
|
REVIEW: Genetic Polymorphism and Variability of Chemical CarcinogenesisG. A. Belitsky* and M. G. YakubovskayaLaboratory of Carcinogen Screening Methods, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; fax: (495) 324-1205; E-mail: gbelitsky@crc.umos.ru; belitsga@gmail.com* To whom correspondence should be addressed. |
Received December 4, 2007
Risk assessment in chemical carcinogenesis involves ratios of several factors. Individual responses of an organism to carcinogenic agents depend on polymorphism of enzymes responsible for metabolic activation/detoxification of carcinogens, DNA repair, and apoptosis, as well as promotion and progression in malignantly transformed cells. The effects of a particular polymorphic variant are manifested only in the case of its high penetrance. An integral effect is formed by the ratio of procarcinogenic and anticarcinogenic effects. The complexity of risk assessment depends on the gene polymorphism mosaic involved, directly or indirectly, in tumorigenesis and upstream/downstream interactions of gene products.
KEY WORDS: chemical carcinogenesis, gene polymorphism, cytochrome P450, DNA repair, apoptosis, promotion, initiationDOI: 10.1134/S0006297908050076
Abbreviations: CYP) cytochrome P450; HPRT) hypoxanthine phosphoribosyl transferases; MMR) mismatch repair; NER) nucleotide excision repair; PAH) polycyclic aromatic hydrocarbons; UDP) uridine diphosphate.
Despite the extreme diversity of carcinogenic agents of physical,
chemical, and biological origin, disturbances in mechanisms of cell
regulation providing autonomous existence of malignantly transformed
cells are very similar.
Chemical carcinogenesis has a number of specific features:
- the formulas of active substances and their physicochemical characteristics are usually known, and their comparison with analogous substances allows identification of molecular structures responsible for carcinogenic effect;
- carcinogens of different classes vary in the type of target cell injury and macromolecular domains, making possible the probing of malignant transformation pathways and programmed cell death;
- the majority of blastogenic compounds, referred to as procarcinogens, do not induce tumorigenesis by themselves, but are activated by enzymatic systems of the cell.
Whatever the mechanism of action of a particular carcinogen, tumor generation requires heritable changes. The latter are provoked by specific mutations and, as a consequence, by uncontrolled proliferation of different cell populations. These changes are induced either directly, e.g. upon interaction of carcinogenic metabolites with DNA, or indirectly, e.g. under conditions of oxidative stress stimulated by them. It is known that malignant transformation is not confined to mutation-related inactivation of tumor growth suppressors or activation of protooncogenes. It can be induced by stable malfunctioning of any link in the signaling pathway stimulating cell division. According to their mechanisms of action, chemical carcinogens are subdivided into genotoxic and nongenotoxic.
Chemical carcinogenesis is a complex multistage process that includes three main steps--initiation, promotion, and progression.
Initiation involves mainly intracellular events, which trigger irreversible changes in the cell genotype and determine its malignant transformation. The promotion stage includes epigenetic processes responsible for the formation of the malignant phenotype and survival of tumor cells. Progression consists of production and selection of cell clones able to compete favorably with the normal cell environment.
By binding to any accessible sites in nucleic acids and proteins, reactive metabolites induce a variety of heritable and nonheritable lesions which trigger all the three stages of carcinogenesis. This process involves an unlimited combination of variants, e.g. production of clones with impaired cell-to-cell contacts and telomerase activation without losing control over cell proliferation. Tumor growth in these cell populations is provoked by new mutations concomitant with inhibition of suppressor activity and stimulation of expression of protooncogene proteins, which occur at much faster rates in comparison with nontransformed cells. In clinical practice, this phenomenon has been coined “cancer fields”.
Tumorigenesis in cancer fields is a recurrent and continuous process. It occurs independently of radical removal of preceding neoplasms and is not a relapse of primary tumors differing from the latter in histological and histochemical characteristics.
The efficiency of each carcinogenesis stage depends on a number of factors conventionally classified as procarcinogenic and those preventing carcinogenesis. Their expression is an independent and highly variable process and determines the multivariability of the chemical carcinogenesis scenario.
In this review, we consider genetic polymorphism and relationships between the main factors of carcinogenesis induced by genotoxic xenobiotics with special reference to polymorphism of enzyme systems responsible for metabolic activation/detoxification of carcinogenic substances, DNA repair, and apoptosis. Along with carcinogen dose and exposure time, these factors determine the outcome of the malignant process.
CHEMICAL CARCINOGENS WITH A GENOTOXIC MECHANISM OF ACTION
Genotoxic carcinogens are compounds capable of inducing DNA lesions. The changes induced by them persist as mutations triggering malignant transformation of damaged cells. Carcinogenic agents are further subdivided into direct carcinogens and procarcinogens, the latter being converted into active carcinogens by cell enzymes.
Direct carcinogens decompose in aqueous solutions into highly reactive components carrying an electrophilic group with a surplus positive charge (Scheme 1). Interactions of electrophilic groups with negatively charged (nucleophilic) groups of DNA yield stable covalent bonds. This group of carcinogens includes N-nitrosoalkylurea, ethyl- and methylmethanesulfonate, N-methyl-N-nitronitrosoguanidine, sulfur mustard, diepoxybutane, beta-propiolactone, ethyleneimine, etc.
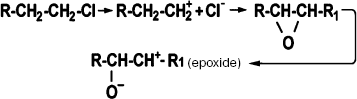
Scheme 1. Nonenzymatic activation of a direct carcinogen in aqueous medium
Procarcinogens are metabolized in the cell in two steps catalyzed by different enzymes. In the first step, procarcinogens are activated and converted into electrophilic derivatives. In the second step, metabolic products are neutralized by conjugation. The main activating enzymes include cytochrome P450 (CYP) isoforms, and, to a lesser degree, oxidases, hydroxylases, reductases, flavin-containing monooxygenases, peroxidases, epoxygenases, dehydrogenases, hydrolases, and some other enzymes. Conjugating enzymes include epoxidases, acyltransferases, sulfotransferases, glutathione-S-transferases, UDP-glucuronyl transferases, and transaminases. Activation and detoxification of carcinogens are characteristic of virtually all mammalian cells.
For some carcinogens, second-stage reactions are a necessary intermediate step and a prerequisite to further activation (Scheme 2).
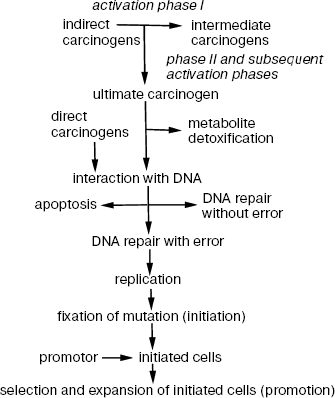
Scheme 2. Sequence of events in chemical carcinogenesis
GENETIC POLYMORPHISM OF CARCINOGEN-METABOLIZING ENZYMES
Cytochrome P450 system. Most currently known procarcinogens are hydrophobic and serve as substrates for enzymes of the monooxygenase superfamily. These enzymes are predominantly localized in the endoplasmic reticulum and contain cytochrome P450 as an effector [2]. According to standard nomenclature, enzyme families within a superfamily are designated by Roman numerals; subfamilies are designated by capital Roman letters, while isoforms are marked by Arabic numerals.
