
|
REVIEW: Tissue-Specific Transcription Factors in Progression of Epithelial TumorsN. L. Lazarevich* and D. I. FleishmanInstitute of Carcinogenesis, Blokhin Russian Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; fax: (495) 324-1205; E-mail: lazarevich@crc.umos.ru* To whom correspondence should be addressed. |
Received January 22, 2008; Revision received February 14, 2008
Dedifferentiation and epithelial-mesenchymal transition are important steps in epithelial tumor progression. A central role in the control of functional and morphological properties of different cell types is attributed to tissue-specific transcription factors which form regulatory cascades that define specification and differentiation of epithelial cells during embryonic development. The main principles of the action of such regulatory systems are reviewed on an example of a network of hepatocyte nuclear factors (HNFs) which play a key role in establishment and maintenance of hepatocytes--the major functional type of liver cells. HNFs, described as proteins binding to promoters of most hepatospecific genes, not only control expression of functional liver genes, but are also involved in regulation of proliferation, morphogenesis, and detoxification processes. One of the central components of the hepatospecific regulatory network is nuclear receptor HNF4alpha. Derangement of the expression of this gene is associated with progression of rodent and human hepatocellular carcinomas (HCCs) and contributes to increase of proliferation, loss of epithelial morphology, and dedifferentiation. Dysfunction of HNF4alpha during HCC progression can be either caused by structural changes of this gene or occurs due to modification of up-stream regulatory signaling pathways. Investigations preformed on a model system of the mouse one-step HCC progression have shown that the restoration of HNF4alpha function in dedifferentiated cells causes partial reversion of malignant phenotype both in vitro and in vivo. Derangement of HNFs function was also described in other tumors of epithelial origin. We suppose that tissue-specific factors that underlie the key steps in differentiation programs of certain tissues and are able to receive or modulate signals from the cell environment might be considered as promising candidates for the role of tumor suppressors in the tissue types where they normally play the most significant role.
KEY WORDS: hepatocyte nuclear factors, tumor progression, differentiation, hepatocyte, hepatocellular carcinoma, epithelial-mesenchymal transition, liver, pancreasDOI: 10.1134/S0006297908050106
Abbreviations: AFP) alpha-fetoprotein; CDK) cyclin-dependent kinase; ECM) extracellular matrix; EGF) epidermal growth factor; EHS-matrix) three-dimensional matrix produced by cells of the Engelbreth-Holm-Swarm mouse sarcoma; EMT) epithelial-mesenchymal transition; FGF) fibroblast growth factor; HCC) hepatocellular carcinoma; HGF/SF) hepatocyte growth factor; HNF) hepatocyte nuclear factor; IGF) insulin-like growth factor; PI[4,5]P2) phosphatidylinositol 4,5-diphosphate; TGFbeta) transforming growth factor beta.
One of the most important characteristics of malignant growth is tumor
progression--gradual change of morphological, biochemical, functional,
and proliferative properties of transformed cells due to selection and
accumulation of genetic alterations which contribute to the development
of more aggressive tumor phenotype.
The key steps of carcinogenesis include failure of proliferation control and increase of cell replicative potential, resistance to apoptosis, stimulation of angiogenesis, alteration of epithelial structure (destruction of intercellular contacts and contacts with extracellular matrix (ECM) and the loss of epithelial polarity) and increase of cell motility, the acquirement of invasion and metastatic capability, dedifferentiation, and genomic instability [1-3]. At the present time the most investigated are alterations that are common for different types of malignancies (they usually affect mechanisms which control proliferation, apoptosis, and genetic stability), while tissue-specific changes, especially disturbance of normal morphology and dedifferentiation, are poorly understood.
The most striking characteristic of malignancy of epithelial cancers is the ability for invasion and metastases, which allows cells to leave the primary tumor and disseminate into new territories with unlimited amount of space and nutrient substances. Distant metastases are the main factor responsible for mortality caused by oncological diseases. To acquire the ability to form metastasis, a cell has to get beyond the control of normal microenvironment and acquire additional motility. The alteration of morphological characteristics and increase of migration properties of transformed cells cause the loss of epithelial morphology and can be qualified as epithelial-mesenchymal transition (EMT).
EMT was first described as morphological rearrangement that underlies key stages of specification and development of embryonic tissues [4, 5]. Later it appeared that EMT plays an important role in epithelial tumor progression towards dedifferentiated, more malignant phenotype.
The basic criteria of EMT are the loss of epithelial cell polarity, the separation into individual cells, and subsequent dispersion after the acquisition of cell motility. These processes are associated with disassembly of tight junctions, adherent junctions, and desmosomes and reorganization of complexes that are normally responsible for cell attachment to the substrate. The loss of cell polarity results in cytoskeletal changes. One of the EMT marker properties is the shift from cytokeratin intermediate filaments to vimentin. EMT is accompanied by alteration of the gene transcription profiles including components of cytoskeleton and ECM, as well as of proteolytic enzymes involved in matrix degradation [4].
The EMT processes at early stages of development and in carcinogenesis are initiated by similar effector mechanisms [4-6]. EMT is induced by signals from the cell environment including, first of all, soluble growth factors (epidermal growth factor (EGF), hepatocyte growth factor (HGF/SF), fibroblast growth factor (FGF), insulin-like growth factors (IGF), as well as transforming growth factors (TGF) beta) and matrix components (fibronectin, laminin 5, collagen). Interaction of growth factors with specific membrane receptors activates intracellular signaling cascades that induce changes of intracellular contacts and cytoskeleton rearrangement.
One of the most general EMT mechanisms which has been described for most types of epithelial cells is downregulation of expression of E-cadherin, a transmembrane glycoprotein responsible for homotypic adhesion in epithelial cells. The E-cadherin insufficiency results in release and translocation into the nucleus of its molecular partner beta-catenin that activates transcription of several genes involved in control of cell proliferation and adhesion. Decrease in E-cadherin production is described in many types of carcinomas; it is a factor of poor prognosis and occurs mainly at the transcriptional level [5]. The most important E-cadherin repressors are transcription factors Snail and Slug regulated by different signal cascades (TGFbeta, FGF, and Wnt) during EMT [7, 8].
The dissociation of intercellular contacts is not sufficient for cells to acquire motility and ability to penetrate into new environments. An important role in this process belongs to integrins, which realize cell interaction with extracellular matrix (ECM), and to proteases responsible for rearrangement or degradation of matrix components. Changes in intracellular ratio of integrin subunits and their affinity can not only confer new substrate specificity to the cell, but also might modulate the activity of proteolytic enzymes, control cytoskeleton organization, and influence cell survival [9].
Changes in local microenvironment and the loss of epithelial morphology may contribute to decrease of differentiation level; failure of specialized tissue functions and deregulation of tissue-specific gene expression are characteristic features of tumor progression [10]. However, the complete loss of tissue-specific properties never takes place and epithelial tumors that undergo dedifferentiation retain at least some features of the original tissue and the capability of redifferentiation [11]. Besides, restoration of synthesis of embryo-specific proteins distinctive for a certain type of immature cells may take place during dedifferentiation. A classical example of such activation is re-expression of alpha-fetoprotein (AFP), specific to embryonic liver, which is observed in the case of hepatocarcinogenesis and is widely used in diagnosis of this kind of tumor [12].
Cell differentiation is defined by architecture of the organ and activity of tissue-specific functional genes. Since these parameters are individual for each tissue and each cell-type forming this tissue, the mechanisms of progression should be considered on the basis of structural and functional properties of the organ where the tumor arose [11, 13]. To understand what mechanisms are responsible for loss of differentiation during tumor progression, it is important to realize the consequence of individual organs and tissues specification during development and understand the mechanisms of maintenance and fine adjustment of the normal cell functional status. As it is clearly seen on the example of liver and pancreas development [14, 15], the key role in this process belongs to tissue-specific transcription factors, their consecutive switching on and off defines the differentiation fate of each cell type at all stages of development of the organism.
One of the best investigated tissue-specific systems of transcriptional regulation is the network of hepatocyte nuclear factors (HNFs) that were described as proteins which bind to promoters of most hepatospecific genes [16]. Although activity of these factors was later revealed in other organs, just in liver expression pattern of all proteins of this group cross and specifically in this organ maximal transcription levels of encoding genes are observed.
In this review, we will consider the role of HNFs in differentiation of liver and some other organs and will try to follow some general regularities that define the contribution of tissue-specific transcription factors to progression of epithelial tumors.
HEPATOCYTE NUCLEAR FACTORS
At the present time HNFs include five unrelated families of transcription factors (HNF1, C/EBP, FoxA, HNF4, and HNF6) that form a common regulatory network. There are hierarchic regulatory interrelations between HNFs, which are significantly modulated during development [17].
The combined activity of the HNF network finely regulated both during embryogenesis and also in the course of normal functioning or in pathological states in adult liver, defines the tissue-specific transcription profile of hepatocytes--the main functional cell type of liver. In addition, certain factors play an important role in differentiation of distinct types of epithelial cells. Below we provide brief characteristics of all families, which are considered in more detail in reviews [13, 18-22]. Some basic features of HNFs expression are listed in Table 1.
Table 1. Hepatocyte nuclear factors
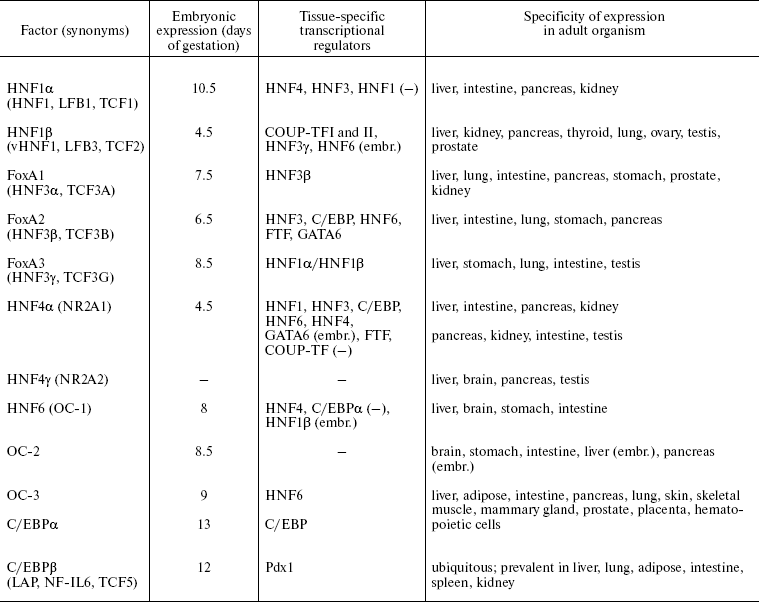
Note: -, no data; (-), negative regulation; (embr.), only at
embryonic stages of development.
HNF1. Proteins of the HNF1 family according to the structure of the DNA-binding domain belong to the superfamily of homeoproteins and contain the POU-like domain responsible for enhancing specificity of binding [16]. Members of this family HNF1alpha and HNF1beta interact with DNA in the form of homo- or heterodimers with different transactivation properties.
Sites for binding of the HNF1 family proteins are identified in most hepatospecific promoters [23], e.g. albumin, AFP, plasminogen, and alpha1-antitrypsin. HNF1 target genes encode proteins involved in intercellular adhesion, metabolism of lipids, glucose, and amino acids, transport of organic anions, and detoxification processes [24].
Expression of HNF1beta starts at the outset of embryonic development and is crucial for the visceral endoderm formation [25, 26]. This factor is essential for formation of intrahepatic bile ducts and gallbladder [27], kidney tubules [25], specification of pancreatic primordium, and further differentiation of various cell types of this organ [28].
Embryonal expression of HNF1alpha starts later and its level is initially below than that of HNF1beta [29]. After birth in all organs except kidneys, this balance changes in favor of HNF1alpha. Expression of HNF1alpha is not crucial for normal embryonic development, but its inactivation causes growth retardation and progressive wasting syndrome due to the disturbance of the liver, kidney, and pancreas functions after birth [30, 31]. This factor controls expression of a variety of tissue-specific genes during differentiation of intestine [32].
Thus, functions of HNF1 family proteins are crucial for different stages of ontogenesis: HNF1beta plays an important role in differentiation of visceral endoderm and development of liver, kidneys, and pancreas during embryogenesis, while HNF1alpha is essential for maintenance of differentiated state of these organs after birth.
