
|
REVIEW: Molecular Epidemiology of CancerD. G. ZaridzeResearch Institute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; fax: (495) 324-2554; E-mail: dgzaridze@crc.umos.ru |
Received January 21, 2008
In this review the role of molecular markers for the assessment of individual exposure to carcinogenic agents was analyzed. Examples of the studies describing mutation patterns related to specific carcinogenic exposures are presented. The results of epidemiological studies of gene polymorphism and its role in the interaction between inheritance, environmental factors, and lifestyles are analyzed in detail. Adequate planning and performance of the epidemiological component of a study is a requirement for obtaining reproducible results reflecting molecular mechanisms of interest. Individual information on lifestyle factors (smoking, alcohol consumption, nutrition, physical activity, reproductive anamnesis) and environmental factors (occupational activity and carcinogen load at workplace), which influence not only the risk of developing cancer, but also the molecular features of a tumor, is crucial for adequate analysis and proper assessment of the results.
KEY WORDS: molecular markers, carcinogen-specific mutations, genetic polymorphism, interaction of genetic and environmental factors in carcinogenesisDOI: 10.1134/S0006297908050064
Abbreviations: CI) confidence interval; GST) glutathione S-transferase; NAT) N-acetyl transferase; PAH) polycyclic aromatic hydrocarbons; RR) relative risk; TSN) tobacco specific nitrosamines; UADO) upper aerodigestive organs.
Molecular epidemiology of cancer studies molecular markers of
distribution of malignant tumors in the population and their effects on
individual risk of developing a disease. Molecular markers can be
detected in tissues and biological liquids and characterize individual
exposure to carcinogens, biological effect of the exposure, genetic
susceptibility to the development of disease, and final result of
carcinogenesis, i.e. tumor. Identification of specific somatic
molecular and genetic changes, so-called fingerprints, is very
important for the molecular characteristics of a tumor and confirmation
of etiology. Identification of early molecular and proteomic markers
preceding clinical manifestation of disease is an important objective
of molecular epidemiology [1-3].
Although present review does not intend to embrace entire field of molecular epidemiology of cancer, it carries an important massage, that obtaining “reproducible” results, reflecting biological or molecular mechanism, requires adequate planning and implementation of both molecular and epidemiological components of the study. Epidemiological study is not only a source of biological material, but is first of all the source of individual information on factors influencing the risk of developing cancer, as well as molecular features of a tumor. Among these are lifestyle factors (smoking, alcohol consumption, nutrition/diet, physical activity,), environmental factors (occupation and exposure to carcinogens at workplace), familial and individual medical history, and many other variables. This information is crucial for adequate analysis and proper evaluation of results of a molecular study [3, 4].
Markers of exposure. They characterize levels of exogenous factors in biological fluids. Among these are chemical substances (dioxins, polychlorinated biphenyls, aromatic amines, polycyclic aromatic hydrocarbons (PAH), aflatoxins, heavy metals) and products of their metabolism, nutrients (vitamins and microelements), and infectious agents (viruses, Helicobacter pylori). Exposure markers can characterize changes in endogenous substrates, for instance, in hormones, resulting from the effects of exogenous factors. Also, a specific immune response to external exposure can serve as a marker of exposure. The markers of exposure are most often used in conjunction with hygienic data or information obtained using a questionnaire, and sometimes, when they are absent, as an independent source of exposure information. All these markers have been successfully used in epidemiological studies in the past [1, 4].
Macromolecular adducts such as carcinogen-protein and carcinogen-DNA are molecular markers of exposure. Carcinogen-DNA adducts characterize biological response to exposure, and particularly reflect the processes of the carcinogen metabolism, their activation and detoxification, DNA repair, and biologically effective dose. Presence and persistence of carcinogen-DNA adducts are early signs (early markers) of carcinogenesis [5].
The majority of studies in this field have been designed to investigate protein and DNA adducts of carcinogenic components of tobacco smoke, particularly adducts of tobacco-specific nitrosamines (TSN) and PAH. The study of hemoglobin adducts of 4-hydroxy-1-(3-pyridyl)-1-butunone, a TSN metabolite, has demonstrated that their concentration is significantly elevated in smokers versus nonsmokers [6]. These results were used further for assessment of exposure to indoor tobacco smoke, i.e. as evidence of “passive smoking”. Adducts of other carcinogens, such as, 4-aminobiphenyl, were used for assessing exposure to tobacco smoke. These adducts have relatively long half-life period, which is very important for identification of exposure in the past [7]. Detection of aflatoxin-DNA and aflatoxin-hemoglobin adducts had a crucial role in determining carcinogenicity of aflatoxin and its role in liver cancer. Aflatoxin is known to enter the human body with various foods, and it is virtually impossible to assess its consumption using questionnaires exclusively [4, 8].
Carcinogen-specific molecular markers in malignant tumors. The most demonstrative example of carcinogen-specific molecular markers is the difference in mutation types in p53 in different tumors, which reflect most probably the effects of carcinogens having different mechanisms of action. For instance, transversions G:C→T:A in codon 249 are typical for hepatocellular cancer etiologically associated with aflatoxin-B [9].
Our group together with the International Agency for Research on Cancer (IARC) has demonstrated a link between smoking and mutations in the p53 gene in lung cancer. The mutation prevalence in tumors of current smokers was significantly higher (75%) than among ex-smokers (56%) and patients who had never smoked (47%). The mutation rate also depends on number of cigarettes smoked per year. Mutations were more often found in tumor cells of squamous cell cancer (77%) and rarely in adenocarcinoma (23%) (Table 1) [10].
Table 1. Relative risk (RR) of p53
mutations in tumor cells of lung cancer depending on smoking status and
histological form of the tumor

*Number of observations is given in parentheses.
**Percentage of total observation number is given in
parentheses.
***95% confidence interval is given in parentheses.
Two types of p53 mutations are found in tumors of current smokers: transversions (G:C→T:A) and transitions (A:T→G:C), which are located in sites of PAH-DNA adduct formations. The transversions G:C→A:T are more often found in tumors of non-smoking patients at non-CpG sites (p = 0.02). This type of mutation results most probably from the formation of promutagenic O6-alkyl adducts of DNA, induced by N-nitrosonornicotine, the concentration of which is high in side-stream smoke. Thus, the type and location of mutations in p53 can serve as markers of exposure to PAH and TSN, as well as active and passive smoking.
In current smokers, mutations in p53 were found in codons 157, 158, 179, 205, 215, 220, 236, 237, 242, 248, 266, and 273. In patients who had never smoked, none of the mutations were localized in these codons. Significant correlation was found between positive staining for p53 protein and smoking. Significantly elevated number of p53-positive cells (p = 0.02) was found in tumors of smoking patients. All the tumors in which all cells were positively stained were from smokers.