For example, CYP2D6 means cytochrome P450, family 2, subfamily D, polypeptide 6. CYP genes of all mammalian species are arranged into 18 families. The number of subfamilies in each family depends on the species.
Each CYP isoform has its own set of metabolized substrates. The same xenobiotic can be metabolized by different isoforms into similar or different metabolic products. Overlapping substrate specificity is a salient feature of enzymes belonging to the CYP1, CYP2, CYP3, and CYP4 gene families. Depending on the predominance of one or another isoform, the metabolic spectrum changes and so does the ratio of activation/detoxification products. Even in individuals belonging to the same species, all CYP isoforms are represented by a combination of variants differing in metabolic activity. Such genetic polymorphism determines, in large measure, the existence of inter- and intraspecies-, sex-, tissue-, and cell-specific differences in the sensitivity of the organism to carcinogens.
All CYP enzymes have one common feature, namely, the presence of a C-terminal conservative peptide motif, Phe-X(6-9)-Cys-X-Gly, where X is a specific amino acid. Cysteine binds to heme iron at the fifth position and takes part in the transfer of one atmospheric oxygen atom to the substrate localized in the pocket of the substrate-binding site in the enzyme active center. When this process is complete, the oxidized substrate leaves the pocket. In some cases, the lifetime of metabolites is very short (several milliseconds) by reason of their high reactivity and fast conversion into stable hydroxylated derivatives. In other cases, slow decomposition of metabolites favors their entry into the nucleus, mitochondria, and other cell organelles.
In a typical monooxygenase reaction catalyzed by CYP isoforms, one oxygen atom interacts with the substrate (RH), while the other is reduced to H2O (Scheme 3).
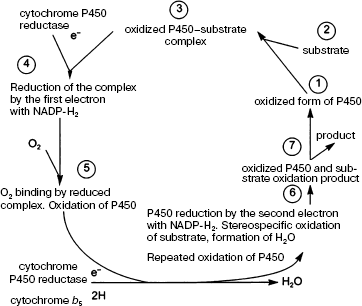
Scheme 3. Substrate oxidation in the cytochrome P450 system [1]
The most hydrophobic substrates, e.g. PAH, polychlorinated biphenyls, and dioxin-like compounds, are oxidized by CYP1 (CYP1A1, CYP1A2, and CYP1B1). More hydrophilic compounds (nitrosoureas, aminoazo dyes, biphenyls, fluorenes, and heterocyclic amines) are metabolized by CYP1A1 and CYP1B1 [2, 3].
Many carcinogens including environmental pollutants are oxidized by CYP2C8, CYP2C9, CYP2C19, CYP2E1, CYP3A4, CYP3A5, CYP2A6, and CYP2B6. CYP2E1 activates predominantly small molecules, e.g. benzene, vinyl chloride, etc., while CYP3A4 and CYP3A5 activate larger molecules such as aflatoxins.
Other CYP families influence carcinogenesis indirectly, i.e. via metabolism of sex hormones, corticosteroids, cholesterol, bile acids, retinoic acid, etc. Mutations in the corresponding genes promote carcinogenesis and progression of tumors (Table 1).
Table 1. Cytochrome CYP isoforms
metabolizing carcinogens in humans
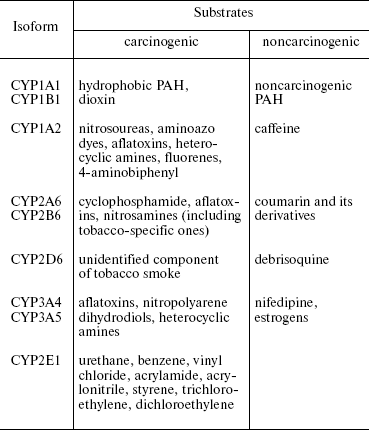
Expression of some alleles of CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP2A6, and CYP2B6 genes correlates with an elevated risk of cancer. However, attempts to identify specific carcinogens or group of carcinogens have failed.
Genetic polymorphism of CYP. The human CYP1A1 gene possesses at least 12 alleles, gene CYP1B1 at least 24, and gene CYP1A2 more than 33. In the human population, the activities of individual isoforms of CYP differ greatly. In liver tissues, individual activities of CYP1A2 isoforms vary more than 100-fold [3].
Increased basal level of CYP is a result of amplification or polymorphism of the gene of receptor protein AhR. The human population contains both metabolizers with superhigh enzymatic activity and up to 12 copies of the same gene and nullers with zero enzymatic activity (Table 2). About 10 mutations in the AhR gene able to modulate CYP activity have been identified. In particular, enhanced CYP activity was observed in case of mutation in the coding region of AhR (G1721A allele). Interestingly, in males this effect is more pronounced than in females [4, 5]. Individual differences between activities of CYP enzymes were established by British pharmacologists in patients with hypertension receiving debrisoquine therapy. In some patients, normal drug doses caused drastic decreases in arterial pressure, while other patients were insensitive even to superhigh doses of debrisoquine. It was found that different sensitivity was a result of different activity of the debrisoquine-oxidizing enzyme (CYP2D6). Later such discrepancies were revealed for the metabolism of other substrates.
Table 2. Polymorphism of some human CYPs [4]

The differences between induced levels are more crucial for carcinogenesis, since the basal (constitutive) level is either very low or indeterminable, as is the case with CYP1A1. Therefore, the inducibility of individual isoforms is a critical factor determining cell-, tissue-, species-specific, and individual differences in cell responses to substrate, stress, or physical factors.
Increasing activity of CYP during substrate exposure is mediated by different mechanisms--transcriptional (de novo synthesis of enzymatic proteins), posttranscriptional (mRNA stabilization), and posttranslational (protein stabilization, enzyme modification). The first, i.e. transcription, entails CYP1 elevation in response to PAH, CYP2B1/2 elevation in response to phenobarbital, and CYP3A1 elevation in response to dexamethasone. The stability of CYP1A2 mRNA is provided by 20-methylcholanthrene, while stabilization and modification of the enzymatic protein CYP2E1 are modulated by ethanol.