FoxA. The FoxA (HNF3) family consists of three proteins (FoxA 1, 2, and 3) that contain “winged helix” DNA-binding domain and interact with DNA as monomers [33]. The essential structural similarity of DNA-binding domain with DNA-packing histones H1 and H5 defines the ability of FoxA factors to bind to specific sites in inactive chromatin regions and initiate the alteration of chromosomal organization which stimulates binding of other factors and activation of gene transcription [34, 35]. This property is of decisive importance at early stages of liver specification when inductive signals from pericardial mesoderm cause alteration of the transcription program in a part of the embryonal endoderm cells that will later give rise to liver and pancreas [36].
Apparently, functions of FoxA proteins are partially overlapped, and inactivation of one member of the family can be compensated in part at the expense of others. The most crucial is the failure of FoxA2 function, which is indispensable already at the outset of embryogenesis for correct gastrulation and formation of notochord, neural tube, and primitive gut [37]. Cooperative action of FoxA 1 and 2 defines development of liver, kidney, lung, and prostate. Inactivation of these genes makes endodermal cells of the primary gut insensitive to environmental stimuli that normally induce organ specification and activation of tissue-specific genes. In an adult organism FoxA family factors regulate expression of serum proteins (albumin, transthyretin, alpha1-antitrypsin, etc.) and enzymes that control glucose metabolism and glycogen accumulation [37].
HNF4. Two members of this family have been identified in mammals, HNF4alpha (NR2A1) [38] and HNF4gamma (NR2A2) (the latter is not expressed in liver and is still poorly studied) [39]. According to their structure, these factors belong to subfamily NR2 of the nuclear receptor superfamily and are most related to retinoid X receptor. Like other proteins of this type, they contain two “zinc finger” DNA-binding domains and a C-terminal region responsible for dimerization and binding with presumptive ligand [40]. Supposed HNF4alpha ligands--Acyl-CoA thioesters of fatty acids--are able to interact with HNF4alpha ligand-binding domain and alter its ability for dimerization and DNA binding in vitro [41]. At the same time, no alterations of HNF4alpha conformation and its ability to bind co-activators, typical of the ligand-receptor interaction upon action of these substances in vivo, were shown [42]. HNF4alpha interacts with DNA as a homodimer. Other nuclear receptors do not form dimers with HNF4alpha [40], but they can be involved in regulation of controlled genes by competition for common binding sites.
Nine isoforms of HNF4alpha originating from alternative splicing are described. Isoforms alpha1-alpha6 transcribed from the main promoter P1 are predominant in adult liver and differentiated hepatomas [43, 44]. Transcription of alpha7-alpha9 isoforms, which differ from other variants by N-terminal sequence, is regulated by an alternative promoter P2 that is active in stem cells, embryonic liver, beta-cells of pancreas, and in cultures of dedifferentiated hepatomas [43-45]. “Adult” (alpha1-alpha6, referred below as HNF4alpha1) and “embryonic” (alpha7-alpha9, referred below as HNF4alpha7) isoforms have different transactivation properties, namely, form alpha7 more efficiently activates promoters of early hepatocyte genes (such as AFP), whereas isoform alpha1 have a more significant impact on the genes of the main hepatic differentiation markers [46].
The HNF4alpha inactivation in mouse embryos results in impaired gastrulation associated with abnormal development of visceral endoderm [47]. HNF4alpha is not crucial for formation of liver primordium but it is essential for hepatocyte differentiation and epithelial morphogenesis during development [48, 49]. HNF4alpha regulates expression of a broad spectrum of hepatospecific genes involved in metabolism of amino acids, lipids, carbohydrates, cholesterol, and xenobiotics, as well as in transport of ions, lipids, and components of the blood coagulation system and of system of proteolysis regulation [40, 50]. HNF4alpha is involved in proliferation control and maintenance of epithelial morphology in some cell types. In addition, HNF4alpha is involved in differentiation of pancreatic cells [51, 52], crypt formation, maturation of mucin-producing goblet cells, and regulation of expression of a number of tissue-specific genes during embryonic colon development and enterocyte differentiation [53, 54]. In beta-cells of pancreas, HNF4alpha regulates genes involved in proliferation control [52], glucose metabolism, and insulin secretion [55]. Differential expression of HNF4alpha in kidney epithelium (cells of proximal kidney tubules) and its involvement in regulation of tissue-specific genes suggests the requirement of HNF4alpha for the differentiation of this organ [56, 57].
HNF6. Factors of the HNF6 family also can interact with FoxA binding sites in promoters of several genes [58]. HNF6 (OC-1), a nuclear protein interacting with DNA as monomer, became the first characterized member of a new class of transcription regulators called ONECUT that contain a homeobox and one DNA-binding domain of the CUT type. Besides HNF6, the family includes OC-2 and OC-3 [59, 60].
Factors of HNF6 family are involved in early stages of liver and pancreas development. HNF6 is necessary for initiation of expression of the earliest marker of pancreatic cells, the pancreatic and duodenal homeobox 1 (Pdx1). HNF6 expression in embryonic endoderm depends on activity of HNF1beta, so the signal cascade HNF1beta-HNF6-Pdx1 underlies the early specification of pancreatic cell progenitors [61]. At later stages of development, HNF6 controls differentiation of endocrine and duct cells of the pancreas [62]. In embryonic liver HNF6 regulates transcription of HNF1beta; this regulatory cascade defines differentiation of cholangiocytes that form intrahepatic bile ducts [63]. In hepatocytes, HNF6 influences the main metabolic processes (for example, by regulation of gluconeogenesis) and, probably, cell proliferation (due to regulation of TGFalpha and cell cycle regulatory genes) [64], adhesion, and migration [65].
C/EBP. Six C/EBP proteins have been characterized. They contain a main DNA-binding bZIP domain, N-terminal transactivation domain, and a helical structure of “leucine zipper” type that provides dimerization [66]. These factors have a similar structure, form homo- and heterodimers, and bind to a common DNA sequence. For C/EBPalpha and beta shortened forms are described which are deprived of the transactivation domain and are negative regulators of transcription. Thus, due to heterodimerization, activation properties of C/EBP factors may undergo significant modulation in different type cells.
Different combinations of the C/EBP family proteins play an important role in organogenesis and differentiation of many types of epithelial cells. In hepatocytes C/EBPalpha, C/EBPbeta, and C/EBPdelta are expressed. They regulate processes of energy metabolism, gluconeogenesis, inflammatory reaction, and xenobiotic, urea, and bilirubin detoxification [66]. C/EBPalpha also plays an important role in differentiation of adipose tissue, lung, skin, and hematopoietic cells [67, 68]. C/EBPbeta is crucial for functional differentiation of mammary gland cells, macrophages, and ovarian follicles [69, 70].
Proteins of the C/EBP family take part in control of proliferation and apoptosis in different tissues [70]. The ratio of the C/EBPalpha and beta expression levels is associated with the cell proliferative status. In quiescent hepatocytes, constant balance of C/EBPalpha and beta is maintained. Liver tissue damage accompanied by regeneration is characterized by a rapid fall of the C/EBPalpha level and activation of C/EBPbeta; in this case total intracellular amount of C/EBP does not change [71]. Inactivation of C/EBPbeta in mice significantly decreases proliferation rate during liver regeneration [72]; in C/EBPalpha-/- mice, on the contrary, liver hyperplasia is observed [73].
Several possible mechanisms of action of C/EBPalpha have been proposed including stabilization of the cyclin-dependent kinase (CDK) 2 inhibitor p21Waf1/Cip1, interaction with pRB-like factors, block of the E2F factor activities and inhibition of c-myc transcription, interaction with the chromatin-remodeling complex SWI/SNF, with Max transcription factor, and binding to complexes CDK2-cyclin E and CDK4-cyclin D and inhibition of their activity [74, 75]. Most likely, due to the ability of C/EBPalpha to interact with many regulatory proteins, the mechanism of its involvement in mitotic arrest induction depends on the particular cell context. So, the coordinated regulation of differentiation and proliferation processes in different cell types can be carried out via modulation of activity of the C/EBP family factors.
Thus, the hepatocyte nuclear factors play an important role in formation and differentiation of a number of epithelial tissues. As is seen in Table 1, expression of all known HNFs can be observed only in liver at different stages of ontogenesis; the direction and level of liver differentiation are unambiguously formed by their consecutive activation and cooperative action.
Consecutive activation of hepatocyte nuclear factors and strict specificity of their expression define key steps of liver development during embryogenesis [14, 76, 77]. Liver bud is formed by a part of the foregut ventral endoderm cells in response to signals from pericardial mesoderm. The competence of certain cells for receiving such inductive signals is provided by activation of FoxA2 that changes nucleosomal organization and facilitates binding to regulatory regions of other transcription factors.
Later hepatoblasts that form liver primordium are gradually differentiated into two types of cells--hepatocytes, the major functional liver cells, and cholangiocytes, which later line bile ducts. Gradually there takes place a formation of a complex organ structure resulting in generation of liver trabeculae, sinusoids, bile ducts, and the whole liver blood supply complex. Under formation of liver trabeculae, hepatocytes acquire polarity and form tight and gap junctions. These processes are accompanied by further complication of the HNFs regulatory network. HNF4alpha plays the crucial role in hepatocyte differentiation and morphogenesis of embryonic liver. The tissue-specific transcription profile of mature hepatocytes is largely defined by activity of HNF1alpha, HNF4alpha, and C/EBPalpha factors. Differentiation of cholangiocytes is controlled by the HNF6 => HNF1beta signaling cascade.
There are complex regulatory relationships between HNFs, which makes it possible to speak about the existence of a complex tightly controlled regulatory network [16, 18-21]. The transcriptional hierarchy between its individual components is actively studied but is not completely solved yet. Besides HNFs, at different stages of ontogenesis additional transcription factors (GATA4 and 6, FTF, COUP-TFI and II, and some others), contributing to stage-specificity of functional gene expression, become involved in the hepatospecific regulatory network. Depending on the stage of development both the spectrum of HNFs expressed in liver and the hierarchy of their regulatory interrelationships changes [17]. These differences are defined both by the spectrum of HNFs expressed at a certain moment in a cell and by activity of others, including not tissue-specific signaling pathways, expression levels of cofactors and antagonists, and by post-translational protein modifications. The additional complexity of the regulatory system refers to autoregulatory mechanisms, reciprocal regulation of HNFs expression, the possibility of heterodimerization between proteins of the same family with different transactivation potential, and compensation of dysfunction of one family member at the expense of activity of related factors.
ROLE OF HNFs IN DIFFERENTIATION OF OTHER ORGANS
A similar situation is also observed in other organs that express the described proteins. Tissue specificity of expression in this case is modulated by combined action of expressed HNFs and other tissue-specific regulators.
Pancreas is the organ “closest” to liver both by the origin during embryogenesis and by the set of expressed HNFs, but in pancreas, instead of “adult” isoforms HNF4alpha1, the “embryonic” HNF4alpha7 isoforms controlled by the alternative promoter P2 are expressed [78]. The pancreas consists of two functional compartments--endocrine composed of four types of hormone-secreting cells that are specialized on production of insulin, glucagon, somatostatin, or pancreatic polypeptides and exocrine that defines secretion of digestive enzymes and consists of acinar and duct cells [79]. Pancreas is formed by dorsal and ventral buds of the primitive gut endoderm and undergoes a series of complex morphological transitions in the course of organogenesis. Specification of the pancreatic progenitor cells and differentiation of all types of pancreas cells are defined by the consequence of activation of pancreas-specific factors and HNFs [79-82], which can be thus considered in this case as a part of general tissue-specific regulatory network.
Early steps of pancreas development are defined by activity of homeoproteins Pdx1, Ptf1alpha, Hlxb9, Isl1, and Hex, while further specification of different cell types is associated with neurogenin-3, factors Pax4, Pax6, NeuroD/beta2, and Nkx2.2 activity [80, 81]. HNFs not only cooperate with these proteins in regulation of pancreas-specific genes, but directly control expression of a number of pancreatic transcription factors Pdx1, Pax4, neurogenin-3, Nkx2.2, etc. Thus, for example, HNF1beta, HNF6, and FoxA2 are involved in transcriptional regulation of Pdx1 gene, which encodes the key factor responsible for pancreas formation, differentiation of various pancreatic lineages, and functional activity of insulin-producing beta-cells [82]. Among HNFs the most crucial role in pancreatic morphogenesis belongs to HNF1beta that is essential both for its formation and specification of different cell types [28].