The rate of mutations in p53 also depends on exposure to carcinogens at workplace, although to considerably less extent than on smoking. Relative risk (RR) of p53 mutation is increased 2-fold in tumor cells of patients with lung cancer who were exposed to PAH at their workplaces. However, this elevation of risk is not significant. The RR increases with increase in the level of exposure to PAH. The risk of mutations is elevated at a high level of exposure to PAH, but this elevation is not significant.
It was suggested that the higher prevalence of p53 mutations in lung cancer of smokers may reflect increased clonal selection of cells with random p53 mutations and that smoking (exposure of lung cancer to carcinogens in tobacco smoke) only stimulates clonal expansion of p53 mutated cells. However, the specificity of mutation patterns for smokers described in our work, which is different from mutation patterns in nonsmokers, and the fact that different mutation types correspond to different components of tobacco smoke support the hypothesis of mutagenic effect of tobacco smoke on p53 [10].
Mutations in the Kras oncogene (codon 12) were found in only 15% of all studied lung cancer cases, and only in 10% of lung cancer in smoking patients. Wild type Kras was found in 90% of tumors containing p53 mutations, whereas wild type p53 was found in 55% of tumors with mutated Kras. Mutations of both genes were more frequently found in relatively young patients who have lung adenocarcinoma [10].
Mutations in epidermal growth factor receptor (EGFR) are often found in women with lung cancer [11]. Mutations in HER2, which is related to the EGFR gene, are also found in some patients [12]. These molecular alterations attract attention due to their critical role in sensitivity of tumor to tyrosine kinase inhibitors, particularly to so-called target pharmaceuticals erlotinib and gefitinib. Mutations in EGFR have been found in 17% of all studied cases of lung cancer in the study of lung cancer carried out in Russia in cooperation with IARC. Mutations were more often found in adenocarcinoma (75%) than in squamous cell (15%) and small-cell (10%) cancer. None of the patients who smoked at the time of the diagnosis had EGFR mutations. However, mutations in EGFR were found in 48% of patients who never smoked. Mutations in EGFR were present also in tumor cells of ex-smokers (10%). The fact that EGFR mutations are more often found in women can be explained by prevalence of non-smokers among them, but not by gender differences in the mechanism of carcinogenesis. Mutations in HER2 were found in only 4% of all studied cases of lung cancer. They were detected exclusively in non-smoker or ex-smokers [13]. These observations point to differences in molecular mechanisms of lung cancer in smokers, ex-smokers, and persons who had never smoked. Mutations in p53, Kras, EGFR, and in other genes can be markers of exposure to specific carcinogens as well as markers of tumors etiologically linked to these carcinogens. Finding such mutations in non-tumor tissue using sensitive methods, which molecular biologists already have at hand, can point to the presence of exposure to a specific carcinogen or group of carcinogens and, correspondingly, to elevated risk of cancer in an individual in whom this marker has been detected.
Markers of genetic susceptibility to cancer. The epidemiological studies have demonstrated that the causes of 90-95% of malignant tumors are environmental and lifestyle factors, such as smoking, infectious agents, exogenous hormones, obesity, low physical activity, professional carcinogens, ultraviolet and ionizing radiation, alcohol consumption, etc. Hence, the vast majority of tumors in humans are not hereditary. Some rare genetic syndromes are exceptions. Genes have been identified which cause the development of hereditary and familial forms of malignant tumors. Germ-line mutation in retinoblastoma gene (Rb) is associated with the development of hereditary retinoblastoma. Germ-line mutations in p53 tumor suppressor gene cause Lee-Fraumeni syndrome, for which familial clustering of multiple primary tumors is typical. The inherited damage of breast cancer BRCA1 and BRCA2 genes is associated with increase in the risk not only of breast and ovary cancer, but also number of other forms of cancer. The list of cancer syndromes caused by germ-line mutations of corresponding genes includes adenomatous polyposis coli (APC), non-polypoid hereditary colon cancer (HMLH1), neurofibromatosis (NF1), etc. [14]. The RR for the development of a given tumor syndrome in persons with germ-line mutations is very high, and in some cases the probability of cancer development reaches 100%. However, the occurrence of the phenomenon itself, i.e. the presence of germ-line mutations, is extremely low (=1-5 cases per 100,000 live births). Thus the number of malignant tumors etiologically linked to these genetic events is low [9].
The vast majority of tumors in humans develops as a result of combined effect of a wide range of genes with low penetration, i.e. are of polygenic etiology. Exogenous factors play an important role in etiology of these tumors. Tumors develop as a result of interaction between endogenous (hereditary) and exogenous (lifestyle and environmental) factors.
Determining the role of gene polymorphism in the etiology of “sporadic” (non-hereditary) tumors in humans and effects of relationships between hereditary susceptibility and environmental factors is an important field of research in molecular epidemiology. Progress in this field might lead to identification of individual risk of cancer based on assessment of gene polymorphism and exposure to carcinogens in the environment and lifestyle.
The case-control study is the method of choice for investigation of the effects of gene polymorphism and the interaction between genetic and environmental factors on the risk of cancer. The size of sample of study subjects (patients and controls) is of critical importance. This is due to the extreme low probability of detection of a commonly occurring gene variant with large effect on cancer risk, or alternatively, rare genes with large effect on cancer development. To unveil such associations one needs very large studies, which include many thousands of patients and controls.
Many studies have investigated the effects of gene polymorphism on the risk of cancer in humans. However, the majority of findings from these studies have not been replicated, while the replication of results is essential for confirmation that observed associations reflect a biological process of interest. The inconsistencies in results of these studies can be explained by inadequate epidemiological design and especially by small sample size and inappropriate control group. Many of these studies lack the information on environmental and lifestyle factors. Ethnic and racial differences of study subjects have been ignored in most studies, although the majority of these studies most probably include persons of different ethnicity and race, which affects the patterns of genetic polymorphism [15, 16].
Single nucleotide polymorphisms (SNPs) are the most common type of gene polymorphism. Several millions of SNP variants have been identified. The risk of cancer associated with this type of polymorphism probably is not high, and the proportion of malignant tumors associated with a distinct polymorphism depends on the frequency of occurrence of this variant in the human population.
The early studies on the association between gene polymorphism and risk of cancer were hypothesis-oriented. They studied polymorphism of 1-2 genes, hypothetically influencing the susceptibility to cancer. Such hypothesis oriented studies have shown that polymorphism of glutathione S-transferase (GST) and N-acetyl transferase (NAT) genes affects risk of bladder cancer [17]. The progress in genetic technologies permits simultaneous analysis of many thousands of gene variants and evaluation of combined effects of rare allele variants of genes participating in various biological mechanisms of carcinogenesis on cancer risk. Introduction of new technologies, i.e. availability of whole genome scan, broadens the possibilities of molecular epidemiology. As mentioned above, “sporadic” tumors have most probably polygenic etiology, and the effect of polymorphism in 1-2 genes can scarcely play any significant role in etiology of a distinct tumor. However, the combined effect of polymorphism of genes participating in various mechanisms of carcinogenesis can be substantial.