MECHANISM OF CYP1 INDUCTION
The mechanism of induction of de novo synthesized CYP1 following penetration of a hydrophobic ligand into the membrane lipid bilayer and binding to the Ah receptor (AhR) has been studied in rather great detail. AhR is a transcription factor activated by a vast variety of ligands, e.g. PAH, dioxin, polyhalogenated aromatic hydrocarbons, indoles, plant benzoflavonones, some endogenous tryptophan derivatives, and low-density lipoproteins [6]. In endoplasmic reticulum, the Ah receptor exists in the inactive form within a complex with heat shock protein HSP90 and the src gene product pp60 and has an optimum configuration for ligand binding. After the binding is complete, HSP90 and pp60 are cleaved off, and the receptor-ligand complex undergoes phosphorylation with concomitant binding of the transport protein ARNT (Ah receptor nuclear translocator). After transfer of the complex to the nucleus, positively charged protein groups come into a reaction with the xenobiotic-responsive element (XRE enhancer) thus initiating the transcription of CYP1 mRNA. Activation of procarcinogens can also occur independently of CYP induction via the Ah receptor. In particular, in knockout Ahr(-/-) mice, in which benz(a)pyrene-induced CYP1 synthesis is absent, the content of benz(a)pyrene metabolites and DNA adducts markedly exceeds that in Ahr(+/-) and Ahr(+/+) mice [7]. Paradoxically, Ahr(-/-) mice are resistant to carcinogenic effects of PAH and dioxin [3]. Quite probably, along with activation of ligand-metabolizing enzymes, this receptor is involved in other carcinogenesis-inducing processes, e.g. regulation of the cell cycle and apoptosis, suggesting that its knockout results in inhibition of some essential steps of carcinogenesis (Scheme 4) [8].
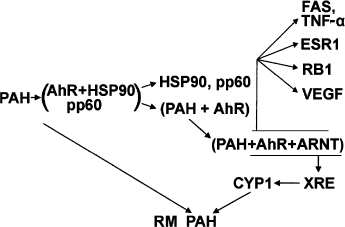
Scheme 4. Mechanism of induction of CYP1
Another paradoxical situation is related to the knockout of Cyp1a2. It is known that CYP1A2 activates many human carcinogens including 4-aminobiphenyl. Cultured cells expressing this enzyme produce large amounts of carcinogenic adducts and manifest pronounced mutagenic activity. It would be natural to suppose that CYP1A2 knockout mice are resistant to these carcinogens, but, curiously enough, they were found to be sensitive to it [3]. The responses of Cyp1a1(-/-) and Cyp1a1,Cyp1b1(-/-) mice to benz(a)pyrene were also found to be paradoxical. Instead of showing resistance to the carcinogen, these mouse strains were more sensitive to its immunotoxic effect and accumulated much more benz(a)pyrene adducts than their wild strain counterparts [9]. Further studies showed that at the organismic level CYP1A1 whose activity is the highest in liver and intestine is responsible for metabolic detoxification, while CYP1B1, which is active in spleen and bone marrow, is responsible for metabolic activation of this carcinogen. In the absence of CYP1A1, CYP1B1 expression causes death of immune cells. These findings suggest that the level of toxic and, probably, carcinogenic effects of benz(a)pyrene is controlled by the balance of tissue-specific expression of CYP1A1 and CYP1B1. The results of experiments with tissue cultures where both isoforms converted benz(a)pyrene into genotoxic metabolites do not fully reproduce the real situation in vivo, which suggests that in vivo testing of compounds for carcinogenic activity has an advantage compared to those in vitro and in tissue culture.
Endogenous substrates, e.g. steroids, are also used as a source of carcinogenic metabolites, especially during the formation of mutant forms of CYP enzymes. The latter metabolize steroids by the epoxide-diol pathway with the formation of intermediate epoxide possessing mutagenic activity. Genotoxic derivatives of CYP1 can also be formed via lipid peroxidation.
ENZYMES AND CONJUGATION REACTIONS
Reactions of the second stage of xenobiotic metabolism are characterized by formation of hydrophilic conjugates that are easily excreted from the organism. These reactions are catalyzed by specific conjugation enzymes predominantly localized in the cytoplasm. Metabolic activation products of hydrophobic procarcinogens undergo a series of intracellular conversions resulting in complex conjugates. As mentioned previously, for the majority of xenobiotics this pathway includes detoxification, whereas for certain procarcinogens it is an essential link in the subsequent activation cascade.
Epoxide hydrolases catalyze the conversion of epoxides into transdihydrodiols. The latter bind to glucuronic acid, glutathione, or sulfate and are excreted from the organism as neutral complexes. In contrast, benz(a)pyrene 7,8-oxide hydration to dihydrodiol and CYP1-catalyzed formation of diol-epoxide result in activation of the molecule and its conversion into a carcinogenic metabolite, (+/-)trans-7,8-dihydrodiol-9,10-epoxide, and quinones inducing single-stranded breaks in DNA (Scheme 5). Under the action of epoxyhydrolases, benzene is converted into o- and p-benzoquinones--metabolites causing leukemia [10, 11]. The activities of these enzymes in the human population differ tenfold and more. This circumstance is of crucial importance for individual sensitivity to these carcinogens.
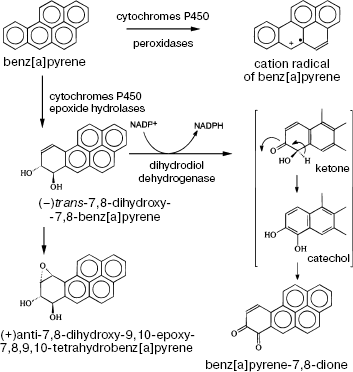
Scheme 5. Metabolic activation of benz[a]pyrene
Glutathione-S-transferases catalyze glutathione transport to electrophilic compounds, e.g. oxyarenes, alkene epoxides, nitronium and carbonium ions, and free radicals, with subsequent neutralization of active metabolites (benzene, aflatoxin, PAH, etc.) and activation of methylene chloride or dibromoethylene [12].
UDP glucuronyltransferases inhibit the activity of many electrophilic compounds, but activate heterocyclic derivatives of amines. In the first step of their metabolism, they are oxidized at the nitrogen atom to be further converted into N-glucuronides. In the gastrointestinal tract, these complexes are hydrolyzed by p-glucuronidase to highly reactive electrophiles--N-acetoxyarylamines. It is thought that these compounds play a crucial role in the etiology of colon carcinomas in humans.
Acetyltransferases acetylate amino groups of arylamines and hydrazines and play a crucial role in their activation and/or detoxification. Under these conditions, 2-aminofluorene is activated, while 2-naphthylamine and benzidine undergo detoxification. Its isoforms are formed in the course of mutations in NAT2 or NAT1 and vary in activity tens of times. The slow acetylation phenotype is a result of mutations in NAT2; its activity and stability decrease under these conditions. Specific mutations in NAT1 increase the intensity of the acetylation process; therefore, within the human population one can distinguish individuals with fast and slow acetylation capacity. In the former, occupation-related 2-naphthylamine- and benzidine-induced urinary bladder carcinomas occur much less frequently than in the latter.