HNF expression is necessary for normal functioning of mature pancreatic beta-cells, in particular, for regulation of insulin secretion. The HNF1 family factors are able to bind directly to the insulin gene promoter, whereas HNF4alpha, FoxA2?, and HNF6 most likely regulate insulin expression via modulation of HNF1, Pdx1, or NeuroD/beta2 activities [28, 31, 61, 81]. C/EBPbeta, on the contrary, is a negative regulator of insulin gene transcription. FoxA factors control activity of the pancreatic alpha-amylase--a marker of exocrine cells [83]--and regulate both differentiation and glucagon expression in alpha-cells [37].
Besides regulation of tissue-specific genes, HNFs influence cell morphology. Expression of dominant-negative HNF1alpha form in beta-cells not only results in disturbance of insulin secretion, but causes dissociation of adhesion contacts due to inhibition of E-cadherin transcription [84]. Lowering of the HNFs transcription target Pdx1 expression is essential for activation of cytokeratin 19 gene, which is specific for the pancreatic duct cells [85].
The pancreas-specific network of transcription factors is the best-studied but not unique system, where particular HNFs appear to be the key elements. Thus, proteins of the FoxA family hold one of the central places in formation of the regulatory network that defines embryonic morphogenesis, proliferation, and differentiation of respiratory epithelium [86]. Later stages of lung differentiation require participation of the C/EBP family factors [68]. In the near future the detailed characterization of signal cascades that specify normal development and differentiation of distinct epithelial cell types will probably facilitate identification of key elements in these regulatory systems. It is likely these will be factors located at the crossroads of signaling cascades controlling proliferation, morphogenesis, and expression of tissue-specific functional genes.
IMPAIRMENT OF HNFs FUNCTION IN GENETIC DISEASES
Additional evidence of the importance of normal HNFs function for homeostasis of liver and some other organs are data concerning the role of these factors in pathogenesis of some genetic diseases. Mutations of HNFs binding sites in promoter regions of genes regulated by these factors result in development of chronic diseases of the respiratory tract and liver (the deficiency of alpha1-antitrypsin), phenylketonuria (absence of phenylalanine hydroxylase), hemophilia B (absence of coagulation factor IX), and the susceptibility to alcohol-induced liver damage (defects of cytochrome P450 function) [87].
Mutations altering the expression or functional activity of HNFs per se are responsible for several forms of insulin-independent maturity onset diabetes of the young (MODY). This type of diabetes is characterized by hyperglycemia caused by derangement in insulin secretion and action. As mentioned above, a crucial role in normal functioning of adult hepatocytes and pancreatic beta-cells belongs to HNF1alpha, HNF1beta, and HNF4alpha involved in the control of normal glucose homeostasis. Mutations in promoter or coding regions of HNF4alpha, HNF1alpha, and HNF1beta genes are the immediate cause of the development of MODY1, MODY3, and MODY5 forms, respectively, that are characterized by distortion of glucose-stimulating insulin secretion, early onset of disease, and autosomal dominant inheritance [81, 88]. Mutations of the HNF1beta gene are also associated with a number of diseases associated with defects of kidney development [88, 89].
TISSUE-SPECIFIC FACTORS AND TRANSDIFFERENTIATION
The precise regulation of the tissue-specific transcription network activity is of principal significance for realization and maintenance of various differentiation programs. More data are appearing showing that deregulation of one or several master-genes is able to change completely the fate of a cell. Thus, ectopic expression of Pdx1 is sufficient for induction of pancreatic transdifferentiation and activation of insulin production in liver or HepG2 hepatoma cells [90-92]. In parallel, Pdx1 induces hepatocyte dedifferentiation, i.e. inhibits transcription of the adult liver-specific genes and of transcription factor C/EBPbeta, and also activates expression of the onco-fetal protein AFP. Inhibition of C/EBPbeta activity is essential for Pdx1-dependent transdifferentiation in this system [90].
Activation of C/EBPbeta expression in the culture of exocrine pancreatic cells AR42J-B13, on the contrary, induces transdifferentiation towards the hepatocyte phenotype, which is characterized by repression of pancreas-specific genes, nuclear translocation of HNF4alpha, and induction of the hepatocyte differentiation markers [93]. However, the Pdx1-independent inhibition of C/EBPbeta activity is not enough to induce pancreatic differentiation of liver cells [90].
Apparently, transdifferentiation upon the change in activity of one of the key tissue-specific transcription factors is possible not only for such ontogenetically related cell types as hepatocytes and exocrine cells of the pancreas. Transdifferentiation of the hepatocyte progenitor cells to those synthesizing prolactin, growth hormone, and thyroid-stimulating hormone beta was described in the case of adenovirus-mediated expression in mouse liver of hypophysial (pituitary) transcription factor 1 (Pit-1) that defines differentiation of several types of pituitary cells [94].
IMPAIRMENT OF HNF FUNCTION IN HEPATIC CARCINOGENESIS
Investigations of the structure and dynamic hierarchy of tissue-specific regulatory networks in various types of epithelial cells present a vast field for studies. However, most likely, in each such regulatory cascade at a certain step of development there are one or very few key components whose damage causes an irreversible failure of the whole differentiation program. It is reasonable to suppose that such elements are factors able to integrate signals from the cell environment and to transform them into alterations of metabolic, proliferative, and morphological properties of cells. Recent studies have shown that for mature hepatocytes HNF4alpha is the central regulator of a variety of processes that control cellular homeostasis.
Dedifferentiation observed during progression of epithelial tumors can be caused by disturbed normal functioning of tissue-specific regulatory cascades controlled by transcription factors. Since HNFs are the central components of tissue-specific regulation in a number of organs and tissues, deregulation of this network may be an important step in progression of corresponding tumor types.
Hepatocellular carcinoma (HCC) is the most frequent malignant tumor of liver and one of the most widespread forms of cancer in the world [95]. The main risk factors for the development of HCCs are chronic infections with hepatitis B and C viruses as well as the prolonged exposure to carcinogens. By now numerous signal pathways important for control of liver functions and hepatocyte proliferation are described, but the molecular basis of HCC progression are still obscured [96, 97].
We have studied some mechanisms of development of liver tumor malignant phenotype on the experimental model of one-step progression of mouse HCC consisting of slowly growing differentiated tumor, obtained during chemical carcinogenesis induction by diethyl nitrosamine and phenobarbital (sgHCC), and of the fast growing dedifferentiated variant (fgHCC) that was derived from sgHCC in vivo during subcutaneous transplantation [98]. The interval between transplantations in vivo was 5-7 months for sgHCC and was shortened to two weeks for fgHCC.
Progression from sgHCC to fgHCC was characterized by significant morphological changes like the loss of cell polarity, weakening of intercellular contacts, and association with ECM [99] that allowed qualifying the above-mentioned changes as EMT. These events were accompanied by changes in expression of some genes encoding components of intercellular contacts and adhesion molecules--overexpression of integrin alpha3 subunit characteristic mainly of immature and transformed hepatocytes, inhibition of expression of the major component of gap junctions in liver, connexin-32, and E-cadherin, which occurred in parallel with a significant increase in the level of its transcriptional repressor Snail [8]. Also, in the course of progression fgHCC cells acquired ability to form metastases and grow in vitro. The fgHCC cell culture was characterized by the loss of epithelial morphology, weakening of intercellular contacts, and tendency toward three-dimensional growth [99].
In addition to the loss of epithelial features during progression in fgHCC coordinated repression of a broad spectrum of hepatospecific genes [100], corresponding to simplification of carcinoma-specific antigenicity, also occurred [11]. However, the dedifferentiated variant retained expression of a number of hepatospecific genes, which is indicative of hepatic origin of the tumor.
Since the described HCC progression occurred very fast but affected a broad spectrum of cell properties, we have supposed that this process may be associated with dysfunction of one or few master-genes that define hepatocyte differentiation. It appeared that in the course of progression from sgHCC to fgHCC, a coordinated decrease of expression levels of entire block of transcription factors influencing the establishment and maintenance of hepatic phenotype (HNF4alpha, HNF1alpha, HNF1beta, FoxA3, HNF6, C/EBPalpha, and FTF) occurred [100]. Thus the observed changes could be associated with the disturbance of differentiation control caused by repression of hepatocyte transcription factors with interrelated regulation.
We consider that the most probable reason for at least some phenotypic changes during progression is the disturbance of HNF4alpha function, reduced expression of which compared to normal liver was earlier described in mouse [101] and rat [102] HCCs and in dedifferentiated hepatoma cultures [103]. Also, HNF4alpha is the only hepatocyte nuclear factor for which morphogenic activity has been demonstrated in vitro [103]. Investigations on mouse embryos deficient in HNF4alpha expression in the liver have shown that this factor is indispensable for the formation of hepatic epithelium in embryonic development [49]. Moreover, it was found that the exogenous expression of HNF4alpha in NIH 3T3 mouse fibroblasts is sufficient to induce mesenchymal-epithelial transition characterized by acquiring polygonal cell shape and membrane localization of E-cadherin and the tight junction marker ZO-1.
Exogenous expression of HNF4alpha1 in fgHCC cell culture resulted in significant changes of cell morphology, namely, cells began to form epithelial islets with tight junctions marked by membrane localization of ZO-1 protein [100]. Restoration of epithelial morphology due to HNF4alpha re-expression was characterized by normalization of cell contacts with ECM, i.e. localization of matrix components characteristic of epithelial cell cultures was restored: fibrils of entactin, fibronectin, laminin, and type IV collagen were located along intercellular contacts. Thus, the HNF4alpha expressing cells created a better formed basal membrane compared to that in the parental cell culture.
Also, HNF4alpha1 re-expression caused induction of a number of hepatospecific genes encoding transcription factors, metabolic enzymes, and molecules of intercellular adhesion. In particular, clear HNF4alpha-dependent activation of hepatospecific regulators HNF1alpha, FTF, HNF6, HNF4alpha7, and FoxA3 was revealed, along with restoration of transcription of the major component of liver gap junctions--connexin 32.
HNF4alpha1 expression decreased the proliferation rate of dedifferentiated HCC cells in vitro and significantly retarded tumor growth after subcutaneous transplantation. Moreover, unlike the control line, in animals with HNF4alpha1 expressing cell transplants no tumor cell metastases into lungs were observed.
Thus, restoration of HNF4alpha1 function in the dedifferentiated HCC cell culture resulted in reversion of the highly malignant tumor cell phenotype, which was revealed in partial restoration of epithelial morphology, redifferentiation, and suppression of proliferation. These data are indicative of the key role of HNF4alpha in coordination of processes associated with HCC progression.
General character of the revealed regularities was confirmed in analysis of the gene expression levels in a panel of chemically induced transplanted mouse HCCs of independent origin with different level of differentiation and growth rate as well as in clinical specimens of human HCCs (Fleishman et al., in preparation). Among 50 genes encoding HNFs, markers of hepatocyte differentiation, adhesion molecules, growth factors, candidate oncogenes and anti-oncogenes for this tumor type, the clearest association with differentiation status and reverse correlation with growth rate were revealed for HNF4alpha1. Similar consistent patterns of expression are revealed for transcription factors that are direct or mediated targets for HNF4alpha (HNF1alpha, HNF1beta, FoxA3, etc.). In differentiated tumors, induction of expression of HNF4alpha “embryonic” isoforms compared to normal liver was observed, which is indicative of partial activation of dedifferentiation programs at this stage of carcinogenesis. In the most malignant HCC variants, the coordinated inhibition of the adult and embryonic HNF4alpha isoform transcription was observed. Among human HCCs not associated with hepatitis virus infection, the frequency of HNF4alpha1 downregulation in tumor compared to the specimens of surrounding non-tumor tissue was about 70%.
These results point to the existence of a clear relationship between differentiation status of the tumor, its morphological characteristics, growth rate, and expression pattern of tissue-specific transcription regulators. Our investigations have shown that HNF4alpha1 repression is a frequent event in hepatocarcinogenesis and confers a number of selective advantages to tumor cells. Together with earlier results, these data are indicative of an important role of HNF4alpha inactivation in progression of liver tumors.
