Mutirangura, A. (2004) Oncogene,
23, 8841-8846.
197.Grimaud, C., Bantignies, F., Pal-Bhadra, M.,
Ghana, P., Bhadra, U., and Cavalli, G. (2006) Cell, 124,
957-971.
198.Schuettengruber, B., Chourrout, D., Vervoort,
M., Leblanc, B., and Cavalli, G. (2007) Cell, 128,
735-745.
199.Hutvagner, G., Simard, M. J., Mello, C. C., and
Zamore, P. D. (2004) PLoS Biology, 2, e98.
200.Esau, C., Kang, X., Peralta, E., Hanson, E.,
Marcusson, E. G., Ravichandran, L. V., Sun, Y., Koo, S., Perera, R. J.,
Jain, R., Dean, N. M., Freier, S. M., Bennett, C. F., Lollo, B., and
Griffey, R. (2004) J. Biol. Chem., 279, 52361-52365.
201.Lecellier, C. H., Dunoyer, P., Arar, K.,
Lehmann-Che, J., Eyquem, S., Himber, C., Saib, A., and Voinnet, O.
(2005) Science, 308, 557-560.
202.Jannot, G., and Simard, M. J. (2006)
Oncogene, 25, 6197-6201.
203.Jay, C., Nemunaitis, J., Chen, P., Fulgham, P.,
and Tong, A. W. (2007) DNA Cell Biol., 26, 293-300.
204.Yanaihara, N., Caplen, N., Bowman, E., Seike,
M., Kumamoto, K., Yi, M., Stephens, R. M., Okamoto, A., Yokota, J.,
Tanaka, T., Calin, G. A., Liu, C. G., Croce, C. M., and Harris, C. C.
(2006) Cancer Cell, 9, 189-198.
205.O'Rourke, J. R., Swanson, M. S., and Harfe, B.
D. (2006) Birth Defects Res. C. Embryo. Today, 78,
172-179.
206.Roldo, C., Missiaglia, E., Hagan, J. P.,
Falconi, M., Capelli, P., Bersani, S., Calin, G. A., Volinia, S., Liu,
C. G., Scarpa, A., and Croce, C. M. (2006) J. Clin. Oncol.,
24, 4677-4684.