Genes belonging to the cytochrome P450 (CYP) family are recognized as influencing the susceptibility to cancer. The enzymes of phase I (cytochromes P450) activate pro-carcinogens (PAH, heterocyclic amines, aflatoxins, nitrosamines, etc.) into reactive, i.e. carcinogenic forms. The enzymes of phase II are responsible for their detoxification and removal from the organism. Active metabolites are decomposed mainly due to their interaction with GST. Steady-state concentration of active metabolites determines the probability of cell transformation and tumor development. There are many isoforms of cytochrome P450 determining activation and deactivation of different xenobiotics. Carcinogenic effect is a result of interaction between metabolic processes resulting in activation or detoxification of carcinogens [18].
CYP1A1, CYP1A2, CYP2A6, and CYP1B1 are the main isoforms of cytochrome P450 oxidizing PAH and other xenobiotics. Polymorphism of these genes influences the activity of the enzyme and, respectively, the activity of carcinogen. A significant amount of CYP1A1 is found only in smokers. CYP1A2 is expressed in liver tissue and is induced by the same xenobiotics as CYP1A1. CYP1A2 is necessary for metabolic activation of pro-carcinogen aryl amines and heterocyclic amines formed under thermal treatment of food. CYP2A6 activates some carcinogenic nitrosamines, including TSN. Besides PAH oxidation with the formation of active metabolite, the CYP1B1 isoform oxidizes estrogens with the formation of 4-oxy-derivative, which can be easily converted to highly active metabolite inducing cell dysfunction and possibly transformation. The level of CYP1B1 isoform is relatively high in estrogen-dependent organs in women. The GST and NAT families are the most studied phase II genes [18, 19].
Apart from the genes of the cytochrome P450, genes involved in alcohol metabolism, genes of DNA repair, cell cycle, angiogenesis, apoptosis, inflammation, and many other key events of carcinogenesis can influence the susceptibility to cancer [19].
Several studies have investigated the causal association between cytochrome P450 gene polymorphism and risk of malignant tumors etiologically linked to smoking. The role of smoking in lung cancer and many other forms of cancer has been proven. However, it is known that not all smokers develop lung cancer. Cumulative risk of lung cancer in smokers (<75 years old) is 17%. Therefore, individual risk of lung cancer, in addition to smoking and other less important risk factors, is most likely influenced by genetic factors, in particular by genes responsible for metabolism of carcinogens. At equal “external” dose of carcinogen in tobacco smoke, the “internal” dose varies and depends on metabolism of pro-carcinogens, their activation and detoxification, and DNA repair [1, 9, 18].
Polymorphism of CYP1A1 leading to increase in enzymatic activity is shown to be associated with increase in the risk of lung cancer in smokers. Meta-analysis of 22 epidemiological studies (2451 patients with lung cancer and 3358 controls) revealed an increased risk of cancer in white Americans with homozygote Msp1 variant (polymorphism) of CYP1A1 (RR = 2.3, confidence interval (CI) 1.2-4.8) [20].
Meta-analysis of 14 epidemiological studies of lung cancer in non-smokers showed an increase in the risk in individuals with Ile/Val-Val/Val allele variant (RR = 3.0, CI 1.5-5.9). The influence of this allele variant was stronger for adenocarcinoma than for squamous cell cancer. No links were found between Msp1 polymorphism of CYP1A1 and risk of lung cancer in this study. An anticipated elevation of risk of lung cancer in individuals with null GSTM1 genotype was also not found. However, a combined effect of CYP1A1 (Ile/Val, Val/Val) polymorphism and null GSTM1 genotype on risk of lung cancer was discovered (RR = 4.7, CI 2.0-10.9). Risk associated with gene polymorphism did not depend on passive smoking or influence of other carcinogens. In connection with this, the authors came to the conclusion that the described gene polymorphism is most probably important in lung cancer etiology in nonsmokers [21].
A multicenter epidemiological study provided data of the effect of gene polymorphism on the risk of lung cancer in patients younger than 50 years old. Young patients were chosen because hereditary cancers are seen more frequently in young patients. SNPs in genes of phase I and II of xenobiotic metabolism were studied. Forty-five SNPs in 15 genes of phase I, 46 SNPs in 17 genes of phase II, and nine SNPs in four genes encoding nucleic acid metabolism were studied. A significant increase in the risk of lung cancer was observed in heterozygote carriers of 164 C > A and 1545 T > C alleles of CYP1A2 (RR = 1.8, CI 1.1-3.0). An elevated risk of lung cancer was observed also in persons having 47 A > C allele variant of CYP2A6 gene. The risk of lung cancer was higher in heterozygote carriers of two “high risk” alleles (CYP1A2 and CYP2A6). The risk of lung cancer was significantly decreased in the individuals having at least one “fast acetylator” allele of the NAT1 gene (RR = 0.7, CI 0.5-1.0). Similar association was found for the NAT2 gene. Both heterozygote and homozygote carriers of “fast acetylator” alleles had the decreased risk of lung cancer (RR = 0.8, CI 0.5-1.1) in comparison with homozygote carriers of “slow acetylator” alleles. Phenotype reconstruction as a result of haplotype analysis demonstrated twofold decrease in risk of lung cancer (RR = 0.5, CI 0.3-0.9) in the carriers of the combined NAT1 “fast” + NAT2 “fast” phenotype compared to those with the combined NAT1 “slow” + NAT2 “slow” acetylator phenotypes [22].
The most reproducible results were related to polymorphism of NAT1 and NAT2. Virtually all studies that demonstrated a link between the risk of bladder cancer and rate of “acetylation” [17] showed a link between the allele variants NAT1 and NAT2 and the risk of bladder cancer [17, 23], lung cancer [24, 25], and tumors in upper aerodigestive organs (UADO) [25].
It is important to note that these studies included large samples of patients. In particular, an epidemiological study of McKey et al. [25] included 2250 patients with lung cancer, 811 patients with UADO cancer, and 2700 control persons. Genotyping has been carried out of several gene groups responsible for xenobiotic metabolism in phase I and II, of which NAT1 and NAT2 genes proved to be the most important regarding their influence on the risk of malignant tumor. This study has demonstrated the effect of CYP1B1 gene polymorphism on the risk of lung cancer.
The null GSTM1 genotype occurs in 45-55% of the white population and is associated with lack of enzyme involved in detoxification and removing carcinogens and other xenobiotics from human body [26]. The influence of GSTM and GSTT polymorphism on the development of lung and UADO cancer was demonstrated at the end of last century [27, 28] and confirmed by subsequent studies [26]. The effect of null genotype was stronger for squamous and small-celled lung cancer (RR = 2.3, CI 0.9-6.1) and was weak for adenocarcinoma.