The loss of functional activity of HNF4alpha1 and its transcription target HNF1alpha has also been described in human renal cell carcinomas [104, 105]. Thus, these factors can be considered as potential tumor suppressors in liver and kidney cells, i.e. in organs where HNF4alpha1/HNF1alpha signalization plays the most essential role in embryonic development and tissue-specific differentiation. This observation is probably also valid for other HNFs as will be illustrated below.
MECHANISMS OF THE EFFECT OF HNF4alpha ON BIOLOGICAL
PROPERTIES OF TUMOR CELLS
How can the dysfunction of tissue-specific transcription factor contribute to the development of the epithelial cell malignant phenotype? New targets of HNF4alpha and other tissue-specific transcription factors, recently revealed in various model systems, significantly expanded the knowledge about the involvement of the HNF regulatory network in control of differentiation, proliferation, and maintenance of epithelial morphology.
Obviously, the most evident is correlation between HNF functional activity and the maintenance of differentiation markers expression in tumor cells. As mentioned above, HNF4alpha controls expression of a broad spectrum of serum proteins and enzymes synthesized in liver [40], both directly and by regulation of activity of other transcription factors like HNF1alpha [106]. Importantly, HNF4alpha is involved in regulation of genes responsible for detoxification and metabolism of drugs, particularly cytochromes P450 [107]. It can be supposed that loss of HNF4alpha function during HCC progression may decrease tumor sensitivity to therapeutic treatment.
An important characteristic of HNF4alpha as a probable tumor suppressor is its antiproliferative function. Exogenous expression of HNF4alpha1 decreases the growth rate of fast growing dedifferentiated mouse HCCs both in vitro and in vivo [100]. These data were also confirmed for other cell types. Antiproliferative activity of HNF4alpha is described in HEK293 embryonic kidney [57], F9 mouse embryonic carcinoma, and RLE rat lung endothelium [108] cells. Transduction of HNF4alpha expressing constructs into INS-1 rat insulinoma cells resulted not only in morphological alterations and activation of expression of several genes characteristic of differentiated alpha-cells of pancreas, but also in decrease of the cell growth rate due both to retardation of proliferation and induction of apoptosis [109].
In F9 mouse embryonic carcinoma and RLE rat lung endothelium [108], as well as in HuH7 [110] and HepG2 [111] human hepatoma cells, exogenous HNF4alpha activates expression of the p21/Waf1 gene, a main inhibitor of CDK-2-dependent transition from G1 to S phase of the cell cycle. These findings indicate that in rodent and human cells HNF4alpha is able to inhibit proliferation due to p21 gene activation. Experiments with reporter constructs, in which the luciferase gene was driven by p21 promoter, had shown that HNF4alpha is able to directly activate the promoter of this gene [108].
The mechanism of p21 activation by HNF4alpha is supposed to be p53-independent [108, 111]. Exogenous expression of HNF4alpha in HCT116 human colorectal carcinoma cells results in hyperexpression of p21, while HNF4alpha inactivation by small interfering RNA in HepG2 human hepatoma cells decreases p21 expression [111]. HNF4alpha binding sites were detected in the p21 regulatory region, but it was supposed that regulation of the p21 gene activity is due to the interaction of HNF4alpha with another transcription factor, Sp1. It is likely that besides direct activation of p21 promoter, formation of this complex prevents c-myc-dependent repression of the p21 gene: c-myc is also able to bind Sp1, and, unlike HNF4alpha, to block the transcription. So, competition between these two factors for interaction with p21 promoter-bound Sp1 is the additional the mechanism of HNF4alpha-dependent proliferation control [111].
The decrease in the tumor cell growth rate caused by HNF4alpha can be also mediated by additional mechanisms. In Caco-2 colorectal carcinoma cells RSK4 (ribosomal S6 kinase 4) and PAK5 (p21-activated kinase 7) genes were identified as direct targets for HNF4alpha [112]. Members of the RSK family are intracellular mediators of MAP-kinase cascades, which control cell division, survival, and differentiation due to phosphorylation of various intracellular proteins and transcription factors. Unlike the other proteins of this family, RSK4 is a presumable inhibitor of signal cascades induced by receptor tyrosine kinases in response to growth factors. PAK kinases are involved in signal transduction from the MAP-kinase cascade, and also participate in the regulation of cell motility due to stabilization of microtubules and disassembly of actin stress fibers and focal adhesions. It is supposed that PAK5 is able to migrate between the nucleus and mitochondria, promoting cell survival under stress conditions. In kidney and brain of rats with experimentally induced diabetes, the coordinated inhibition of HNF4alpha, RSK4, and PAK5 is observed. These results with HNF4alpha point again to the probable significance of this protein in control of proliferation and, possibly, of apoptosis, as well as in regulation of cell motility [112].
So, being a key factor of differentiation of several types of epithelial tissues, HNF4alpha is involved in the control of cell proliferation. Its antiproliferative function was noted particularly in the cell types where HNF4alpha is involved in regulation of tissue-specific gene expression.
The significance of HNF4alpha for maintenance of the epithelial cell morphology is defined by its involvement in regulation of genes whose products form tight, gap, and adhesion junctions and participate in interaction with extracellular matrix.
Exogenous HNF4alpha expression in cells of H5 dedifferentiated rat hepatoma causes re-expression of the adhesion molecule E-cadherin [104]. HNF4alpha inactivation in mouse embryonic liver causes disturbance of morphogenesis due to alteration of membrane localization of E-cadherin, adhesion molecule CEACAM1, and the tight junction protein ZO-1, as well as transcriptional repression of genes encoding the gap junction proteins (connexin 32 and 26) [49].
Curiously, in Caco-2 colorectal carcinoma enterocytes, an influence of E-cadherin membrane localization on HNF4alpha transcription activity was noted (see below), which is indicative of the existence of a feedback loop between cell adhesion and expression of tissue-specific regulatory factors [113]. However, in some model systems, particularly during one-step progression of mouse HCC, HNF4alpha re-expression is not sufficient for restoration of E-cadherin synthesis and/or membrane localization, and the restoration of cell adhesion is probably due to different proteins of this family.
Analysis of transcription changes caused by HNF4alpha inactivation in mouse embryonic liver has shown that this factor defines the expression of 27 genes encoding a broad spectrum of adhesion proteins: components of tight, adhesion, and gap junctions, desmosomes, and other proteins involved in intercellular interactions and interactions with ECM, and in regulation of cytoskeleton and also targets of the MAP-kinase and Rho signaling pathways. Regulatory sequences of most of these genes contain HNF4alpha binding sites; for eight of them (E-cadherin, connexin 32, occludin, claudin 1, Jam-A, epiplaquin 1, galectin 9, and Crb3) in vivo HNF4alpha binding with corresponding regulatory regions was already shown [50].
It is necessary to note that the HNF4alpha-dependent regulation of a major protein of the liver gap junctions (connexin 32) can be carried out not only due to HNF4alpha direct binding to the promoter of this gene, but can also be mediated via activation of another transcription factor, HNF1alpha, which is also able to activate this promoter [114, 115]. One more HNF1alpha target is the tight junction component claudin-2 gene. The effect of HNF1alpha on this gene promoter is tissue-specific: it increases the level of claudin-2 transcription in Caco-2 colorectal carcinoma cells and is necessary for the expression of this gene in liver; claudin-2 activation in kidney epithelium is HNF1alpha-independent [116]. It is interesting that the decrease of claudin-2 transcription was also registered in embryonal intestinal cells upon HNF4alpha_inactivation [53].
In F9 mouse embryonic carcinoma cells exogenous expression of HNF4alpha results in cell polarization associated with localization of the tight junction proteins occludin, claudin-6, claudin-7, and ZO-1 on the apical part of the lateral membrane, while the expression of occludin, claudin-6, and claudin-7 genes was shown to be HNF4alpha-dependent [117]. Later it was demonstrated on the same model that HNF4alpha induced expression of the tight junction protein JAM-A (receptor of F11), and it was confirmed that the HNF4alpha expression promotes localization of the tight junction proteins on the apical part of the membrane and so provides for the cells acquiring a more differentiated phenotype [118]. Also, in F9 embryonic carcinoma cells exogenous expression of HNF4alpha induces formation of microvilli, specialized protrusions of plasma membrane localized on apical surfaces of epithelial cells. These data is consistent with observation concerning the absence of microvilli on the surface of hepatocytes forming bile canaliculi in the liver of HNF4alpha-/- mice [49]. Evidently, at least in some types of epithelial cells formation of microvilli is defined by the HNF4alpha-dependent expression of the gene of ezrin/radixin/moesin-binding phosphoprotein 50 [119].
In HEK293 human embryonic kidney cells, expression of HNF4alpha, on the contrary, results in loss of epithelial morphology and weakening intercellular contacts. In HEK293 cells a number of HNF4alpha-responsive genes were detected that encode adhesion molecules plakophilin 2, desmocollin 2, type XXI collagen, chondroitin sulfate proteoglycan 2, as well as a number of cytoskeleton proteins, but mechanisms of morphological changes induced by this transcription factor are still unclear [57].
Thus, data obtained on different model systems clearly demonstrate the important role of HNF4alpha in regulation of expression of cell adhesion molecules, ECM components, and cytoskeleton proteins that define the basic morphological characteristics of epithelial cells.
A number of recent investigations show that the number of genes directly or indirectly regulated by HNF4alpha significantly exceeds the number of targets of other tissue-specific regulatory factors [120]. This is an additional argument indicating that HNF4alpha occupies a principally important place in coordination of various processes that control homeostasis of hepatocytes and some other types of epithelial cells. It can be expected that detailed analysis of regulatory mechanisms and biological functions of new HNF4alpha targets will make possible more complete understanding of the role of this factor in normal development and in malignancy.
WHAT MECHANISMS CAN DETERMINE THE IMPAIRMENT OF HNFs FUNCTION IN
HEPATOCARCINOGENESIS?
As mentioned above, there is a complex system of mutual regulation between HNFs (and, probably, between components of other tissue-specific networks), which undergoes significant modification depending on the stage of development and tissue-specific context. Table 1 shows the combined data concerning transcriptional regulators of each HNF obtained during investigation of various experimental systems and stages of ontogenesis as described in the literature. Sometimes these data are controversial and are indicative of the regulation possibilities rather than of exact hierarchic relationships for a concrete cell context. The central place of HNF4alpha in the control of hepatocyte differentiation is probably defined not only by the broad spectrum of its targets, but also by a variety of factors influencing its expression and/or transcriptional activity.
As also mentioned above, transcription of HNF4alpha gene is driven from two independently regulated promoters [43]. In P1 promoter, regulating expression of “adult” isoforms, binding sites for factors of HNF1, FoxA, HNF6, and GATA6 families were revealed [17, 121]. The existence of two major alternative mechanisms of activation of HNF4alpha1 expression is supposed, namely, the cooperative action of HNF1beta and GATA6 factors at early stages of ontogenesis or synergistic activation by HNF1alpha and HNF6 factors in differentiated cells [121]. The regulation of P1 activity may also involve FTF and COUP-TFII, the latter being a negative regulator [121, 122]. The enhancer in which binding sites for C/EBP, HNF4, FoxA, and HNF1 are mapped along with those for nuclear receptors is necessary to provide the maximal level of HNF4alpha1 expression in differentiated cells [123].
The alternative promoter P2 is located 45.6 kb upstream from P1. It can be activated by HNF1alpha, HNF1beta, HNF6, and by pancreatic regulatory factor Pdx1, which is likely critical for the expression of “embryonic” isoforms in the pancreas. Inhibition of P2 activity in differentiated hepatocytes is probably due to repressor activity of HNF4alpha1 [17, 45, 124].
Our experiments on the dedifferentiated mouse HCC have shown that exogenous expression of none of the HNFs, which, according to the literature, can activate HNF4alpha transcription, is sufficient to restore its expression in that system (Lazarevich, unpublished). This observation does not deny a possible role of these factors in tumor progression, but support the idea that the derangement of expression of any HNF cannot be considered as the primary cause of HNF4alpha repression in the studied system.
ECM and growth factors are probably the most significant elements of the microenvironment capable of influencing hepatocyte differentiation and maintenance of epithelial morphology [13]. A growing number of recently obtained data indicates that these factors to a considerable extent control the activity of HNF regulatory network in normal and tumor cells.