Sulfotransferases are represented by multiple isoforms that vary in activity. In the majority of cases, sulfate formation is accompanied by suppression of pharmacological and carcinogenic activities of xenobiotics. The opposite effect is observed less frequently and is accompanied by the formation of unstable sulfate conjugates whose decomposition yields strongly electrophilic products. Sulfoconjugation is an indispensable step in the activation of many procarcinogens and promutagens, e.g. acetylaminofluorenes, arylamines, etc.
POLYMORPHISM OF FACTORS CONTROLLING GENETIC LESIONS. APOPTOSIS
AND REPAIR
The binding of direct carcinogens and electrophilic procarcinogenic metabolites to all kinds of nucleophilic centers is accompanied by disturbances in various cell structures essential for their vital activity. The ability of these compounds to bind to DNA and to form adducts, DNA crosslinks, and modified bases and to hydrolyze N-glycoside and phosphodiester bonds [13] plays an important role in carcinogenesis. In particular, O6-methylguanine formation is induced by tobacco smoke, endogenous peroxidation products, chemotherapeutic drugs (e.g. procarbazine and streptozocin), etc. Very often, alkylating agents methylate bases at positions 3 and 7 of the nitrogen atom resulting in N3-methyladenine and N7-methylguanine. O6-Chloroethylation is largely mediated by antitumor monofunctional chloroethylamines. Benz(a)pyrene diol-epoxides forms bulky adducts with a guanine at position N7; cisplatin analogs and bifunctional chloroethylamines produce DNA crosslinks.
In cells exposed to intensive attacks of chemical carcinogens, one out of two processes is initiated: apoptosis (programmed cell death) associated with severe lesions of DNA; repair, which restores the damaged structure of DNA. However, in case of insufficient activity of repair enzymes, perfect DNA repair may be impossible and mutations will occur.
APOPTOSIS
Almost all lesions induced by chemical carcinogens result in apoptosis. The level of intensity of DNA lesions required for triggering apoptosis is determined by components of the p53-dependent pathway; their polymorphism determines susceptibility to cancer. With a decrease in intensity of apoptosis initiation, a greater number of cells with sufficiently intensive DNA lesions are switched to repair. The efficiency of this system is inversely proportional to the number of damaged DNA sites [14, 15].
Analysis of 82 genes coding p53-dependent signaling pathway proteins established single-nucleotide polymorphism at more than 1000 positions [16]. The mechanisms whereby polymorphism influences chemical carcinogenesis in the majority of these positions are poorly understood. However, the relatedness of some of these polymorphisms to cancer has been established. Thus, single-nucleotide polymorphism of the 309th codon in the first intron of Mdm2 is associated with enhanced expression and high risk of esophageal, uterine, lung, and renal cancer [16].
Programmed cell death can be induced not only by direct attack of DNA by some carcinogen. Along with DNA lesions, UV irradiation and alkylating reagents activate epidermal growth factor receptors. The latter activate signaling pathways, e.g. ras-MAPK, PI3K, SAPK, or STAT and induce DNA-independent responses to DNA lesions triggering the MAP kinase cascade resulting in apoptosis [15].
The initial polymorphism of p53 and its signaling pathways plays a crucial role in chemical carcinogenesis. Inhibition of its regulatory mechanisms and inactivation of p53 induced by carcinogen metabolites favor enhanced proliferation and accumulation of genetically modified cells. The pattern of p53 mutations is determined by the carcinogen type. Mutations in the 249th codon are specific for aflatoxin B1; mutations in the 145th codon are more typical of tobacco smoke carcinogens [14, 17].
POLYMORPHISM OF DNA REPAIR ENZYMES
The type of DNA damage by carcinogens determines the mechanism of its repair (Scheme 6). Single-stranded breaks are repaired via restoration of broken phosphodiester bonds by DNA polynucleotide ligases. Several different enzyme isoforms have been identified, but their relatedness to susceptibility to cancer-associated diseases is not established yet [18].
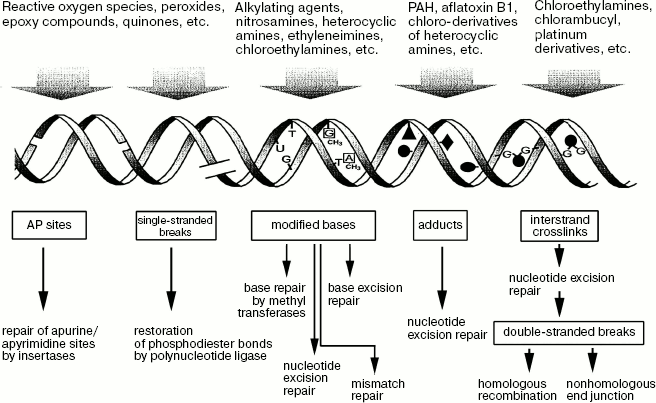
Scheme 6. Variants of DNA repair
Repair of apurine and apyrimidine (AP) sites is accomplished by special enzymes called insertases [19, 20] or by nucleotide excision [19], which involves recognition of DNA lesions by deoxyribonuclease I and disruption of neighboring phosphodiester bonds. This is followed by restoration of the excised fragment by DNA polymerase with the undamaged complementary chain used as a template. Then polynucleotide ligase restores phosphodiester bonds in the repaired fragment. Five human deoxyribonuclease I isoforms have been identified; the presence of two of them correlates with susceptibility to gastric and colon cancer [21].
Attacks of electrophilic compounds result in various modifications of bases. The formation of N7-methylguanine, O6-methylguanine, and O4-alkylthymine in response to alkylating agent attacks is an illustrative example. Carcinogenic effects of methylating agents correlate with concentration and persistence of O6-methylguanine; the lack of repair stimulates the GC → AT transition. O6-Methylguanine and O4-alkylthymine repair is provided by methyltransferases able to entrap methyl groups and restore the damaged structure of DNA. However, the methyl groups entrapped by methyltransferases are retained on the protein molecules; therefore, each repair cycle utilizes a new protein molecule synthesized by the cell. If DNA damage occurs faster than the synthesis of new protein molecules, cell mutations may occur. Polymorphism of O6-methyltransferase and correlation between susceptibility to cancer and protein type were recently established for the human population [22].
Excision repair, the most frequent type of the repair of base modifications, entails recognition of a modified base by various glycosylases cleaving an N-glycoside bond of this base with a sugar-phosphate backbone of a DNA molecule. Some glycosylases can specifically recognize modified bases of DNA, e.g. oxymethyluracyl, hypoxanthine, 5-methyluracyl, 3-methyladenine, 7-methylguanine, etc. Polymorphisms of many currently known glycosylases involve substitution of one of the nucleotides in the coding sequence of the corresponding gene and are described in the literature. Some isoforms of these enzymes are associated with a high risk of cancer [23]. Genetic polymorphism of enzymes responsible for the terminal step of DNA repair, e.g. deficiency of DNA polymerase beta or ligase III, strongly increases the cytotoxic activity of these adducts [24, 25]. Glycosylases initiate the formation of AP sites; their repair mechanisms are described in the foregoing sections.