A link has been demonstrated between GSTM and GSTT gene polymorphism and risk of UADO tumors. Meta analysis of 31 case-control studies, which included 4635 cases with UADO cancer and 5770 healthy controls, demonstrated that individuals with GSTM1, GSTT1, and GSTP1 null genotypes are at increased risk of developing cancer. A significant increase in risk (p of the trend = 0.04) was found with increase in number of GST null genotypes. The individuals with null variants of all three genes, GSTM1, GSTT1, and GSTP1, had significantly elevated risk of UADO cancer (RR = 2.1, CI 1.1-3.8) [29].
The role of GSTM and NAT gene polymorphism is confirmed by the results of epidemiological case-control study of bladder cancer carried out in Italy. Smoking and, as a result, exposure to PAH and occupational exposure to aromatic amines are the main risk factors for bladder cancer. Thus polymorphism of genes involved in metabolism of these carcinogens, namely NAT, CYP, GSTM, and GSTT, may influence the risk of developing this disease. Significant elevation of bladder cancer risk was seen in individuals with null GSTM1 genotype (RR = 1.7, CI 1.1-2.6) and GSTT1 genotype (RR = 1.7, CI 1.0-3.0). The effect of this polymorphism was especially high in smokers exposed to aromatic amines at their workplaces (RR = 2.8, CI 1.1-7.1). Individuals with slowly acetylating variant of NAT2 were also at elevated risk of bladder cancer. The combination of this genotype with the exposure to aromatic amines triples the risk of bladder cancer (RR = 3.3, CI 1.1-10.0) [30].
Several studies have investigated the link between mutation in CYP1B1 gene and the risk of breast and endometrial cancer. It has been shown that there is some increase in the risk of these cancers associated with specific polymorphism of CYP1B1 in obese women [31, 32]. Some authors attribute a low incidence of breast cancer in Japan to significantly lower frequency of CYP1B1 mutation in Japanese women compared with European women [32].
The role of excessive consumption of alcohol in etiology of UADO cancer (cancer of oral cavity, pharynx, larynx, and esophagus) is confirmed by many epidemiological studies. Alcohol consumption, as well as tobacco smoking, is carcinogenic to humans (group 1) according to IARC (WHO) classification [33, 34]. The mechanism of carcinogenic effect of alcohol is unclear. One hypothesis suggests that acetaldehyde, the alcohol metabolite, is carcinogenic. In connection with this, the risk of cancer in these organs probably depends on individual patterns of alcohol metabolism. Alcohol dehydrogenases (ADH) are the enzymes catalyzing the conversion (oxidation) of alcohol to acetaldehyde. Subsequent conversion of acetaldehyde to acetate is catalyzed by aldehyde dehydrogenase (ALDH). The rate of ethanol conversion to acetaldehyde and then to acetate depends substantially on polymorphism of these genes.
Polymorphism of genes encoding alcohol dehydrogenase (ADH1B, ADH1C) and aldehyde dehydrogenase (ALDH2) and a link between polymorphism of these genes and the risk of malignant UADO tumor were analyzed in a case-control study (811 patients with cancer of oral cavity, pharynx, larynx, and esophagus and 1083 healthy controls) carried out in Russia and several East European countries (Table 2) [35].
Table 2. ADH/ALDH genotype and
risk of head and neck cancer
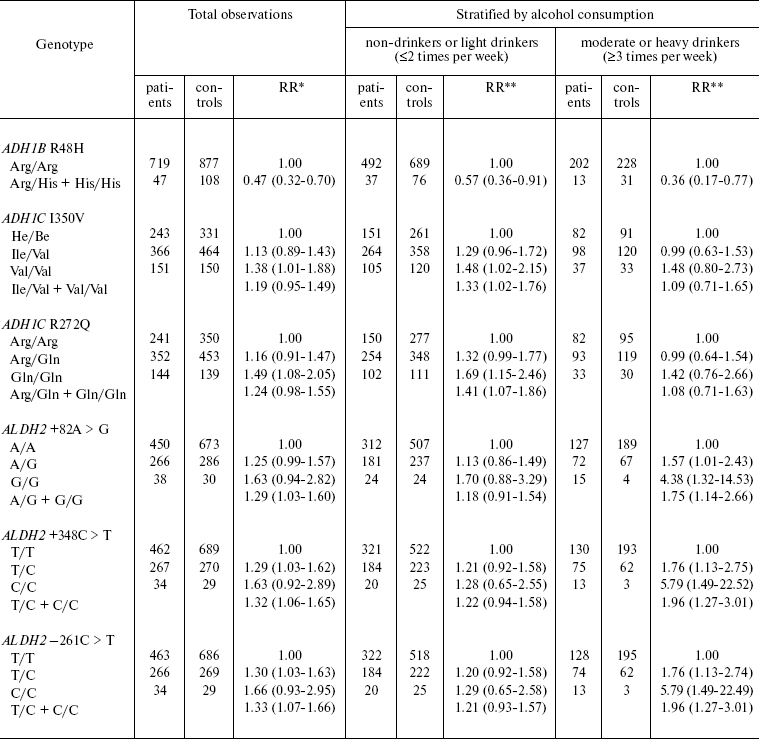
*Relative risk adjusted for gender, age, smoking, and alcohol
consumption duration. The 95% confidence interval is given in
parentheses.
**Relative risk adjusted for gender, age, and smoking. The
95% confidence interval is given in parentheses.
The carriers of rare allele ADH1B R48H Arg/His + His/His (“fast” metabolizing variant) are at decreased risk of UADO cancer (RR = 0.4, CI 0.2-0.8). The protective effect is most prominent for esophageal cancer (RR = 0.2, CI 0.1-1.5) (not shown in Table 2). Persons homozygous for ADH1C I350V Val/Val (“fast metabolizing” variant) have elevated risk of UADO cancer (RR = 1.4, CI 1.0-1.9). The risk is elevated in persons homozygous in ADH1C R272Q Gln/Gln (“fast” metabolizing variant), compared with Arg/Arg (“slow” metabolizing variant). Further analysis by alcohol consumption has shown that increase in the risk associated with the described polymorphisms is evident only in non-drinkers and light drinkers, but not in moderate or heavy drinkers.
Allelic variants ALDH2 +82 A > G, ALDH +348 C > T, ALDH2 -261 C > T are associated with increased risk of UADO cancer. The highest risk was detected in moderate and heavy drinkers homozygous for these alleles. The RR of UADO cancer in individuals homozygous for +82 G/GA > G variant is 4.4 (CI 1.3-14.5), for 348 C/C variant - 5.8 (CI 1.5-22.5), for -261 C/C variant - 5.8 (CI 1.5-22.5) (Table 2).