Disruption of liver architecture during isolation of primary hepatocyte cultures is accompanied by destruction of intercellular contacts and cell-matrix interaction, and causes massive changes in the cell transcription program. Even in the presence of growth factors, required for this cell type, in primary hepatocytes the loss of polarity is observed along with alteration of cell morphology and rapid repression of numerous tissue-specific genes. This dedifferentiation can be reversed at least in part by cultivation of hepatocytes in the three-dimensional matrix produced by cells of the Engelbreth-Holm-Swarm mouse sarcoma (EHS) [125]. This matrix contains laminin, type IV collagen, heparan sulfate proteoglycan, entactin, and some growth factors. During reversible dedifferentiation in culture, transcription levels of key factors responsible for the transcription program of mature hepatocytes (C/EBPalpha, HNF4alpha. and its direct target HNF1) decrease, whereas cultivation in the EHS matrix restores HNF4alpha and HNF1alpha transcription and enhances the C/EBPalpha DNA-binding activity [126-128]. Thus, it can be supposed that HNF4alpha and C/EBPalpha mediate matrix-dependent redifferentiation of primary hepatocytes.
C/EBPalpha is perhaps not the key link in this process because its absence does not influence the ECM-dependent induction of albumin gene expression in primary hepatocytes isolated from C/EBPalpha-/- mice [129]. Simultaneously, HNF4alpha inactivation in primary rat hepatocytes by interfering RNA prevents the EHS-induced upregulation of the hepatocyte differentiation markers but has no effect on the organization of the actin cytoskeleton [128]. Adenovirus-mediated exogenous expression of HNF4alpha in these cells contributes to the long-term maintenance of morphological and functional hepatocyte differentiation [130].
Early steps of liver specification and induction of hepatospecific gene expression are defined by the activity of FoxA family factors [15]. In some model systems the influence of ECM on the activity of this family members and albumin gene transcription was described [131]. However, FoxA1 expression in primary hepatocytes increases during differentiation, whereas cultivation in EHS gel is accompanied by decrease of FoxA DNA-binding activity [126, 132]. Thus, it is unlikely that induction of the hepatic differentiation program in this system is governed by the activity of FoxA family factors. This is consistent with data indicating that the impact of FoxA factors in the control of differentiation in mature hepatocytes markedly decreases compared to early steps of embryogenesis [133].
Similar mechanisms were described on another experimental model in which “small hepatocytes” (probably oval liver cells or their derivatives) under ECM influence undergo differentiation, accompanied by acquiring of mature hepatocyte-specific morphological features and induction of functional gene transcription [134]. These alterations are accompanied by activation of HNF4alpha, HNF6, C/EBPalpha, and C/EBPbeta transcription. It is important to note that neither individual growth factors nor distinct matrix components of the EHS gel are able to induce the described morphological and functional differentiation.
Analysis of gene expression profiles in hepatocytes, cultivated in EHS gel or without it, made it possible to reveal the significant ECM-mediated induction of phosphatidylinositol 4,5-diphosphate (PI[4,5]P2) phosphatase. This decreases PI(4,5)P2 level, which affects actin organization via alteration of different actin-regulating protein activities and actin polymerization in response to extracellular stimuli. Actin depolymerization observed in EHS gel can be reversed using inhibitors by increasing intracellular level of PI(4,5)P2 and is accompanied by lowering expression of HNF4alpha and markers of hepatocyte differentiation [135].
In Caco-2 enterocytes cell culture, the dependence of HNF4alpha intracellular distribution and transcription activity upon enhancement or weakening of E-cadherin-mediated intercellular adhesion was described [113].
All these data point to HNF4alpha as the central element that determines the dependence of the transcription program of normal hepatocytes on such elements of microenvironment as ECM, intercellular contacts, and growth factors produced by different cell types.
From the first stages of embryonic development cytokines and growth factors produced by mesenchymal cells play an essential role in activation of tissue-specific differentiation programs [15]. These factors are able both to regulate directly intracellular HNFs levels and modulate their activities depending on the position of the cell in the total architecture of the organ [136]. Further realization of the hepatocyte transcription program, maintenance of their functional and structural differentiation, and proliferation control are also mediated by the action of different factors produced by stromal cells. During the progression of malignant phenotype, tumor cells acquire the capability of autocrine secretion of growth factors that not only activate proliferation but provide those cells additional independence from microenvironment [137].
Growth factors are capable of modulating HNF expression in cooperation with ECM alterations. Thus, the expression of HNF4alpha in the cultures of fetal mouse hepatocytes can be induced by ECM and the interleukin-6-related factor oncostatin M [138]. This differentiation effect can be suppressed by tumor necrosis factor alpha [139].
The transforming growth factor beta (TGFbeta) plays a dual role in the control of proliferation and differentiation of liver and other epithelial tissues. In normal hepatocytes, it inhibits proliferation and contributes to cell differentiation. In transformed cells, activation of TGFbeta-induced signaling cascades is able to induce EMT through regulation of genes encoding ECM components, integrins, and metalloproteinases. Recent data show that the TGFbeta signaling substantially affects the HNF4alpha-dependent differentiation of hepatocytes. HNF4alpha was shown to directly interact with transcription factors Smad3 and Smad4 being the key effectors of TGFbeta-dependent signalization [140].
In immortalized mouse hepatocytes which express constitutively active c-Met (MMH), TGFbeta induces EMT and dedifferentiation accompanied by suppression of HNF1alpha and HNF4alpha transcription [141]. Inhibition of HNF4alpha expression is also described upon the TGFbeta-induced EMT in cultures of fetal rat hepatocytes [142] and in MMH hepatocytes transformed by activated Ha-Ras oncogene [137]. In all these cases, the authors reported overexpression of the Snail gene. It was shown that Snail overexpression in MMH cells is sufficient for EMT induction, inhibition of expression of a number of epithelial adhesion proteins, activation of matrix metalloproteinase 2, and inhibition of HNF4alpha transcription due to the direct binding of Snail to the HNF4alpha1 promoter [143]. Thus, at least some of the effects caused by activation of the TGFbeta signaling in this system are determined by the Snail-dependent inhibition of the HNF4alpha1 gene activity.
It was shown in another study that the TGFbeta treatment of primary rat hepatocytes causes, along with dedifferentiation, inhibition of HNF4alpha transcription due to activation of transcription factor WT1 [144]. Additional putative mechanism of TGFbeta impact on HNF4alpha transcriptional activity is the TGFbeta-dependent proteosomal degradation of this protein described in HepG2 human hepatoma cells [145].
Alterations of HNF4alpha transcription can be observed upon activation of MAP-kinase signalization. Treatment of HepG2 human hepatoma cells with the protein kinase C activator phorbol-12-myristate-13-acetate activates the Ras-Raf-MEK-Erk signaling cascade, which inhibits HNF4alpha transcription. This effect is mediated by inhibition of the C/EBPalpha expression and breaking the interaction between HNF4alpha1 promoter and enhancer regulatory regions [146].
Certainly HNF4alpha activity is regulated not only at the level of transcription. The activity of this factor depends on its interaction with different cofactors, differentially expressed in various cell types, on acetylation and phosphorylation carried out by various kinases, as well as on the rate of protein degradation [21, 40]. It is also supposed that the HNF4alpha transcriptional activity can be modulated by presumable ligands, but up to now there is no clear indication of ligand-dependent regulation of HNF4alpha function in vivo. Most likely, mechanisms of HNF4alpha inactivation and its consequences can vary in different cell types depending on the presence of different transcription factors and activity of various signal cascades. Probably these differences also affect the range of the HNF4alpha target genes in each particular system.
The data described above obtained on different model systems including normal and tumor cell cultures, in which many of regulatory relationships realized in vivo are distorted, do not yet allow reconstruction of a general controlling mechanism of HNF4alpha-dependent transcription. However, presented examples clearly indicate the important role of the cellular microenvironment in regulation of activity of tissue-specific factors controlling differentiation of normal and tumor hepatocytes. Apparently, similar regulatory mechanisms can be realized in other types of epithelial cells. For example, factors of C/EBP family playing an important role in mammary gland epithelium differentiation are clearly involved in the ECM-induced tissue-specific activation of alphas1- and beta-casein genes [147, 148]. An additional exemplification of the suggested thesis is the Snail-dependent repression of HNF1beta, essential for differentiation of kidney epithelium, which, in turn, defines the disturbance of epithelial homeostasis and inhibition of expression of specific for this cell type cadherin-16 [149].
HNFs DYSFUNCTION IN OTHER TYPES OF EPITHELIAL TUMORS
Recently reported data concerning HNFs dysfunction in other types of epithelial tumors are summarized in Table 2. One can see that mutations, deletions, and reduced expression of transcription factors are observed mainly in tumors originated from tissues which differentiation critically depends on particularly these transcription factors. For example the decrease in expression of the antiproliferative protein C/EBPalpha was observed during carcinogenesis of mammary gland, skin, and lung [74, 75]. In lung cancer, the direct correlation between the differentiation state and expression of this gene was shown. Moreover, the possibility of partial reversion of malignant phenotype upon restoration of C/EBPalpha expression is described [150]. Apparently, the impact of C/EBPalpha inactivation in this type of tumors is not limited by the direct antiproliferative activity of the factor. One of the consequences of derangement of the normal function of the gene is inhibition of transcription of FoxA2 gene, which was shown to be direct transcriptional target of C/EBPalpha in this cell type. Restoration of FoxA2 expression in tumor cells causes proliferation arrest and induction of apoptosis [151]. As noted above, both FoxA2 and C/EBPalpha are crucial for specification and differentiation of respiratory epithelium during embryonic development [152]. A similar situation is described for the HNF1 family factors that are essential for kidney morphogenesis and differentiation, regulate a number of tissue-specific genes, and are frequently inactivated in renal carcinogenesis and a number of other kidney pathologies [153].
Table 2. Impairment of HNFs function in
epithelial tumors
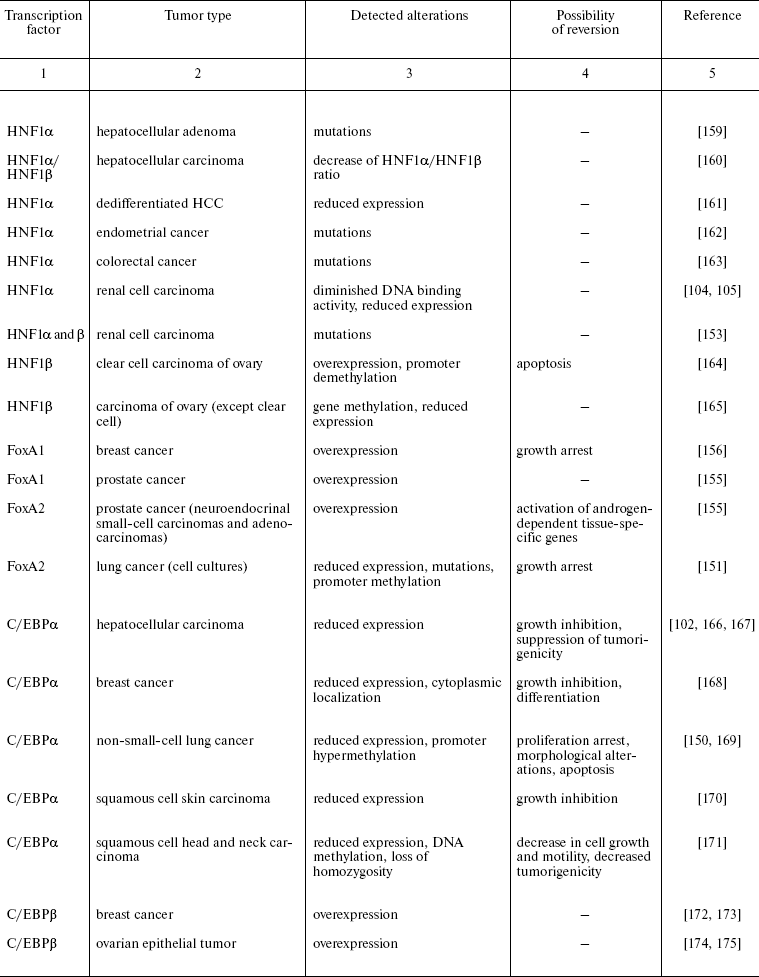
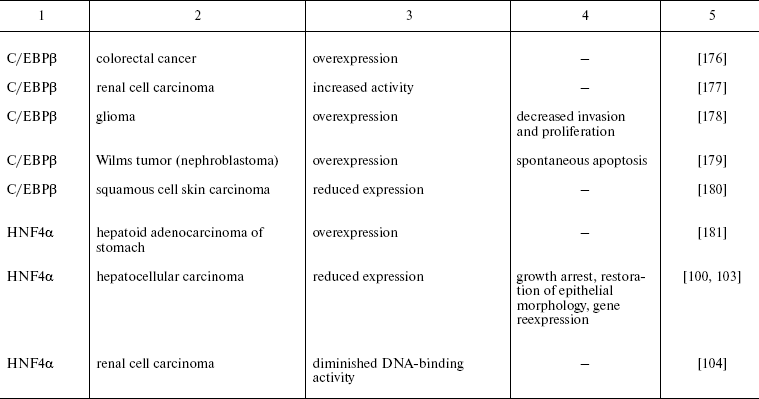
Another variant of functional impairment of transcription factors--overexpression in the tissues where their level is normally low--may result in alteration of activity of the array of genes involved in the control of proliferation and morphogenesis. Particularly such situation was described in estrogen-dependent luminal type A breast cancer and in FoxA1 hyperexpressing prostate cancer [154, 155]. As discussed above, FoxA transcription factors are able to modulate conformation of chromatin regions making them accessible for other regulatory proteins with closely located binding sites. In hormone-dependent tumor cells, FoxA factors facilitate interaction of nuclear receptors with corresponding binding sites frequently located close to the sequences recognized by FoxA [156]. For example, this mechanism is realized upon FoxA1-dependent activation of the promoter of cyclin D1 oncogene in breast tumors expressing estrogen receptor, which results in accelerated proliferation [157]. In prostate cells, FoxA1 is necessary for androgen-dependent activation of a number of tissue-specific genes such as that of prostate-specific antigen [158]. FoxA1 and 2 are differentially expressed in different types of prostate tumors, and in this case FoxA2 that is detected mainly in malignant tumors evidently can activate androgen-responsive genes in receptor- and ligand-independent manner [155]. These observations not only suppose the prognostic significance of HNF overexpression in some types of tumors but also allow considering them as promising targets for antitumor therapy.
