
|
REVIEW: Transport Proteins of the ABC Family and Multidrug Resistance of Tumor CellsA. A. Stavrovskaya* and T. P. StromskayaInstitute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; E-mail: astavrovskaya@yahoo.com; stromsktat@mail.ru* To whom correspondence should be addressed. |
Received November 24, 2007; Revision received February 1, 2008
Some new data concerning the role of transport proteins of the ABC family in multidrug resistance (MDR) of human tumor cells, and problems connected with regulation of these proteins are considered. MDR is a complex phenomenon that may be caused simultaneously by several mechanisms functioning in one and the same cell. Among them there may be the alterations of activity of several transport proteins. Activation of these proteins may be associated with alterations of activities of different cell protective systems and of the signal transduction pathways involved in regulation of proliferation, differentiation, and apoptosis. Clinical significance of multifactor MDR is discussed.
KEY WORDS: ABC family proteins, P-glycoprotein, multidrug resistance of tumorsDOI: 10.1134/S0006297908050118
Abbreviations: AP) acceleration phase; CML) chronic myelogenous leukemia; CP) chronic phase; MDR) multidrug resistance; MDT) transporters of multiple drugs; NBD) nucleotide binding domains; Pgp) P-glycoprotein; PI3K) phosphoinositol-3-kinase; PTEN) phosphatase and tensin homolog; Rh123) Rhodamine 123; RT-PCR) reverse transcription polymerase chain reaction; TMD) transmembrane binding domains.
The response of tumor cells to drugs can be defined by a number of
various molecular mechanisms operating at different stages (from the
penetration of the substance into the cell to cell death or arrest at
any stage of the cell cycle). Recently the set of mechanisms able to
define drug resistance has expanded, and our concepts of organization
of tumor cell protection have become more complicated. An example of
this complication is the detection during the last decade of a
significant number of cellular transport proteins of the ABC family
(below - ABC transporters), some of them operating at the first
step of the action of the toxic substance (at the stage of drug
penetration through the cell membrane and its intracellular
accumulation to effective concentrations) [1, 2]. These proteins determine multidrug resistance
(MDR) of tumor cells. MDR is a system of protection of the cell
population against numerous compounds, including drugs characterized by
different chemical structure and by different mechanisms of
intracellular activity.
SUPERFAMILY OF ABC PROTEINS
The ABC (ATP Binding Cassette) superfamily now includes ~300 proteins, among them transporters of quite different compounds [3-6]. Proteins of this family are characterized by the presence of an ATP-binding domain of specific structure. The Human Genome program and sequencing of different genomes led to a large number of ABC transporters being discovered during the last two years of the XXth century. The greatest number of ABC family transporters (129) was detected in plants owing to deciphering of the genome of Arabidopsis. Proteins of this family are present in all living organisms. About fifty ABC proteins have been found in man and approximately the same number in mouse [5]. Since a great number of ABC proteins were discovered only recently, many of them are still poorly studied. Studies of ABC proteins are important both for medicine and biology because they concern problems of protection of all living cells.
Human proteins of the ABC family are divided to seven subfamilies (Table 1) [5, 7]. The affiliation of each protein to a subfamily is determined by its domain organization, namely by the number and combination of transmembrane domains (TMDs) and ATP-binding domains (NBDs, nucleotide-binding domains). The NBDs of all proteins of this family are alike (they have 30-40% homology) independently of the transporter substrate specificity (which is quite different) and species affiliation [8].
Domain organization of ABC proteins is described in detail in a number of reviews [1, 3, 6, 8]. Briefly, at least four domains are necessary for the ABC transporter functioning. Two cytoplasmic NBDs bind and hydrolyze ATP. Each of two TMDs is represented by several (most often by six) transmembrane alpha-helices. Multidomain polypeptides formed by these four domains can be organized differently. Thus, bacterial transporters (for example, Sav1866) most often are homodimers incorporating one NBD and one TMD (two homodimers usually function together). In mammalian P-glycoprotein, all four domains are fused in one polypeptide. So, in the ABCB (MDR) subfamily proteins ABCB1 (P-glycoprotein, below Pgp), ABCB4 (Pgp3 or MDR2/3), and ABCB11 (BCEP or SPGP) are represented by the (TMD-NBD)2 structure, i.e. these proteins have two parts, each of which contains a TMD and a NBD. In the same subfamily proteins ABCB2 (TAP1) and ABCB3 (TAP2) have the (TMD-NBD)1 structure. Some transporters of ABCC (MRP) subfamily have at the N-terminus a third TMD (designated as TMD0) containing five transmembrane helices [9, 10]. These are proteins ABCC1 (MRP1), ABCC2 (MRP2), ABCC3 (MRP3), ABCC6 (MRP6), ABCC8 (SUR1), and ABCC9 (SUR2). Proteins included in the same subfamily by their homology to other MRP transporters (ABCC4 (MRP4) and ABCC5 (MRP5)) have no TMD0. Other ABC subfamilies (ABCE and ABCF) include some proteins that have only NBD [11]. Thus, domain organization of ABC proteins is various.