Statistical analysis with stratification by the level of alcohol consumption has shown that a substantial portion of UADO cancer in moderate and heavy drinkers can be attributed to these allele variants (polymorphism) of the ALDH gene. The attributable proportion of UADO cancer in individuals bearing ALDH 348 C > T allele, independently of the level of alcohol consumption, is 10% for UADO tumors and 31% for esophageal cancer. The attributable proportions for UADO tumors and esophageal cancer are 20 and 38% among moderate and heavy drinkers, respectively. In individuals with this type of polymorphism (ALDH2 348 C > T), which is present in 30% of the population studied, the attributable proportion of UADO and esophageal cancer, independently on the level of alcohol consumption, is 24 and 59%, respectively.
Based of these results a hypothesis on the mechanism of carcinogenic effect of alcohol could be formulated. Most probably carcinogenic effect is exerted by alcohol metabolite acetaldehyde. It is suggested that ADH variants encoding fast conversion of ethanol to acetaldehyde and its high concentration can result in switch-on of additional mechanism for its elimination and, respectively, low level of exposure of UADO to this putatively carcinogenic substance. On the other hand, slow conversion of ethanol to acetaldehyde favors slow accumulation of acetaldehyde and high level of exposure to this substance, respectively. While ALDH variants causing elevated risk of UADO cancer most probably prevent the conversion of carcinogenic acetaldehyde to acetate [35].
Consumption of cruciferous vegetables is known to reduce the risk of malignant tumor, particularly, the risk of lung cancer [9]. These vegetables contain isothiocyanites, substances which have the chemopreventive effect. Isothiocyanites are eliminated by GSTM and GSTT enzymes. Both GSTM1 and GSTT1 genes have null alleles with homozygous null genotypes, resulting in no enzyme being produced. Individuals who are homozygous for the inactive form of either or both genes probably have higher isothiocyanite concentrations because of their reduced elimination capacity. Thus protective effect of cruciferous vegetables must be stronger in individuals with GSTM1 null genotype, than in individuals with wild type. Brennan with coauthors [36] reported the results of case-control study of lung cancer carried out in Russia and other East European countries. A total of 2141 patients with lung cancer and 2168 controls were studied. The risk of lung cancer associated with high consumption of cruciferous vegetables was significantly decreased in individuals with null GSTM1 (RR = 0.7, CI 0.5-0.9) and GSTT1 (RR = 0.6, CI 0.4-1.0) alleles. The protective effect is stronger in individuals with null variants of both genes (RR = 0.3, CI 0.1-0.7) and respectively low level of the enzymes in circulation. The protective effect of cruciferous vegetables was absent in individuals with active GSTM and GSTT alleles (Table 3).
Table 3. Influence of GSTM1- and
GSTT1-null genotypes on the protective effect of cruciferous
vegetables

*The 95% confidence interval is given in parentheses.
Similar results were obtained in large epidemiological study of kidney cancer (including 925 patients with kidney cancer and 1247 controls). The individuals with low consumption of cruciferous vegetables (less than once per month) have elevated risk of kidney cancer (RR = 1.3, CI 1.0-1.6) in comparison with the individuals consuming more cruciferous vegetables (no less than once per week). After stratification by GSTM and GSTT variants, it was shown that largest increase in the risk was observed in persons with low consumption of cruciferous vegetables and null genotypes of GSTT1 (RR = 1.9, CI 1.1-3.2) and GSTM1 (RR = 2.5, CI 1.1-5.8). These results support the notion about the role of interaction between environmental factors and heredity, particularly gene polymorphism in etiology of cancer (“gene-environment interaction or nature-nurture”) [37].
Exposure to carcinogens results in the damage of DNA. The latter can affect cell cycle, ability of cells to grow and multiply, and finally, the transformation of normal to malignant cell. However, the majority of DNA damages are repaired. The study of the fifth codon of O6-alkylguanine-DNA-alkyltransferase gene involved in DNA repair demonstrated that the risk of lung cancer is associated with polymorphism of this gene [38].
Of four most studied gene variants participating in excision DNA repair (OGG1 Ser326Cys, XCC1 Arg194Trp, XRCC1 Arg280His, XRCC1 Arg399Gin), strongest effect on the risk of lung cancer is exerted by polymorphism of OGG1. Individuals with OGG1 Cys/Cys genotype have the elevated risk of lung cancer (RR = 1.3, CI 1.0-1.9) in comparison with those having OGG1 Ser/Ser genotype. The effect is most prominent for lung adenocarcinoma (RR = 1.7, CI 1.0-2.7). Polymorphism of other studied genes generally had no influence on lung cancer. Heavy-smokers were an exception. In this group, XCC1 Arg194Trp and XRCC1 Arg280His variants were associated with decrease in lung cancer risk [39]. Similar results were obtained in Japan and Hawaii [40, 41]. However, in two other relatively small studies carried out in Germany and Japan, no influence of OGG1 polymorphism on the risk of lung cancer was observed [42, 43].
Analysis of 102 SNP in 34 key genes regulating DNA repair demonstrated that polymorphism of genes encoding proteins involved in mismatch repair (LIG1, LIG3, MLH1, MSH6), as well as genes involved in DNA damage sensing (ATM), affects the risk of lung cancer in young persons. The strongest effect on lung cancer risk was detected in heterozygote carriers of LIG1-7 C > T variants (RR = 1.7, CI 1.1-2.6) and homozygote carriers of LIG3 rs1052536 variants (RR = 2.1, CI 1.3-3.4) [44]. Thus, genotyping of a large number of genes involved in DNA repair has demonstrated that these genes have no strong effect on the risk of cancers induced by smoking.
Evidence has been accumulated on the role of inflammation in the etiology of malignant tumors. Genotoxic free radicals and active oxygen forms are generated as a result of inflammation leading to DNA damage and, respectively, elevated probability of developing cancer. The evidence of the role of inflammation in carcinogenesis has been supported by the studies, which have shown a protective effect of non-steroid anti-inflammatory drugs, particularly aspirin, against cancer of colon, stomach, and some other organs [45, 46]. Polymorphism of genes regulating inflammation response influences the risk of some malignant tumors. Particularly, polymorphism of IL1, IL2, IL8 interleukin genes modifies the risk of stomach cancer [47-49] and UADO cancer [50], and polymorphism of cyclooxygenase (COX2) and ornithine decarboxylase (ODC) genes influences the risk of colon cancer [51].