In the last few years, considerable progress in understanding the regulatory events that govern specification and differentiation of epithelial tissues and organs during development has been achieved. Factors which play critical roles in these processes together with the consequence of their activation and hierarchic relationships within tissue-specific transcription networks are actively investigated. In one of the most explored networks of transcription regulation, HNF regulatory cascade that specifies differentiation of hepatocytes, the central role is played by nuclear receptor HNF4alpha that controls processes of differentiation, proliferation, and morphogenesis. This factor is able to modulate the transcription program of cells both directly and also through activation of other tissue-specific transcription factors. At the same time, expression of HNF4alpha gene is to a great extend controlled by the local microenvironment. It can be supposed that HNF4alpha plays the role of specific sensor that receives signals from the outside of the cell (growth factors and alterations of tissue architecture) and transforms them into alterations of the transcription program.
Inhibition of HNF4alpha expression in tumor cells results in acceleration of cell proliferation, loss of epithelial morphology, dedifferentiation, and finally, in acquiring the ability for invasion and metastasis. It is not surprising that the inactivation of HNF4alpha, conferring a number of selective advantages to the tumor cell, is frequent event during HCC progression and correlates with more malignant tumor phenotype. The reason for the loss of HNF4alpha function during HCC progression might be not only the aberrations of the gene structure but also changes in different upstream signaling cascades. The variety of pathways of HNF4alpha activity regulation defines the high frequency of decrease of its expression during HCC progression, since the failure in the function of this regulatory network can be caused by diverse events, from distortion of extracellular signalization to damage of effector mechanisms.
Our investigations on the model system of the mouse HCC one-step progression have shown that restoration of HNF4alpha function causes at least partial reversion of malignant phenotype both in vitro and in vivo [100]. These results suggest that tissue-specific transcription factors, especially those that define key steps of realization of certain differentiation programs and are able to receive or modulate signals from the cell's microenvironment, can be considered as promising candidates for the role of tumor suppressors in the tissue types where they normally play the important role. This suggestion points to the possibility for development of new approaches to therapy of tumors through forced increase of their level of differentiation.
Experimental data cited in this review were obtained with a financial support from the US Civilian Research and Development Foundation for the Independent States of the Former Soviet Union (No. RUB1-2662-MO-05), the Russian Foundation for Basic Research (Nos. 07-04-01422 and 07-04-12151-ofi), and grants of the President of Russian Federation for support of young Russian scientists and scientific schools.
REFERENCES
1.Hanahan, D., and Weinberg, R. A. (2000)
Cell, 100, 57-70.
2.Hahn, W. C., and Weinberg, R. A. (2002) Nature
Rev. Cancer, 2, 331-341.
3.Kopnin, B. P. (2004) in Encyclopedia of Clinical
Oncology (Davydov, M. I., ed.) [in Russian], RLS-Press, Moscow, pp.
34-53.
4.Savagner, P. (2001) BioEssays, 23,
912-923.
5.Thiery, J. P. (2002) Nature Rev. Cancer,
2, 442-454.
6.Thiery, J. P., and Sleeman, J. P. (2006) Nat.
Rev. Mol. Cell. Biol., 7, 131-142.
7.Nieto, M. A. (2002) Nat. Rev. Mol. Cell.
Biol., 3, 155-166.
8.Peinado, H., Portillo, F., and Cano, A. (2004)
Int. J. Dev. Biol., 48, 365-375.
9.Hood, J. D., and Cheresh, D. A. (2002) Nature
Rev. Cancer, 2, 91-100.
10.Abelev, G. I. (2000) Biochemistry
(Moscow), 65, 107-116.
11.Abelev, G. I. (2003) Mol. Biol. (Moscow),
37, 4-11.
12.Abelev, G. I., and Eraiser, T. L. (1999)
Semin. Cancer Biol., 9, 95-107.
13.Abelev, G. I., and Lazarevich, N. L. (2006)
Adv. Cancer Res., 95, 61-113.
14.Zaret, K. (2002) Nat. Rev. Genet.,
3, 499-512.
15.Lemaigre, F., and Zaret, K. (2004) Curr. Opin.
Genet. Dev., 14, 582-590.
16.Tronche, F., and Yaniv, M. (1992)
Bioessays, 14, 579-587.
17.Kyrmizi, I., Hatzis, P., Katrakili, N., Tronche,
F., Gonzalez, F. J., and Talianidis, I. (2006) Genes Dev.,
20, 2293-2305.
18.Cereghini, S. (1996) FASEB J., 10,
267-282.
19.Lazarevich, N. L. (2000) Biochemistry
(Moscow), 65, 117-133.
20.Locker, J. (2001) in Transcription Factors
(Locker, J., ed.) BIOS Scientific Publishers, Oxford, pp. 237-262.
21.Schrem, H., Klempnauer, J., and Borlak, J. (2002)
Pharmacol. Rev., 54, 129-158.
22.Costa, R. H., Kalinichenko, V. V., Holterman, A.
X., and Wang, X. (2003) Hepatology, 38, 1331-1347.
23.Tronche, F., Ringeisen, F., Blumenfeld, M.,
Yaniv, M., and Pontoglio, M. (1997) J. Mol. Biol., 266,
231-245.
24.Shih, H. H., Xiu, M., Berasi, S. P., Sampson, E.
M., Leiter, A., Paulson, K. E., and Yee, A. S. (2001) Mol. Cell.
Biol., 21, 5723-5732.
25.Coffinier, C., Barra, J., Babinet, C., and Yaniv,
M. (1999) Mech. Dev., 89, 211-213.
26.Barbacci, E., Reber, M., Ott, M. O., Breillat,
C., Huetz, F., and Cereghini, S. (1999) Development, 126,
4795-4805.
27.Coffinier, C., Gresh, L., Fiette, L., Tronche,
F., Schutz, G., Babinet, C., Pontoglio, M., Yaniv, M., and Barra, J.
(2002) Development, 129, 1829-1838.
28.Haumaitre, C., Barbacci, E., Jenny, M., Ott, M.
O., Gradwohl, G., and Cereghini, S. (2005) Proc. Natl. Acad. Sci.
USA, 102, 1490-1495.
29.Ott, M. O., Rey-Campos, J., Cereghini, S., and
Yaniv, M. (1991) Mech. Devel., 36, 47-58.
30.Pontoglio, M., Barra, J., Hadchouel, M., Doyen,
A., Babinet, C., and Yaniv, M. (1996) Cell, 84,
575-585.
31.Pontoglio, M., Sreenan, S., Roe, M., Pugh, W.,
Ostrega, D., Doyen, A., Pick, A. J., Baldwin, A., Velho, G., Froguel,
P., Levisetti, M., Bonner-Weir, S., Bell, G. I., Yaniv, M., and
Polonsky, K. S. (1998) J. Clin. Invest., 101,
2215-2222.
32.Walters, J. R. (2004) Curr. Opin.
Gastroenterol., 20, 70-76.
33.Clark, K. L., Halay, E. D., Lai, E., and Burley,
S. K. (1993) Nature, 364, 412-420.
34.McPherson, C. E., Shim, E. Y., Friedman, D. S.,
and Zaret, K. S. (1993) Cell, 75, 387-398.
35.Cirillo, L. A., and Zaret, K. S. (2007) J.
Mol. Biol., 366, 720-724.
36.Cirillo, L. A., Lin, F. R., Cuesta, I., Friedman,
D., Jarnik, M., and Zaret, K. S. (2002) Mol. Cell, 9,
279-289.
37.Friedman, J. R., and Kaestner, K. H. (2006)
Cell Mol. Life Sci., 63, 2317-2328.
38.Sladek, F. M., Zhong, W., Lai, E., and Darnell,
J. E., Jr. (1990) Genes Dev., 4, 2353-2364.
39.Drewes, T., Senkel, S., Holewa, B., and Ryffel,
G. U. (1996) Mol. Cell. Biol., 16, 925-931.
40.Sladek, F. M., and Seidel, S. D. (2001) in
Nuclear Receptors and Genetic Diseases (Burris, T. P., and
McCabe, E., eds.) Academic Press, London, pp. 309-361.
41.Hertz, R., Magenheim, J., Berman, I., and
Bar-Tana, J. (1998) Nature, 392, 512-516.
42.Ruse, M. D., Jr., Privalsky, M. L., and Sladek,
F. M. (2002) Mol. Cell. Biol., 22, 1626-1638.
43.Nakhei, H., Lingott, A., Lemm, I., and Ryffel, G.
U. (1998) Nucleic Acids Res., 26, 497-504.
44.Torres-Padilla, M. E., Fougere-Deschatrette, C.,
and Weiss, M. C. (2001) Mech. Dev., 109, 183-193.
45.Thomas, H., Jaschkowitz, K., Bulman, M.,
Frayling, T. M., Mitchell, M. S., Roosen, S., Lingott-Frieg, A., Tack,
C. J., Ellard, S., Ryffel, G. U., and Hattersley, A. T. (2001) Hum.
Mol. Gen., 10, 2089-2097.
46.Torres-Padilla, M. E., Sladek, F. M., and Weiss,
M. C. (2002) J. Biol. Chem., 277, 44677-44687.
47.Duncan, S. A., Nagy, A., and Chan, W. (1997)
Development, 124, 279-287.
48.Li, J., Ning, G., and Duncan, S. A. (2000)
Genes Dev., 14, 464-474.
49.Parviz, F., Matullo, C., Garrison, W. D.,
Savatski, L., Adamson, J. W., Ning, G., Kaestner, K. H., Rossi, J. M.,
Zaret, K. S., and Duncan, S. A. (2003) Nat. Gen., 34,
292-296.
50.Battle, M. A., Konopka, G., Parviz, F., Gaggl, A.
L., Yang, C., Sladek, F. M., and Duncan, S. A. (2006) Proc. Natl.
Acad. Sci. USA, 103, 8419-8424.
51.Nammo, T., Yamagata, K., Tanaka, T., Kodama, T.,
Sladek, F. M., Fukui, K., Katsube, F., Sato, Y., Miyagawa, J. I., and
Shimomura, I. (2008) Gene Exp. Patterns, 8, 96-106.