If a modified base is not excised from DNA and replication induces the appearance of new noncanonical nucleotide pairs, the mismatch repair (MMR) mechanism is turned on by enzymes responsible for recognition of duplexes with impaired structure and their excision at special sites. This repair system includes several functionally important proteins--MLH1, MSH2, MSH3, MSH6, and hPSM2. The carcinogenic activity of some mutagens depends on functional activity of MMR enzymes and can be attributed to different abilities of some protein components of this enzymatic system to trigger apoptosis [15]. The polymorphism of some MMR enzymes is also manifested in increased mutagenic activity of some carcinogens with a decrease in MLH1. Furthermore, the restoration of DNA structure by modified bases can occur as nucleotide excision repair (NER). The latter is mediated by enzymes and involves about 30 proteins and protein complexes. The most important of them are XPA, XPB, XPC, XPD, and XPG proteins named from the hereditary pigment xeroderma syndrome. The latter is manifested in hypersensitivity of the skin to carcinogenic effects of UV irradiation due to ability of repair enzymes to suppress the activity of cyclobutane dimers. In healthy donors, the activity of these proteins varies widely. For example, after UV irradiation the repair capacity of leucocytic DNA varies more than 5-fold [26].
Covalent binding of some carcinogen derivatives to DNA is concomitant with pronounced structural disturbances in the duplex and transcription and replication arrest. Adduct levels in a tissue and duration of their existence can be used as parameters characterizing individual sensitivity to carcinogens. The highest content of carcinogenic adducts in tobacco smoke was found in lymphocytes and lung tissues of cancer patients with a short smoking history. In female smokers, the adduct content is higher than in males, which is consistent with provisional epidemiological data on higher sensitivity of female smokers to tobacco smoke carcinogens. Individual differences are such that the difference between the content of benz(a)pyrene adducts reaches 75-fold in bronchial tissue, 70-fold in the urinary bladder wall, and 100-fold in the esophagus [27]. Monitoring of DNA adduct levels in individuals with high occupational risk of cancer is necessary for predicting morbidity from cancer and strict control over observance of safety rules. Interestingly, in nurses in charge of infusion of chemotherapeutic drugs to cancer patients, blood lymphocyte levels of DNA adducts vary widely depending on professional skill, neatness, and discretion.
Under the action of bifunctional alkylating reagents, DNA forms so-called crosslinks; their excision repair results in double-stranded breaks. Repair of these DNA lesions is achieved via homologous somatic recombination or junction of nonhomologous ends. The first approach enables complete restoration of the original sequence using homologs or sister chromatids; small recombination initiation sites subjected to genetic conversions are excluded from the process. However, if the recombination occurs with the involvement of nonsister chromatids, e.g. during inactivation of suppressor genes responsible for tumor growth, heterozygosity can be lost, giving rise to recessive mutations in interacting loci. The main components of this enzymatic system are RAD51, XRCC2, and XRCC3 [19]. Single-nucleotide polymorphism of RAD51 and XRCC2 is more common; effects of their isoforms on the incidence of urinary bladder tumors were strongly supported by experimental data [28]. So-called nonhomologous end junction (NHEJ) is yet another alternative approach to the repair of double-stranded breaks; in this case, cell viability is preserved at the expense of a new combination of genes. However, the use of this technique often results in malignant transformation [19].
CARCINOGENESIS PROMOTERS AND NONGENOTOXIC CARCINOGENS
The concepts “carcinogenesis promoters” and “nongenotoxic carcinogens” often coincide. The former came into use in the late 1940s and takes its origin from the Berenblum model of two-phase carcinogenesis [29]. In this model, noncarcinogenic crotonic oil induces skin tumors in mice preliminarily treated with low doses (inefficient in separate treatment) of 7,12-dimethylbenz(a)anthracene. Promoters must be used in high doses and, what is especially important, continuously, without intervals, since long-term discontinuance results in carcinogenesis arrest and even regression of primary tumors. Promoters stimulate cell proliferation, inhibit apoptosis, and interfere with cell-to-cell contacts.
Mass-scale screening of novel chemical substances for carcinogenic activity revealed a new heterogeneous group of compounds that induce tumorigenesis in chronic experiments with rodents, but were inactive in prescreening genotoxic tests. The recently introduced term “nongenotoxic carcinogens” indicates lack of characteristic features in genotoxic carcinogens rather than change in their properties. The main distinguishing feature of this class of carcinogens is inability for covalent binding to DNA and induction of DNA lesions or mutations. The lack of mutagenic or clastogenic effects of model systems does not imply their absence in target tissues. More recent studies based on the use of other experimental procedures established that in addition to other capabilities, some representatives of this class of carcinogens are weakly mutagenic. Their mutagenicity is a result of genomic injuries induced by reactive oxygen species generated from phorbol esters, organic peroxides, or CYP inducers [2, 30]. In the latter case, hydrogen peroxide and hydroxyl radicals are formed upon substrate oxidation during decomposition of the cytochrome P450-substrate-superoxide complex; the superoxide anion liberated thereupon undergoes dismutation [1, 31]. Protection against peroxides is provided by peroxyredoxins; their depletion in the course of peroxide inactivation is compensated by sestrins controlled by p53. Inactivation of p53 results in disturbances in sestrin expression, which strongly enhances the genotoxic effect of peroxides generated by CYP and other sources [32]. All other mechanisms entail spontaneous initiation promotion, i.e. mutations. They include inhibition of apoptosis, disturbance of intercellular gap junctions, cytotoxicity with stable mitogenic effect, damage of signaling cascade molecules, carcinogen-receptor complex formation, and direct stimulation of division of cells of various types. The latter also refers to reactive oxygen species known as triggers of transmission of mitogenic signals either at the level of the c-src gene product or indirectly (via NF-kappaB).
MicroRNA is a novel factor whose role in carcinogenesis is in the focus of attention of investigators. Recent studies demonstrated the ability of this novel class of noncoding RNAs to control growth, differentiation, and apoptosis by repression or derepression of the corresponding genes. As microRNAs control the activity of more than one gene, they might regulate the activity of up to 30% of protein-coding genes. Some microRNAs, e.g. let-7 gene cluster transcripts, function as suppressors. Thus, microRNA-84 inhibits the expression of RAS by direct binding to Ras RNA. MicroRNA-15a and microRNA-16-1 are widely known as tumor growth suppressors in patients with leukemias. These compounds inhibit the expression of the antiapoptotic protein BCL2 responsible for viability of tumor cells. MicroRNAs manifest oncogenic activity during inhibition of functional activity of suppressor cells or inactivation of oncogene inhibitors [33, 34].
Obviously, mutations induced by chemical carcinogens in microRNA-encoding gene clusters may have a pronounced effect at all the stages of carcinogenesis.