Obesity is known as a risk factor of some cancers such as cancer of the kidney, breast, and gall bladder cancer. On the other hand, obesity was found to have a protective effect against cancers associated with smoking. Polymorphism of FTO gene has been shown to influence the body-mass index and risk of obesity. A multicenter case-control study carried out in six East European countries including Russia have shown that FTO gene polymorphism, namely its variant rs9939609, is associated with the risk of lung, kidney, and UADO cancer. A total of 7000 patients and an equal number of controls were studied. Homozygous A/A variant of this gene correlates significantly with overweight and obesity in the control group (p < 0.00001). In carriers of this genotype the decrease in the risk of lung cancer (RR = 0.8, CI 0.7-1.0) was observed. This effect was strongest for squamous cell lung cancer (RR = 0.7, CI 0.5-0.9), especially for non-smokers (RR = 0.5, CI 0.3-0.9). The carriers of A/A genotype, especially those younger than 50 years, had increased risk of kidney cancer (RR = 2.3, CI 1.1-3.4). There was no link between A/A genotype and the risk of UADO cancer. Thus, A/A variant of FTO gene is a genetic marker of obesity, which enables more precise evaluation of the role of obesity in etiology of various cancers [52].
The investigation of polymorphism of genes participating in folic acid metabolism has demonstrated that homozygote C677T variant of the gene encoding methylene tetrahydrofolate reductase (MTHFR) is associated with elevated risk of lung cancer (RR = 1.4, CI 1.1-1.7). The risk elevation is strongest in patients younger than 50 years old (RR = 1.9, CI 1.1-3.3). Stratification by the consumption of products enriched by folic acid has shown that the risk of lung cancer is higher in individuals with the described variant of MTHFR gene and with low consumption of folic acid (RR = 2.6, CI 1.4-4.9). This result confirms a hypothesis about protective effect of folate against malignant tumors associated with smoking [53].
The gene encoding checkpoint kinase (CHEK2) is one of the components of the signal transduction system from damaged DNA to various effectors. This gene product can bind to and activate p53 protein as well as other proteins. Germ-line mutations in CHEK2 have been detected in patients with Lee-Fraumeni syndrome not bearing mutations in p53. However, germ-line mutations in CHEK2 increase the risk of developing cancers associated with smoking.
The influence of CHEK2 gene polymorphism on the risk of developing cancers of the lung, kidney, and UADO (oral cavity, pharynx, esophagus, and larynx) was investigated in a case-control in Russia, Poland, and other countries of central and Eastern Europe [54]. Rare A/G genotype was revealed in 5% of control individuals (totally 2934 persons). However, the frequency of this genotype substantially varied. It was high in Russian controls (7.6%) and low in Romania (3.2%). Individuals having A/G genotype were at significantly decreased risk of lung (RR = 0.4, CI 0.3-0.6) and UADO (RR = 0.4, CI 0.3-0.7) cancers. On the other hand, they had an elevated risk of kidney cancer (RR = 1.4, CI 1.0-2.0). Significant decrease in the risk was observed only among smokers, while prevalence of smoking was very high among the patients with cancer of lung, oral cavity, pharynx, larynx, and esophagus (Table 4). The effect of having rare A/G genotype on the risk of cancer of the lung and UADO was strong for Russian (RR = 0.3, CI 0.1-0.6), Polish (RR = 0.5, CI 0.3-0.9), and Hungarian populations (RR = 0.4, CI 0.1-1.0), but was weak or absent among Czechs and Romanians.
Table 4. CHEK2 (A/G)-genotype
occurrence frequency among patients with cancer and persons of the
control group
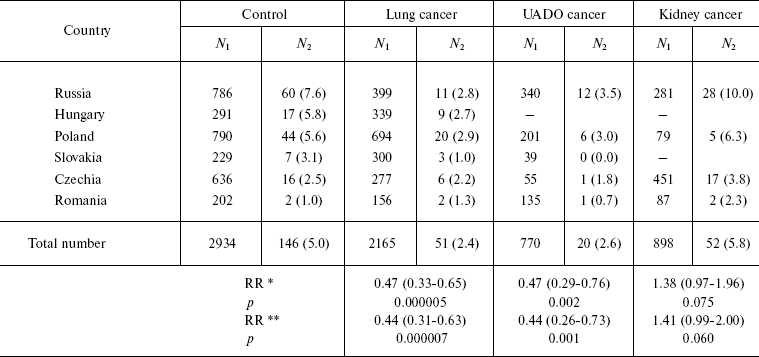
Notes: N1, total case number; N2,
number of cases with A/G; percent values of N1 and
N2 are given in parentheses.
*Adjusted to age, gender, and country; 95% confidence interval is
given in parentheses.
**Adjusted to age, gender, country, and smoking; 95% confidence
interval is given in parentheses.
One of the most promising avenues for early detection and individualization of treatment of cancer patients is the discovery of molecular and proteomic markers which are specific for certain types of cancer. Identification of such markers will favor the “isolation” of less heterogeneous molecular subcategories of tumors with identical outlook and clinical response to the therapy. Substantial progress has already been achieved in this field. Markers are identified (genes and proteins) which are typical for distinct molecular forms of tumors. These molecular forms differ from tumors of the same site and histological structure in terms of clinical outcome and response to chemotherapy. The methods of target therapy and the pharmaceuticals “targeting” specific genes have been developed. They inhibit gene function or bind to protein encoded by the gene, thus blocking the tumor growth and progression.
New technologies, such as availability of total genome scan, application of mRNA expression arrays, and new mass-spectrometry methods in proteomics enable the analysis of complex biological responses to environmental factors and modification of this response by genetic variants in the whole genome. It also becomes possible to characterize and classify tumors at chromosomal, mRNA, and proteomic levels.
REFERENCES
1.Zaridze, D. G. (1996) Arkh. Patol.,
58, 45-49.
2.Perera, F. P. (2000) J. Natl. Cancer Inst.,
92, 602-612.
3.Rothmans, N., Wacholder, S., Caporaso, N. E.,
Garcia-Closas, M., Buetoco, K., and Fraumeni, J. F. (2001) Biophys.
Acta, 1471, C1-10.
4.Garcia-Closas, M., Vermenlen, R., Sherman, M. E.,
Meore, L. E., Smith, M. T., and Roman, N. (2006) in Cancer
Epidemiology (Schottenfeld, D., and Fraumeni, J. F., eds.) 3rd
Edn., Oxford University Press, Oxford, pp. 70-88.
5.Barr, D. B., and Needham, L. L. (2002) J.
Chromatogr. B. Analyt. Technol. Biomed. Life Sci., 778,
5-29.
6.Atawodi, S. E., Lea, S., Nyberg, F., Mukeria, A.,
Constantinescu, V., Ahrens, W., Brueske-Hohlfeld, I., Fortes, C.,
Boffetta, P., and Friesen, M. D. (1998) Cancer Epidemiol. Biomarkers
Prev., 7, 817-821.
7.Godschalk, R. W. L., Feldker, D. E. M., Borm, P. J.