52.Gupta, R. K., Gao, N., Gorski, R. K., White, P.,
Hardy, O. T., Rafiq, K., Brestelli, J. E., Chen, G., Stoeckert, C. J.,
Jr., and Kaestner, K. H. (2007) Genes Dev., 21,
756-769.
53.Garrison, W. D., Battle, M. A., Yang, C.,
Kaestner, K. H., Sladek, F. M., and Duncan, S. A. (2006)
Gastroenterology, 130, 1207-1220.
54.Stegmann, A., Hansen, M., Wang, Y., Larsen, J.
B., Lund, L. R., Ritie, L., Nicholson, J. K., Quistorff, B.,
Simon-Assmann, P., Troelsen, J. T., and Olsen, J. (2006) Physiol.
Genomics, 27, 141-155.
55.Wang, D., Yamamoto, S., Hijiya, N., Benveniste,
E. N., and Gladson, C. L. (2000) Oncogene, 19,
5801-5809.
56.Jiang, S., Tanaka, T., Iwanari, H., Hotta, H.,
Yamashita, H., Kumakura, J., Watanabe, Y., Uchiyama, Y., Aburatani, H.,
Hamakubo, T., Kodama, T., and Naito, M. (2003) Nucl. Recept.,
1, 5.
57.Lucas, B., Grigo, K., Erdmann, S., Lausen, J.,
Klein-Hitpass, L., and Ryffel, G. U. (2005) Oncogene, 24,
6418-6431.
58.Lemaigre, F. P., Durviaux, S. M., Truong, O.,
Lannoy, V. J., Hsuan, J. J., and Rousseau, G. G. (1996) Proc. Natl.
Acad. Sci. USA, 93, 9460-9464.
59.Jacquemin, P., Lemaigre, F. P., and Rousseau, G.
G. (2003) Dev. Biol., 258, 105-116.
60.Vanhorenbeeck, V., Jacquemin, P., Lemaigre, F.
P., and Rousseau, G. G. (2002) Biochem. Biophys. Res. Commun.,
292, 848-854.
61.Poll, A. V., Pierreux, C. E., Lokmane, L.,
Haumaitre, C., Achouri, Y., Jacquemin, P., Rousseau, G. G., Cereghini,
S., and Lemaigre, F. P. (2006) Diabetes, 55, 61-69.
62.Vanhorenbeeck, V., Jenny, M., Cornut, J. F.,
Gradwohl, G., Lemaigre, F. P., Rousseau, G. G., and Jacquemin, P.
(2007) Dev. Biol., 305, 685-694.
63.Clotman, F., Lannoy, V. J., Reber, M., Cereghini,
S., Cassiman, D., Jacquemin, P., Roskams, T., Rousseau, G. G., and
Lemaigre, F. P. (2002) Development, 129, 1819-1828.
64.Tan, Y., Yoshida, Y., Hughes, D. E., and Costa,
R. H. (2006) Gastroenterology, 130, 1283-1300.
65.Margagliotti, S., Clotman, F., Pierreux, C. E.,
Beaudry, J. B., Jacquemin, P., Rousseau, G. G., and Lemaigre, F. P.
(2007) Dev. Biol., 311, 579-589.
66.Schrem, H., Klempnauer, J., and Borlak, J. (2004)
Pharmacol. Rev., 56, 291-330.
67.McKnight, S. L. (2001) Cell, 107,
259-261.
68.Cassel, T. N., and Nord, M. (2003) Am. J.
Physiol. Lung Cell Mol. Physiol., 285, L773-781.
69.Zahnow, C. A. (2002) Breast Cancer Res.,
4, 113-121.
70.Ramji, D. P., and Foka, P. (2002) Biochem.
J., 365, 561-575.
71.Greenbaum, L. E., Cressman, D. E., Haber, B. A.,
and Taub, R. (1995) J. Clin. Invest., 96, 1351-1365.
72.Greenbaum, L. E., Li, W., Cressman, D. E., Peng,
Y., Ciliberto, G., Poli, V., and Taub, R. (1998) J. Clin.
Invest., 102, 996-1007.
73.Flodby, P., Barlow, C., Kylefjord, H.,
Ahrlund-Richter, L., and Xanthopoulos, K. G. (1996) J. Biol.
Chem., 271, 24753-24760.
74.Schuster, M. B., and Porse, B. T. (2006)
Biochim. Biophys. Acta, 1766, 88-103.
75.Fuchs, O. (2007) Folia Biol. (Praha),
53, 97-108.
76.Zaret, K. S. (2004) Nature, 429,
30-31.
77.Zhao, R., and Duncan, S. A. (2005)
Hepatology, 41, 956-967.
78.Boj, S. F., Parrizas, M., Maestro, M. A., and
Ferrer, J. (2001) Proc. Natl. Acad. Sci. USA, 98,
14481-14486.
79.Slack, J. M. (1995) Development,
121, 1569-1580.
80.Edlund, H. (2001) Diabetes, 50,
S5-S9.
81.Habener, J. F., Kemp, D. M., and Thomas, M. K.
(2005) Endocrinology, 146, 1025-1034.
82.Murtaugh, L. C. (2007) Development,
134, 427-438.
83.Cockell, M., Stolarczyk, D., Frutiger, S.,
Hughes, G. J., Hagenbuchle, O., and Wellauer, P. K. (1995) Mol.
Cell. Biol., 15, 1933-1941.
84.Yamagata, K., Nammo, T., Moriwaki, M., Ihara, A.,
Iizuka, K., Yang, Q., Satoh, T., Li, M., Uenaka, R., Okita, K.,
Iwahashi, H., Zhu, Q., Cao, Y., Imagawa, A., Tochino, Y., Hanafusa, T.,
Miyagawa, J., and Matsuzawa, Y. (2002) Diabetes, 51,
114-123.
85.Deramaudt, T. B., Sachdeva, M. M., Wescott, M.
P., Chen, Y., Stoffers, D. A., and Rustgi, A. K. (2006) J. Biol.
Chem., 281, 38385-38395.
86.Costa, R. H., Kalinichenko, V. V., and Lim, L.
(2001) Am. J. Physiol. Lung Cell. Mol. Physiol., 280,
L823-838.
87.Lazarevich, N. L. (2004) Uspekhi Biol.
Khim., 44, 365-418.
88.Ryffel, G. U. (2001) J. Mol. Endocrinol.,
27, 11-29.
89.Ulinski, T., Lescure, S., Beaufils, S., Guigonis,
V., Decramer, S., Morin, D., Clauin, S., Deschenes, G., Bouissou, F.,
Bensman, A., and Bellanne-Chantelot, C. (2006) J. Am. Soc.
Nephrol., 17, 497-503.
90.Meivar-Levy, I., Sapir, T., Gefen-Halevi, S.,
Aviv, V., Barshack, I., Onaca, N., Mor, E., and Ferber, S. (2007)
Hepatology, 46, 898-905.
91.Sapir, T., Shternhall, K., Meivar-Levy, I.,
Blumenfeld, T., Cohen, H., Skutelsky, E., Eventov-Friedman, S.,
Barshack, I., Goldberg, I., Pri-Chen, S., Ben-Dor, L., Polak-Charcon,
S., Karasik, A., Shimon, I., Mor, E., and Ferber, S. (2005) Proc.
Natl. Acad. Sci. USA, 102, 7964-7969.
92.Li, W. C., Horb, M. E., Tosh, D., and Slack, J.
M. (2005) Mech Dev., 122, 835-847.
93.Shen, C. N., Slack, J. M., and Tosh, D. (2000)
Nat. Cell Biol., 2, 879-888.
94.Lee, E. J., Russell, T., Hurley, L., and Jameson,
J. L. (2005) Mol. Endocrinol., 19, 964-971.
95.Bosch, F. X., Ribes, J., and Borras, J. (1999)
Semin. Liver Dis., 19, 271-285.
96.Thorgeirsson, S. S., and Grisham, J. W. (2002)
Nat. Gen., 31, 339-345.
97.Buendia, M. A. (2000) Cancer Biol.,
10, 185-200.
98.Engelhardt, N. V., Kudrjavtseva, E. I., Morozova,
O. V., Rudinskaya, T. D., and Turusov, V. S. (2000) Arkh.
Patol., 62, 24-29.
99.Kudrjavtseva, E. I., Morozova, O. V., Rudinskaya,
T. D., and Engelhardt, N. V. (2001) Arkh. Patol., 4,
33-37.
100.Lazarevich, N. L., Cheremnova, O. A., Varga, E.
V., Ovchinnikov, D. A., Kudrjavtseva, E. I., Morozova, O. V.,
Fleishman, D. I., Engelhardt, N. V., and Duncan, S. A. (2004)
Hepatology, 39, 1038-1047.
101.Kalkhul, A., Kaestner, K., Buchmann, A. B., and
Schwarz, M. (1996) Carcinogenesis, 17, 609-612.
102.Flodby, P., Liao, D. Z., Blanck, A.,
Xanthopoulos, K. G., and Hallstrom, I. P. (1995) Mol. Carcinog.,
12, 103-109.
103.Spath, G. F., and Weiss, M. C. (1998) J.
Cell. Biol., 140, 935-946.
104.Sel, S., Ebert, T., Ryffel, G. U., and Drewes,
T. (1996) Cancer Lett., 101, 205-210.
105.Anastasiadis, A. G., Lemm, I., Radzewitz, A.,
Lingott, A., Ebert, T., Ackermann, R., Ryffel, G. U., and Schulz, W. A.
(1999) Anticancer Res., 19, 2105-2110.
106.Kuo, C. J., Conley, P. B., Chen, L., Sladek, F.
M., Darnell, J. E., Jr., and Crabtree, G. R. (1992) Nature,
355, 457-461.
107.Jover, R., Bort, R., Gomez-Lechon, M. J., and
Castell, J. V. (2001) Hepatology, 33, 668-675.
108.Chiba, H., Itoh, T., Satohisa, S., Sakai, N.,
Noguchi, H., Osanai, M., Kojima, T., and Sawada, N. (2005) Exp. Cell
Res., 302, 11-21.
109.Erdmann, S., Senkel, S., Arndt, T., Lucas, B.,
Lausen, J., Klein-Hitpass, L., Ryffel, G. U., and Thomas, H. (2007)
Biol. Chem., 388, 91-106.
110.Naiki, T., Nagaki, M., Shidoji, Y., Kojima, H.,
Imose, M., Kato, T., Ohishi, N., Yagi, K., and Moriwaki, H. (2002)
J. Biol. Chem., 277, 14011-14019.
111.Hwang-Verslues, W. W., and Sladek, F. M. (2008)
Mol. Endocr., 22, 78-90.
112.Niehof, M., and Borlak, J. (2005) Mol.
Pharm., 67, 604-611.
113.Peignon, G., Thenet, S., Schreider, C.,
Fouquet, S., Ribeiro, A., Dussaulx, E., Chambaz, J., Cardot, P.,
Pincon-Raymond, M., and le Beyec, J. (2006) J. Biol. Chem.,
281, 3560-3568.
114.Piechochki, M. P., Toti, R. M., Fernstorm, M.
J., Burk, R. D., and Ruch, R. J. (2000) Biochim. Biophys. Acta,
1491, 107-122.
115.Koffler, L. D., Fernstorm, M. J., Akiyama, T.
E., Gonzalez, F. J., and Ruch, R. J. (2002) Arch. Biochem.
Biophys., 407, 160-167.
116.Sakaguchi, T., Gu, X., Golden, H. M., Suh, E.,
Rhodas, D. B., and Reinecker, H. C. (2002) J. Biol. Chem.,
277, 21361-21370.
117.Chiba, H., Gotoh, T., Kojima, T., Satohisa, S.,
Kikuchi, K., Osanai, M., and Sawada, N. (2003) Exp. Cell Res.,
286, 288-297.
118.Satohisa, S., Chiba, H., Osanai, M., Ohno, S.,
Kojima, T., Saito, T., and Sawada, N. (2005) Exp. Cell Res.,
310, 66-78.
119.Chiba, H., Sakai, N., Murata, M., Osanai, M.,
Ninomiya, T., Kojima, T., and Sawada, N. (2006) J. Cell Biol.,
175, 971-980.