PROMOTER EFFECTS OF CYP AND THEIR RECEPTORS
It is well known that genotoxic carcinogens induce tumor growth without additional stimulation and regulate the three stages of carcinogenesis--initiation, promotion, and progression. Suggestively (though not sufficiently experimentally proved), this is mediated by carcinogen derivatives formed during its metabolism. At the same time, activation of CYP-specific enzymes plays a crucial role in carcinogenesis, particularly in mitogenesis, inflammation, cell-to-cell contacts, chemotaxis, etc. [2]. Many enzymes of the CYP1, CYP2, CYP3, and CYP4 gene families are involved in eicosanoids metabolism--arachidonic acid conversions into prostaglandins, metabolism of omega- and omega-1 fatty acids, leukotrienes, etc. This is significant for the inflammation process, which frequently manifests the first stage of chemical carcinogenesis [35, 36]. By hydroxylating estrogens, CYP1A2 and CYP1B1 enzymes modulate their activity and stimulate tumorigenesis in hormone-dependent tissues. Moreover, some isoforms of enzymes expressed by CYP2, CYP3, and CYP4 genes are actively involved in metabolic conversions of testosterone, bile acids, cholesterol, and other derivatives of endogenous substrates endowed with promoter activity.
Specific receptors of xenobiotic-metabolizing enzymes are directly involved in regulation of intracellular processes (Scheme 4). The activated AhR complex (AhR + ligand + transport protein) regulates apoptosis in different parts of the signaling chain via the proapoptogenic receptor FAS, retinoblastoma protein RB1, tumor necrosis factor TNF, etc. [37]. By binding to RB1, AhR increases its ability to inhibit E2F-dependent transcription and prevents the transition from the G1 to the S phase. The binding of the hypophosphorylated form of RB protein to AhR takes place only after its activation and transfer to the cell nucleus. In this case, AhR acts as a sensor triggering signals of cell division arrest during interaction of the cell with exogenous xenobiotics [38].
Independently of CYP1 induction, binding of activated AhR to the estrogen receptor (ESR1) changes the hormonal status of the organism and regulates the growth of hormone-dependent tumors [39, 40]. In addition, AhR inhibits hypoxia-induced expression of vascular endothelium growth factor (VEGF). Ligation of the femoral artery in mice with Ah receptor knockout results in enhanced (in comparison with wild strains) production of the hypoxia inducible factor 1alpha (HIF-1alpha) and the transport protein ARNT and stimulates the binding of the HIF-1alpha-ARNT complex to the gene promoter VEGF. Seemingly, the Ah receptor can also be a negative growth regulator of tumors, which depend on vascularization [41]. All CYP receptors (except AhR) belong to the superfamily of nuclear hormone receptors, which mediate transcription and regulate gene expression in target cells and tissues. Thus, the CYP2B system is functionally coupled with tumor necrosis factor alpha (TNF-alpha) cytokine [2]. The farnesoid X-receptor, regulating CYP7A1- and CYP8A1-mediated synthesis and transport of bile acids, participates in immune responses, and its knockout in mice causes hepatomas [42].
In macrophages, which produce mediators of inflammation and activate signaling pathways of the inflammation process (which is important for carcinogenesis), hepatic X-receptors regulate CYP7A1-mediated metabolism of cholesterol. Their agonists inhibit inflammation and growth of prostate and breast adenocarcinomas [43, 44]. A crucial role in immune responsiveness is played by vitamin D3 receptor, whose ligands are metabolized by CYP24A1 and CYP27B1. These ligands inhibit maturation of dendritic cells, activate T-cell responses, and play a role in antigen-specific tolerance [45].
If regulatory pathways of AhR and nuclear receptors somehow intersect, then activation of one of them may change many parameters essential for carcinogenesis, first of all, the immune status of the body.
NONGENOTOXIC CARCINOGENS
The relationship between genetic polymorphism and efficiency of nongenotoxic carcinogens have been studied relatively little. Among a vast array of carcinogens, signaling pathway “venoms”, i.e. inhibitors or activators of enzymatic systems involved in regulation of transmission of mitotic signals, present special interest. For example, carcinogenic activity of okadaic acid is coupled with its ability to inhibit phosphatases PP1 and PP2A phosphorylating major signaling proteins, e.g. Src, c-Jun, p56, and NF-AT. These proteins are inactive in the phosphorylated state, but are activated after dephosphorylation. Inhibition of phosphatases results in generation of a constant cell proliferation signal. The same mechanism is implemented by another nongenotoxic carcinogen--cyclosporin--a selective inhibitor of phosphatase PP2B.
Calcium exchange inhibitors can also provoke uncontrolled cell proliferation, since calcium ions are indispensable components of the cell proliferation cascade. Hormones act as promoters of carcinogenesis by virtue of their specific physiological activity and ability to induce excessive proliferation of sensitive cells.
Cytotoxic agents, by causing mass-scale cell death, thus induce proliferation of surviving cells (which may include transformed ones). If during this process intercellular gap junctions are disturbed, then a potentially malignant cell may escape from proliferative control of surrounding normal cells. Disturbances in intercellular contacts and inhibition of apoptosis are the basic mechanisms whereby phorbol-like promoters exert their effect. Until recently, compounds inducing oxidative stress and formation of reactive oxygen species responsible for DNA damage and 8-hydroxyguanine synthesis were attributed to the group of nongenotoxic substances. However, it would be more appropriate to refer them to the group of genotoxic carcinogens with indirect mechanism of action and low activity. In macrophages, “oxygen burst” is induced by phorbol esters, mineral fibers, CYP inducers, agents increasing the number of mitochondria in cells, peroxisome proliferators, and organic peroxides.
PREDICTION OF INDIVIDUAL SENSITIVITY TO CHEMICAL
CARCINOGENS
As mentioned in the foregoing sections, so-called “high penetrance factors” (carcinogen dose, ratio between activating and detoxifying enzymes, DNA repair, etc.) are the main elements of carcinogenesis [2]. Earlier, it was thought that these processes are determined by single genes. Now, it has become evident that the outcome of carcinogenesis is determined by many genes and modifying agents.
Tobacco smoking is a unique model for the study of polymorphisms of high penetrance factors and their effects on chemical carcinogenesis in human beings. The large body of evidence accumulated thus far allows comprehensive analysis of the role of even very rare genetic polymorphism variants and contributions of their metabolic products.
The 1980s were marked by the discovery of a positive correlation between inducibility of individual CYP and effect of chemical carcinogens. Analysis of PHA- and PAH-treated blood lymphocytes from smoking individuals with a high level of CYP1A1 and CYP2D6 induction revealed that the risk of lung cancer significantly exceeded the control level [46]. Of substantial interest is the situation with CYP2D6: in smoking patients with high activity of this isoform, the incidence of lung cancer is four times higher than in patients with low activity of CYP2D6. Furthermore, in cases where smoking was concomitant with asbestosis this difference was as high as 18-fold [47]. Considering that CYP2D6 metabolizes none of the currently known procarcinogens (including those formed on tobacco burning), one can suggest that tobacco smoke contains a hitherto unknown procarcinogen activated by this isoform.