A., Wouters, E. F. M., and van Schooten, F. J. (2003) Cancer
Epidemiol. Biomarkers Prev., 11, 790-793.
8.Zaridze, D. G. (2004) in Carcinogenesis
(Zaridze, D. G., ed.) [in Russian], Meditsina, Moscow, pp. 29-85.
9.Pfeifer, D. H., and Hainaut, P. (2003) Mutat.
Res., 526, 39-43.
10.Le Calvez, F., Mukeria, A., Hunt, J., Kelm, O.,
Hung, R., Taniere, P., Brennan, P., Boffetta, P., Zaridze, D., and
Hainaut, P. (2005) Cancer Res., 65, 5076-5083.
11.Shigematsu, H., Lin, L., Takahashi, T., Nomura,
M., Suzuki, M., Wistuba, I. I., Fong, K. M., Lee, H., Toyooka, S.,
Shimizu, N., Fujisawa, T., Feng, Z., Roth, J. A., Herz, J., Minna, J.
D., and Gazdar, A. (2005) J. Natl. Cancer Inst., 97,
339-346.
12.Shigematsu, H., Takahashi, T., Nomura, M.,
Majmudar, K., Suzuki, M., Lee, H., Wistuba, I. I., Fong, K. M.,
Toyooka, S., Shimizu, N., Fujisawa, T., Minna, J. D., and Gazdar, A. F.
(2005) Cancer Res., 65, 1642-1646.
13.Mounawar, M., Mukeria, A., le Calvez, F., Hung,
R. J., Renard, H., Cortot, A., Bollart, C., Zaridze, D., Brennan, P.,
Boffetta, P., Brambilla, E., and Hainaut, P. (2007) Cancer Res.,
67, 5667-5672.
14.Kopnin, B. P. (2004) in Carcinogenesis
(Zaridze, D. G., ed.) [in Russian], Meditsina, Moscow, pp. 125-156.
15.Wacholder, S., Chanock, S., Garcia-Closas, M., El
Ghormli, L., and Rottiman, N. (2004) J. Natl. Cancer Inst.,
96, 434-442.
16.Marchini, J., Cardon, L. R., Phillips, M. S., and
Donnelly, P. (2004) Nat. Genet., 36, 512-517.
17.Garcia-Crosas, M., Malats, N., Silverman, D.,
Dosemeci, M., Kogevinas, M., Hein, D. W., Turdon, A., Serra, C.,
Carrato, A., Wacholder, S., and Rothman, N. (2005) Lancet,
366, 649-659.
18.Belitskii, G. A., and Turusov, V. S. (2004) in
Carcinogenesis (Zaridze, D. G., ed.) [in Russian], Meditsina,
Moscow, pp. 225-250.
19.Caporaso, N. E. (2006) in Cancer
Epidemiology (Schottenfeld, D., and Fraumeni, J. F., eds.) 3rd
Edn., Oxford University Press, Oxford, pp. 577-602.
20.Vineis, P., Veglia, F., Benhamou, S., Butkiewicz,
D., Cascorbi, I., Clapper, M. L., Dolzan, V., Haugen, A., Hirvonen, A.,
Ingelman-Sundberg, M., Kihara, M., Kiyohara, C., Kremers, P., le
Marchand, L., Ohshima, S., Pastorelli, R., Rannug, A., Romkes, M.,
Schoket, B., Shields, P., Strange, R. C., Stacker, I., Sugimura, H.,
Garte, S., Gaspari, L., and Taioli, E. (2003) Int. J.
Cancer, 104, 650-657.
21.Hung, R. J., Boffetta, P., Brockmoller, J.,
Butkiewicz, D., Cascorbi, I., Clapper, M. L., Garte, S., Haugen, A.,
Hirvonen, A., Anttila, S., Kalina, I., le Marchand, L., London, S. J.,
Rannug, A., Romkes, M., Salagovic, J., Schoket, B., Gaspari, L., and
Taioli, E. (2003) Carcinogenesis, 24, 875-882.
22.Gemignani, F., Landi, S., Szeszenia-Dabrowska,
N., Zaridze, D., Lissowska, J., Rudnai, P., Fabianova, E., Foretova,
L., Janout, V., Bencko, V., Gaborieau, V., Gioia-Patricola, L.,
Bellini, I., Barale, R., Canzian, F., Hall, J., Boffetta, P., Hung, R.
J., and Brennan, P. (2007) Carcinogenesis, 28,
1287-1293.
23.Hein, D. W. (2006) Oncogene, 25,
1649-1658.
24.Wikman, H., Thiel, S., Jager, B., Schmerer, P.,
Spiegelhalder, B., Edler, L., Dienemann, H., Kayser, K., Schulz, V.,
Drings, P., Bartsch, H., and Risch, A. (2001)
Pharmacogenetics, 11, 157-168.
25.McKay, J. D., Hashibe, M., Hung, R. J.,
Wakefield, J., Gaborieau, V., Szeszenia-Dabrowska, N., Zaridze, D.,
Lissowska, J., Rudnai, P., Fabianova, E., Mates, D., Foretova, L.,
Janout, V., Bencko, V., Chabrier, A., Hall, J., Boffetta, P., Canzian,
F., and Brennan, P. (2008) Cancer Epidemiol. Biomarkers Prev.,
17, 141-147.
26.Malats, N., Camus-Radon, A.-M., Nyberg, F.,
Ahrens, W., Constantinescu, V., Mukeria, A., Benhamou, S.,
Batura-Gabryel, H., Bruske-Hohlfeld, I., Simonato, L., Menezes, A.,
Lea, S., Lang, M., and Boffetta, P. (2000) Cancer Epidemiol.
Biomarkers Prev., 9, 827-833.
27.Jourenkova, N., Reimkanen, M., Bouchardy, C.,
Husgafvel-Pursiamen, K., Dayer, P., Benhamou, S., and Hirvonen, A.
(1997) Pharmacogenetics, 7, 515-518.
28.Jourenkova, N., Reimkainen, M., Bouchardy,
C., Dayer, P., Benhamou, S., and Hirvonen, A. (1998) Cancer
Epidemiol. Biomarkers Prev., 7, 19-23.
29.Hashibe, M., Brennan, P., Strange, R. C., Bhisey,
R., et al. (2003) Cancer Epidemiol. Biomarkers Prev., 12,
1509-1517.
30.Hung, R. J., Boffetta, P., Brennan, P.,
Malaveille, C., Hautefeuille, A., Donate, F., Gelatti, U., Spaliviero,
M., Placidi, D., Carta, A., Scotto di Carlo, A., and Porru, S. (2004)
Int. J. Cancer, 110, 598-604.
31.Kocabas, N. A., Sardas, S., Cholerton, S., Daly,
A. K., and Karakaya, A. E. (2002) Arch. Toxicol.,
76, 643-649.