120.Odom, D. T., Zizlsperger, N., Gordon, D. B.,
Bell, G. W., Rinaldi, N. J., Murray, H. L., Volkert, T. L., Schreiber,
J., Rolfe, P. A., Gifford, D. K., Fraenkel, E., Bell, G. I., and Young,
R. A. (2004) Science, 303, 1378-1381.
121.Hatzis, P., and Talianidis, I. (2001) Mol.
Cell. Biol., 21, 7320-7330.
122.Pare, J. F., Roy, S., Galarneau, L., and
Belanger, L. (2001) J. Biol. Chem., 276, 13136-13144.
123.Bailly, A., Torres-Padilla, M. E., Tinel, A.
P., and Weiss, M. C. (2001) Nucleic Acids Res., 29,
3495-3505.
124.Briancon, N., Bailly, A., Clotman, F.,
Jacquemin, P., Lemaigre, F. P., and Weiss, M. C. (2004) J. Biol.
Chem., 279, 33398-33408.
125.Ben-Ze'ev, A., Robinson, G. S., Bucher, N. L.,
and Farmer, S. R. (1988) Proc. Natl. Acad. Sci. USA, 85,
2161-2165.
126.Oda, H., Imai, Y., Nakatsuru, Y., Hata, J.-I.,
and Ishikawa, T. (1995) Cancer Res., 56, 3320-3323.
127.Runge, D., Runge, D. M., Bowen, W. C., Locker,
J., and Michalopoulos, G. K. (1997) Biol. Chem., 378,
873-881.
128.Kimata, T., Nagaki, M., Ogiso, T., Naiki, T.,
Kato, T., and Moriwaki, H. (2006) Hepatology, 44,
140-151.
129.Soriano, H. E., Kang, D. C., Finegold, M. J.,
Hicks, M. J., Wang, N. D., Harrison, W., and Darlington, G. J. (1998)
Hepatology, 27, 392-401.
130.Naiki, T., Nagaki, M., Asano, T., Kimata, T.,
and Moriwaki, H. (2005) Biochem. Biophys. Res. Commun.,
335, 496-500.
131.Di Persio, C. M., Jackson, D. A., and Zaret, K.
S. (1991) Mol. Cell. Biol., 11, 4405-4414.
132.Runge, D., Runge, D. M., Drenning, S. D.,
Bowen, W. C., Jr., Grandis, J. R., and Michalopoulos, G. K. (1998)
Biochem. Biophys. Res. Commun., 250, 762-768.
133.Sund, N. J., Ang, S. L., Sackett, S. D., Shen,
W., Daigle, N., Magnuson, M. A., and Kaestner, K. H. (2000) Mol.
Cell. Biol., 20, 5175-5183.
134.Sugimoto, S., Mitaka, T., Ikeda, S., Harada,
K., Ikai, I., Yamaoka, Y., and Mochizuki, Y. (2002) J. Cell.
Biochem., 87, 16-28.
135.Kimata, T., Nagaki, M., Tsukada, Y., Ogiso, T.,
and Moriwaki, H. (2006) Hepatol. Res., 35, 3-9.
136.Clotman, F., Jacquemin, P., Plumb-Rudewiez, N.,
Pierreux, C. E., van der Smissen, P., Dietz, H. C., Courtoy, P. J.,
Rousseau, G. G., and Lemaigre, F. P. (2005) Genes Dev.,
19, 1849-1854.
137.Gotzmann, J., Huber, H., Thallinger, C.,
Wolschek, M., Jansen, B., Schulte-Hermann, R., Beug, H., and Mikulits,
W. (2002) J. Cell Sci., 115, 1189-1202.
138.Kamiya, A., Inoue, Y., and Gonzales, F. J.
(2003) Genes Dev., 13, 495-504.
139.Kamiya, A., and Gonzalez, F. J. (2004)
Hepatology, 40, 527-536.
140.Chou, W. C., Prokova, V., Shiraishi, K.,
Valcourt, U., Moustakas, A., Hadzopoulu-Cladaras, M., Zannis, V. I.,
and Kardassis, D. (2003) Mol. Biol. Cell, 14,
1279-1294.
141.Spagnoli, F. M., Cicchini, C., Tripodi, M., and
Weiss, M. C. (2000) J. Cell Sci., 113, 3639-3647.
142.Valdes, F., Alvarez, A. M., Locascio, A., Vega,
S., Herrera, B., Fernandez, M., Benito, M., Nieto, M. A., and Fabregat,
I. (2002) Mol. Cancer Res., 1, 68-78.
143.Cicchini, C., Filippini, D., Coen, S.,
Marchetti, A., Cavallari, C., Laudadio, I., Spagnoli, F. M., Alonzi,
T., and Tripodi, M. (2006) J. Cell Physiol., 209,
230-238.
144.Berasain, C., Herrero, J. I., Garcia-Trevijano,
E. R., Avila, M. A., Esteban, J. I., Maro, J. M., and Prieto, J. (2003)
Hepatology, 38, 148-157.
145.De Lucas, S., Lopez-Alcorocho, J. M.,
Bartolome, J., and Carreno, V. (2004) Biochem. Biophys. Res.
Commun., 321, 688-694.
146.Hatzis, P., Kyrmizi, I., and Talianidis, I.
(2006) Mol. Cell. Biol., 26, 7017-7029.
147.Myers, C. A., Schmidhauser, C.,
Mellentin-Michelotti, J., Fragoso, G., Roskelley, C. D., Casperson, G.,
Mossi, R., Pujuguet, P., Hager, G., and Bissel, M. J. (1998) Mol.
Cell. Biol., 18, 2184-2195.
148.Jolivet, G., Pantano, T., and Houdebine, L. M.
(2005) J. Cell Biochem., 95, 313-327.
149.Boutet, A., de Frutos, C. A., Maxwell, P. H.,
Mayol, M. J., Romero, J., and Nieto, M. A. (2006) EMBO J.,
25, 5603-5613.
150.Halmos, B., Huettner, C. S., Kocher, O.,
Ferenczi, K., Karp, D. D., and Tenen, D. G. (2002) Cancer Res.,
62, 528-534.
151.Halmos, B., Basseres, D. S., Monti, S., D'Alo,
F., Dayaram, T., Ferenczi, K., Wouters, B. J., Huettner, C. S., Golub,
T. R., and Tenen, D. G. (2004) Cancer Res., 64,
4137-4147.
152.Cardoso, W. V., and Lu, J. (2006)
Development, 133, 1611-1624.
153.Rebouissou, S., Vasiliu, V., Thomas, C.,
Bellanne-Chantelot, C., Bui, H., Chretien, Y., Timsit, J., Rosty, C.,
Laurent-Puig, P., Chauveau, D., and Zucman-Rossi, J. (2005) Hum.
Mol. Genet., 14, 603-614.
154.Nakshatri, H., and Badve, S. (2007) Expert
Opin. Ther. Targets, 11, 507-14.
155.Mirosevich, J., Gao, N., Gupta, A., Shappell,
S. B., Jove, R., and Matusik, R. J. (2006) Prostate, 66,
1013-1028.
156.Laganiere, J., Deblois, G., Lefebvre, C.,
Bataille, A. R., Robert, F., and Giguere, V. (2005) Proc. Natl.
Acad. Sci. USA, 102, 11651-11656.
157.Eeckhoute, J., Carroll, J. S., Geistlinger, T.
R., Torres-Arzayus, M. I., and Brown, M. (2006) Genes Dev.,
20, 2513-2526.
158.Gao, N., Zhang, J., Rao, M. A., Case, T. C.,
Mirosevich, J., Wang, Y., Jin, R., Gupta, A., Rennie, P. S., and
Matusik, R. J. (2003) Mol. Endocrinol., 17,
1484-1507.
159.Bluteau, O., Jeannot, E., Bioulac-Sage, P.,
Marques, J. M., Blanc, J. F., Bui, H., Beaudoin, J. C., Franco, D.,
Balabaud, C., Laurent-Puig, P., and Zucman-Rossi, J. (2002) Nat.
Genet., 32, 312-315.
160.Ninomiya, T., Hayashi, Y., Saijoh, K., Ohta,
K., Yoon, S., Nakabayashi, H., Tamaoki, T., Kasuga, M., and Itoh, H.
(1996) J. Hepatol., 25, 445-453.
161.Wang, W., Hayashi, Y., Ninomiya, T., Ohta, K.,
Nakabayashi, H., Tamaoki, T., and Itoh, H. (1998) J. Pathol.,
184, 272-278.
162.Rebouissou, S., Rosty, C., Lecuru, F.,
Boisselier, S., Bui, H., le Frere-Belfa, M. A., Sastre, X.,
Laurent-Puig, P., and Zucman-Rossi, J. (2004) Oncogene,
23, 7588-7592.
163.Laurent-Puig, P., Plomteux, O., Bluteau, O.,
Zinzindohoue, F., Jeannot, E., Dahan, K., Kartheuser, A., Chapusot, C.,
Cugnenc, P. H., and Zucman-Rossi, J. (2003) Gastroenterology,
124, 1311-1314.
164.Tsuchiya, A., Sakamoto, M., Yasuda, J., Chuma,
M., Ohta, T., Ohki, M., Yasugi, T., Taketani, Y., and Hirohashi, S.
(2003) Am. J. Pathol., 163, 2503-2512.
165.Terasawa, K., Toyota, M., Sagae, S., Ogi, K.,
Suzuki, H., Sonoda, T., Akino, K., Maruyama R., Nishikawa, N., Imai,
K., Shinomura, Y., Saito, T., and Tokino, T. (2006) Br. J.
Cancer, 94, 914-921.
166.Xu, L., Hui, L., Wang, S., Gong, J., Jin, Y.,
Wang, Y., Ji, Y., Wu, X., Han, Z., and Hu, G. (2001) Cancer
Res., 61, 3176-3181.
167.Watkins, P. J., Condreay, J. P., Huber, B. E.,
Jacobs, S. J., and Adams, D. J. (1996) Cancer Res., 56,
1063-1067.
168.Gery, S., Tanosaki, S., Bose, S., Bose, N.,
Vadgama, J., and Koeffler, H. P. (2005) Clin. Cancer Res.,
11, 3184-3190.
169.Tada, Y., Brena, R. M., Hackanson, B.,
Morrison, C., Otterson, G. A., and Plass, C. (2006) J. Natl. Cancer
Inst., 98, 396-406.
170.Shim, M., Powers, K. L., Ewing, S. J., Zhu, S.,
and Smart, R. C. (2005) Cancer Res., 65, 861-867.
171.Bennett, K. L., Hackanson, B., Smith, L. T.,
Morrison, C. D., Lang, J. C., Schuller, D. E., Weber, F., Eng, C., and
Plass, C. (2007) Cancer Res., 67, 4657-4664.
172.Zahnow, C. A., Younes, P., Laucirica, R., and
Rosen, J. M. (1997) J. Natl. Cancer Inst., 89,
1887-1891.
173.Bundy, L. M., and Sealy, L. (2003)
Oncogene, 22, 869-883.
174.Sundfeldt, K., Ivarsson, K., Carlsson, M.,
Enerback, S., Janson, P. O., Brannstrom, M., and Hedin, L. (1999)
Br. J. Cancer, 79, 1240-1248.
175.Arnett, B., Soisson, P., Ducatman, B. S., and
Zhang, P. (2003) Mol. Cancer, 2, 21.
176.Rask, K., Thorn, M., Ponten, F., Kraaz, W.,
Sundfeldt, K., Hedin, L., and Enerback, S. (2000) Int. J.
Cancer, 86, 337-343.
177.Oya, M., Horiguchi, A., Mizuno, R., Marumo, K.,
and Murai, M. (2003) Clin. Cancer Res., 9, 1021-1027.
178.Homma, J., Yamanaka, R., Yajima, N., Tsuchiya,
N., Genkai, N., Sano, M., and Tanaka, R. (2006) Oncol. Rep.,
15, 595-601.
179.Li, W., Kessler, P., Yeger, H., Alami, J.,
Reeve, A. E., Heathcott, R., Skeen, J., and Williams, B. R. G. (2005)
Cancer Res., 65, 2592-2601.
180.Oh, H. S., and Smart, R. C. (1998) J.
Invest. Dermatol., 110, 939-945.
181.Yano, T., Kishimoto, T., Tomaru, U., Kawarada,
Y., Kato, H., Yoshiki, T., and Ishikura, H. (2003) Pathology,
35, 75-78.