The risk of lung cancer is the highest in smokers in whom specific combinations of alleles stimulate procarcinogenic activity and inhibit conjugation of active metabolites. In smokers with mutant GSTP1 and CYP1A1 alleles and homozygosity in wild myeloperoxidase alleles, the risk of lung cancer is fivefold, and that in individuals with combinations of CYP1A1*2B or CYP1A1*4 alleles and deletions in GSTM1 and GSTT1 is eightfold [48, 49]. In lung cancer patients the ability of lymphocytic DNA to repair benz(a)pyrene diol-epoxide-induced lesions in culture is lower compared to the adequate control [50]. The development of novel efficient genotyping protocols sheds additional light on the role of individual DNA repair enzymes in this process. Owing to its multicomponent composition, tobacco smoke initiates all possible forms of DNA lesions. The polymorphism of excision repair genes responsible for the recovery of small and crude DNA lesions has been investigated. Unfortunately, epidemiological studies into the role of MMR and NER in tobacco-induced carcinogenesis have given contradictory results, since genetic polymorphism established in these studies either correlated with a high or low risk of lung cancer or was insignificant [51, 52]. Recent studies revealed that APE1 Asp148Glu polymorphism in Europeans significantly increases the risk of non-small cell lung cancer in smokers and XRCC1 Arg399Gln and XPD Lys751Gln polymorphism in nonsmokers and light smokers. Paradoxically, in heavy smokers the last variant of polymorphism did not correlate with the risk of lung cancer. Significant reduction of cancer risk was associated with heterozygosity in XRCC1 Arg194Trp, Arg280His, and OGG1 Ser326Cys. It is not excluded that association of certain types of polymorphism with individual sensitivity to tobacco smoke has racial features. For example, within the Chinese population increased risk of lung cancer is associated with polymorphism of the XRCC1 77 T/C promoter [53, 54].
Some authors consider the effect of mononucleotide polymorphism of repair genes as insignificant, though they admit an increased risk of lung cancer in the case of ERCC2 751Gln/Gln genotype and decreased risk in the case of XPA 23G/G genotype [55]. Apparently, direct correlation between individual polymorphism variants and smoking can be established only under conditions of their high penetrance. In the majority of cases, modification of carcinogenic effects is a result of cooperative interactions of a vast array of genes responsible for mutations or bringing the cell to apoptosis.
A crucial role in carcinogenic effect of tobacco smoke is ascribed to its promoter component whose effect is reversible. Within the first five years after discontinuing smoking, the risk of lung cancer sharply decreases, which points to the crucial role of factors exerting reversible effects. At the same time, the fact that this risk never reaches the nonsmoker level corroborates the hypothesis that effects of genotoxic components of tobacco smoke are irreversible.
In conclusion, the efficiency of carcinogenicity on cells can be thought of as a result of passing some heterogeneous mixture through a system of sieves having different pore size varying independently of their position in the column. At any level a sieve with very small pores may occur and the process will be minimized, and vice versa. Obviously, in an extreme case, when all carcinogenic factors are active and those which prevent carcinogenesis are not inactive, the organism will be particular sensitive to carcinogens, and vice versa. In the human organism, such variants do not exceed 15% and are associated with hereditary syndromes with a high level of penetrance. In the present state-of-the-art, individualization of the degree of potential risk is possible only at the level of integral markers, i.e. in the presence of more profound changes in morphological and biochemical characteristics of the cell. By illustration, in industrial workers engaged in the manufacture of benzidine dyes, where the risk of urinary bladder tumors is especially high, the degree of exposure to the carcinogen correlated with urine levels of benzidine metabolites, concentration of DNA adducts, and risk of carcinogenic effect. In case of monocyclic aromatic compounds, e.g. benzenes and styrenes, changes in these parameters were less apparent. The human leukemogenic carcinogen benzene induces hyperdiploidy in the ninth chromosome in peripheral blood lymphocytes and oral cavity epithelium, while styrene stimulates accumulation of DNA adducts in peripheral blood lymphocytes concomitantly with mutations in the HPRT (hypoxanthine phosphoribosyl transferases) locus, micronuclei, sister chromatid exchange, and chromosome aberrations. Vinyl chloride, widely known as an inducer of liver angiosarcomas in human beings, causes chromosome aberrations in peripheral blood lymphocytes, stimulates the formation of micronuclei, sister chromatid exchange, and mutations in HPRT (but no DNA adducts). Unfortunately, all these changes are detectable only after long-term exposure to the carcinogen inadmissible in screening tests.
Safe and reliable typing of potential sensitivity to carcinogens based on the use of molecular-genetic methods may become a routine procedure after the advent of novel efficient techniques for adequate assessment of factors responsible for carcinogenic effects and their relationships. Microarrays with adequate data processing software seem to be the most promising approach.
REFERENCES
1.Turusov, V. S., Belitsky, G. A., Pylev, L. N., and
Koblyakov, V. A. (2004) in Carcinogenesis [in Russian],
Meditsina, Moscow, pp. 204-224.
2.Nebert, D. W., and Dalton, T. P. (2006)
Nature, 6, 947-960.
3.Nebert, D. W., Dalton, T. P., Okey, A. B., and
Gonzalez, F. J. (2004) J. Biol. Chem., 279,
23847-23850.
4.Nelson, D. R. (1999) Arch. Biochem.
Biophys., 369, 1-10.
5.Smart, J., and Daly, A. K. (2000)
Pharmacogenetics, 10, 1124.
6.McMillan, B. J., and Bradfield, C. F. (2007)
Proc. Natl. Acad. Sci. USA, 104, 1412-1417.
7.Sagredo, C., Ovrebo, S., Haugen, A.,
Fujii-Kuriyama, Y., Baera, R., Botnen, I. V., and Mollerup, S. (2006)
Toxicol. Lett., 167, 173-182.
8.Nebert, D. W., Roe, A. L., Dieter, M. Z., Solis, W.
A., Yang, Y., and Dalton, T. P. (2000) Biochem. Pharmacol.,
59, 65-85.
9.Uno, S. (2004) Mol. Pharmacol., 65,
1225-1237.
10.Conney, A. H., Chang, R. L., Jerina, D. M., and
Wei, S. (1994) Drug Metab. Rev., 26, 125-163.
11.Gillis, B., Gavin, I. M., Arbieva, Z., King, S.
T., Jayaraman, S., and Prabhakar, B. S. (2007) Genomics,
90, 324-333.
12.Hishida, A. (2005) Asian Pac. J. Cancer
Prev., 6, 251-255.
13.Flowers, L., Ohnishi, S. T., and Penning, T. M.
(1997) Biochemistry, 36, 8640-8648.
14.McLure, K. G., and Kastan, M. B. (2005) in 25
Years of p53 Research (Hainaut, P., and Wiman, K. G., eds.)