32.Sasaki, M., Tanaka, Y., Kaneuchi, M., Sakuragi,
N., and Dahiya, R. (2003) Cancer Res., 63, 3913-3918.
33.IARC Monographs of the Evaluation of
Carcinogenic Risk to Humans ''Tobacco Smoke and Involuntary
Smoking'' (2004) Lyon, France, Vol. 83.
34.IARC Monographs of the Evaluation of
Carcinogenic Risk to Humans ''Alcohol Drinking'' (1988)
Lyon, France, Vol. 83.
35.Hashibe, M., Boffetta, P., Zaridze, D., Shangina,
O., Szeszenia-Dabrowska, N., Mates, D., Janout, V., Fabidnovi, E.,
Bencko, V., Moullan, N., Chabrier, A., Hung, R., Hall, J., Canzian, F.,
and Brennan, P. (2006) Cancer Epidemiol. Biomarkers Prev.,
15, 696-703.
36.Brennan, P., Hsu, C. C., Moullan, N.,
Szeszenia-Dabrowska, N., Lissowska, J., Zaridze, D., Rudnai, P.,
Fabianova, E., Mates, D., Bencko, V., Foretova, L., Janout, V.,
Gemignani, F., Chabrier, A., Hall, J., Hung, R. J., Boffetta, P., and
Canzian, F. (2005) Lancet, 366, 1558-1560.
37.Moore, L. E., Brennan, P., Karami, S., Hung, R.
J., Hsu, C., Boffetta, P., Toro, J., Zaridze, D., Janout, V., Bencko,
V., Navratilova, M., Szeszenia-Dabrowska, N., Mates, D., Mukeria, A.,
Holcatova, I., Welch, R., Chanock, S., Rothman, N., and Chow, W. H.
(2007) Carcinogenesis, 28, 1960-1964.
38.Cohet, C., Borel, S., Nyberg, F., Mukeria, A.,
Bruske-Hohlfeld, I., Constantinescu, V., Benhamou, S., Brennan, P.,
Hall, J., and Boffetta, P. (2004) Cancer Epidemiol. Biomarkers
Prev., 13, 320-323.
39.Hung, R. J., Brennan, P., Canzian, F.,
Szeszenia-Dabrowska, N., Zaridze, D., Lissowska, J., Rudnai, P.,
Fabianova, E., Mates, D., Foretova, L., Janout, V., Bencko, V.,
Chabrier, A., Borel, S., Hall, J., and Boffetta, P. (2005) J. Natl.
Cancer Inst., 97, 567-576.
40.Sugimura, H., Kohno, T., Wakai, K., Nagura, K.,
Genka, K., Igarashi, H., et al. (1999) Cancer Epidemiol. Biomarkers
Prev., 8, 669-674.
41.Le Marchand, L., Donlon, T., Lum-Jones, A.,
Seifried, A., and Wilkens, L. R. (2002) Cancer Epidemiol. Biomarkers
Prev., 11, 409-412.
42.Wikman, H., Risch, A., Klimek, F., Schmezer, P.,
Spiegelhalder, B., Dienemann, H., et al. (2000) Int. J.
Cancer, 88, 932-937.
43.Ito, H., Hamajima, N., Takezaki, T., Matsuo, K.,
Tajima, K., Hatooka, S., et al. (2002) J. Epidemiol., 12,
258-265.
44.Landi, S., Gemignani, F., Canzian, F., Gaborieau,
V., Barale, R., Landi, D., Szeszenia-Dabrowska, N., Zaridze, D.,
Lissowska, J., Rudnai, P., Fabianova, E., Mates, D., Foretova, L.,
Janout, V., Bencko, V., Gioia-Patricola, L., Hall, J., Boffetta, P.,
Hung, R. J., and Brennan, P. (2006) Cancer Res., 66,
11062-11069.
45.Zaridze, D., Borisova, E., Maximovitch, D., and
Chkhikvadze, V. (1999) Int. J. Cancer, 82, 473-476.
46.Thun, M. J., Henley, J., and Patrono, C. (2002)
J. Natl. Cancer Inst., 94, 252-263.
47.El-Omar, E. M., Carrington, M., Chow, W.-H.,
McColl, E. E. L., Bream, J. H., Young, H. A., et al. (2002)
Nature, 404, 398-402.
48.El-Omar, E. M., Rabkin, C. S., Gammon, M. D.,
Vaughan, T. L., Risch, H. A., Schroenberg, J. B., et al. (2003)
Gastroenterology, 124, 1193-1201.
49.Taguchi, A., Ohmiya, N., Shirai, K., Mabuchi, N.,
Itoh, A., Hirooka, Y., Niwa, Y., and Goto, H. (2005) Cancer
Epidemiol. Biomarkers Prev., 14, 2487-2493.
50.Campa, D., Hashibe, M., Zaridze, D.,
Szeszenia-Dabrowska, N., Mates, I. N., Janout, V., Holcatova, I.,
Fabianova, E., Gaborieau, V., Hung, R. J., Boffetta, P., Brennan, P.,
and Canzian, F. (2007) Cancer Causes Control, 18,
449-455.
51.Martinez, M. E., O'Brien, T. G., Fultz, K. E.,
Babbar, N., Yerushalmi, H., Qu, N., Guo, Y., Boorman, D., et al. (2003)
Proc. Natl. Acad. Sci. USA, 100, 7859-7864.
52.Brennan, P., McKay, J., Moore, L., Zaridze, D.,
Mukeria, A., Szeszenia-Dabrowska, N., Lissowska, J., Rudnai, P.,
Fabianova, E., Mates, D., Bencko, V., Foretova, L., Janout, V., Chow,
W.-H., Rothman, N., Chabrier, A., Gaborieau, V., Timpson, N., Hung, R.,
and Smith, G. D. (2008) Am. J. Epidemiol., in press.
53.Hung, R. J., Hashibe, M., McKay, J., Gaborieau,
V., Szeszenia-Dabrowska, N., Zaridze, D., Lissowska, J., Rudnai, P.,
Fabianova, E., Mates, I., Foretova, L., Janoyt, V., Bencko, V.,
Chabrier, A., Moullan, N., Canzian, F., Hall, J., Boffetta, P., and
Brennan, P. (2007) Carcinogenesis, 28,
1334-1340.
54.Brennan, P., McKay, J., Moore, L., Zaridze, D.,
Mukeria, A., Szeszenia-Dabrowska, N., Lissowska, J., Rudnai, P.,
Fabianova, E., Mates, D., Bencko, V., Foretova, L., Janout, V., Chow,
W.-H., Rothman, N., Chabrier, A., Gaborieau, V., Odefrey, F., Southey,
M., Hashibe, M., Hall, J., Boffetta, P., Peto, R., and Hung, R. J.
(2007) Hum. Mol. Genet., 16, 1794-1801.