Springer, Dordrecht, pp. 53-73.
15.Roos, W. P., and Kaina, B. (2006) TRENDS Mol.
Med., 12, 440-450.
16.Bond, G. L., Hu, W., and Levine, A. A. (2005)
Cancer Res., 65, 5481-5484.
17.Soussi, T. (2005) in 25 Years of p53
Research (Hainaut, P., and Wiman, K. G., eds.) Springer, Dordrecht,
pp. 255-292.
18.Shen, H., Spitz, M. R., Qiao, Y., Zheng, Y.,
Hong, W. K., and Wei, Q. (2002) Lung Cancer, 36,
243-247.
19.Soifer, V. N. (1997) Soros Obrazovat. Zh.,
8, 4-13.
20.Deutsch, W. A., and Linn, S. (1979) Proc.
Natl. Acad. Sci. USA, 76, 141-144.
21.Tsutsumi, S., Takeshita, H., Yasuda, T., Kuwano,
H., and Kishi, K. (2000) Cancer Lett., 16, 109-112.
22.Tranah, G. J., Bugni, J., Giovannucci, E., Ma,
J., Fuchs, C., Hines, L., Samson, L., and Hunter, D. J. (2006)
Cancer Causes Control., 17, 721-731.
23.Chen, L., Elahi, A., Pow-Sang, J., Lazarus, P.,
and Park, J. (2003) J. Urol., 170, 2471-2474.
24.Sobol, R. W., Horton, J. K., Kuhn, R., Gu, H.,
Singhal, R. K., Prasad, R., Rajewsky, K., and Wilson, S. H. (1996)
Nature, 379, 183-186.
25.Ochs, K., Sobol, R. W., Wilson, S. H., and Kaina,
B. (1999) Cancer Res., 59, 1544-1551.
26.Li, C., Hu, Z., Liu, Z., Wang, L. E., and Strom,
S. S. (2006) Cancer Epidemiol. Biomarkers Prev., 15,
2526-2532.
27.IARC (1994) IARC Scientific Publication No.
125, Lyon.
28.Figueroa, J. D., Malats, N., Rothman, N., Real,
F. X., and Silverman, D. (2007) Carcinogenesis, 28,
1788-1793.
29.Berenblum, I., and Shubik, P. (1949) Br. J.
Cancer, 3, 384-386.
30.Skulachev, V. P. (2006) Apoptosis,
11, 473-485.
31.Archakov, A., and Zhukov, A. (1989) in
Frontiers in Biotransformation (Ruckpaul, K., and Kein, H., eds.)
Berlin, pp. 151-175.
32.Kopnin, P. B., Agapova, L. S., Kopnin, B. P., and
Chumakov, P. M. (2007) Cancer Res., 67, 4671-4678.
33.Espinosa, C. E. S., and Slack, F. J. (2006) J.
Biol. Med., 79, 131-140.
34.Cimmino, A., Calin, G. A., Fabbri, M., Iorio, M.
V., Ferracin, M., and Shimizu, M. (2005) Proc. Natl. Acad. Sci.
USA, 102, 13944-13949.
35.Li, A. G., Lu, S. L., Han, G., Hoot, K. E., and
Wang, X. J. (2006) Mol. Carcinog., 45, 389-396.
36.Wagenlehner, F. M., Elkahwaji, J. E., Algaba, F.,
Bjerklund-Johansen, T., Naber, K. G., Hartung, R., and Weidner, W.
(2007) BJU Int., 100, 733-737.
37.Park, K. T., Mitchell, K. A., Huang, G., and
Elferink, C. J. (2005) Mol. Pharmacol., 67, 612-622.
38.Puga, A., Barnes, S. J., Dalton, T. P., Knudsen,
E. S., and Maier, M. A. (2000) J. Biol. Chem., 275,
2943-2950.
39.Ohtake, F. (2003) Nature, 423,
545-550.
40.Mortensen, A. S., and Arukwe, A. (2007) Comp.
Hepatol., 6, 2-5.
41.Ichihara, S., Yamada, Y., Ichihara, G., Nakajima,
T., Li, P., Kondo, T., Gonzalez, F. J., and Murohara, T. (2007)
Arterioscler. Thromb. Vasc. Biol., 27, 1297-1304.
42.Kim, I., Morimura, K., Shah, Y., Yang, O., Ward,
J. M., and Gonzalez, F. J. (2007) Carcinogenesis, 28,
940-946.
43.Chuu, C. P., Kokontis, J. M., Hiipakka, R. A.,
and Liao, S. (2007) J. Biomed. Sci., 14, 543-553.
44.Zelcer, N., and Tontonoz, P. (2006) J. Clin.
Invest., 116, 607-614.
45.Ayesh, R., Idle, J. R., Ritchie, J. C., Crothers,
M. J., and Hetzel, M. R. (1984) Nature, 312, 169-170.
46.Griffin, M. D., Dong, X., and Kumar, R. (2007)
Arch. Biochem. Biophys., 460, 218-226.
47.Caporaso, N., Hayes, R. B., Dosemeci, M., Hoover,
R., Ayesh, R., Hetzel, M., and Idle, J. (1989) Cancer Res.,
49, 3675-3679.
48.Yang, M., Choi, Y., Hwangbo, B., and Lee, J. S.
(2007) Lung Cancer, 57, 135-142.
49.Vineis, P., Anttila, S., Benhamou, S., and
Spinola, M. (2007) Carcinogenesis, 28, 1902-1905.
50.50.Wei, Q., Cheng, L., Amos, C. I., Wang, L.
E., Guo, Z., Hong, W. K., and Spitz, M. R. (2000) J. Natl. Cancer
Inst., 92, 1764-1772.
51.Zienolddiny, S., Campa, D., Lind, H., Ryberg, D.,
Skaug, V., Stangeland, L., Phillips, D. H., Canzian, F., and Haugen, A.
(2006) Carcinogenesis, 27, 560-567.
52.Hung, R. J., Brennan, P., Canzian, F.,
Szeszenia-Dabrowska, N., Zaridze, D., Lissowska, J., Rudnai, P.,
Fabianova, E., Mates, D., Foretova, L., Janout, K., Bencko, H.,
Chabrier, A., Borel, S., Hall, J., and Boffetta, P. (2005) J. Natl.
Cancer Inst., 97, 567-576.
53.Hu, Z. B., Zhu, J. F., Huo, X., Qian, J., Wei, Q.
Y., and Shen, H. B. (2005) Pharmacogenet. Genomics, 15,
457-463.
54.Hao, B., Miao, X., Li, Y., Zhang, X., Sun, T.,
Liang, G., Zhao, Y., Zhou, Y., Wang, H., Chen, X., Zhang, L., Tan, W.,
Wei, Q., Lin, D., and He, F. (2006) Oncogene, 25,
3613-3620.
55.Kiyohara, C., and Yoshimasu, K. (2007) Int.
J. Med. Sci., 4, 59-71.