Transporters characterized by the (TMD-NBD)2 structure are called complete transporters. They are usually localized in the cell plasma membrane, whereas half-type transporters containing only one TMD-NBD set are usually found in intracellular membranes [11]. Thus, proteins TAP1 and TAP2 are localized in membranes of endoplasmic reticulum. The only exception is the half transporter ABCG2 (also BCRP or MXR) found in the cell plasma membrane [12]. The similarity of domain organization, on one hand, and its variability, on the other, indicates that these proteins are related evolutionarily and that evolution of the human ABC protein family was a very complex process. It is also clear that physiological functions of these proteins should be different. Investigation of physiological functions of the ABC family proteins is now an intensively developed trend.
Although some proteins of the ABC family may have no transport functions, most of them transfer various substances (from inorganic ions to polysaccharides, amino acids, and proteins). Among a few exceptions there is protein ABCC7 (CFTR) that functions as a channel and plays the most important role in regulation of Cl- flow in epithelial cells [13, 14]. Mutations of the CFTR gene are responsible for the severe and frequent (1 : 2000-2500) hereditary disease mucoviscidosis (cystic fibrosis). Mutations in a number of other genes of the ABC family are also responsible for hereditary human diseases [5]. This is indicative of the important role of these proteins in the human body.
DRUG TRANSPORTING PROTEINS OF THE ABC SUPERFAMILY (MULTIDRUG
TRANSPORTERS, MDT)
At the present time MDT, or transporters able to determine MDR, include 10-12 out of 48 members of the human ABC protein family (Table 1) [4, 15]. We have included an additional transporter ABCB4 in Table 1. It is important that these proteins can be divided to two groups: (i) transporters whose ability to impart MDR in patients and in cell cultures is doubtless, and (ii) transporters for which only in some in vitro experiments their ability to confer drug resistance to cells has been demonstrated [4]. The first group includes three human ABC transporters: Pgp (ABCB1), ABCC1 (below MRP1), and ABCG2 (below BCRP). The comparison of drug-resistant cultures and original cells sensitive to cytostatics, as well as analysis of material obtained from patients, has shown that in most cases just these three transporters are MDR inducers [5]. However, different ABC proteins are quite often associated with MDR. First of all, this is ABCC2 (MRP2). We have included these four proteins in Table 2, which shows the results of investigations of their substrates--antitumor preparations [4, 5, 10]. Other transporters listed in Table 1 also eliminate different preparations from cells, though other proteins bind significantly lower amounts of the preparations. A large number of the different class antitumor drugs including new target preparations (kinase inhibitors) are eliminated from cells by ABC transporters (Table 2). It is obvious that problems of drug therapy of malignant tumors are closely associated with those of ABC transporter inheritance.
Table 1. Classification of human proteins of
the ABC family and proteins of the family that determine multidrug
resistance [4, 5]
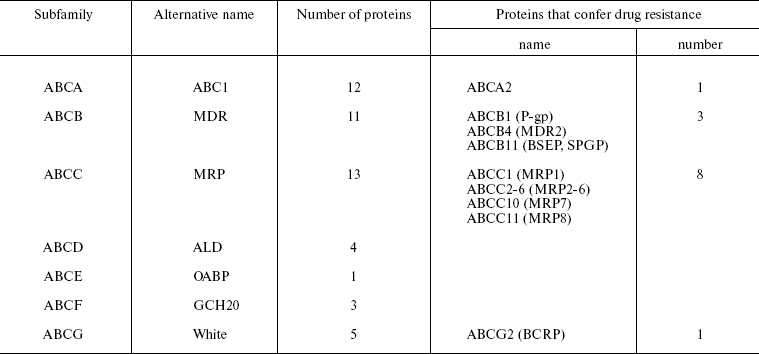
Table 2. Antitumor drugs and their ABC
transporters
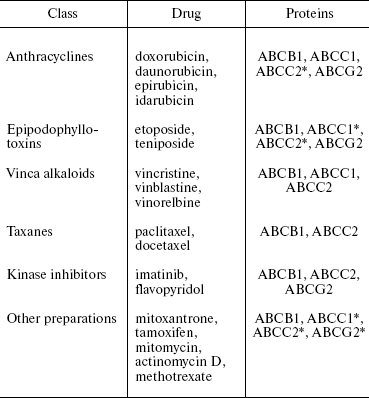
*This ABC transporter binds and removes from the cell not all
substances listed in the corresponding column.
It is seen in Table 2 that Pgp (ABCB1) transports the largest number of drugs. One and the same drug can be a substrate for different transporters, but substrate specificities of different ABC transporters, even of the most similar ones (cf. ABCC1 and ABCC2), are distinct. Thus, several different proteins can determine the resistance of tumor cells to one and the same preparation.
Most data on detection of the ABC transporter substrates was earlier obtained in experiments on cell cultures. Cells were transfected with genes encoding ABC transporters (most often with the MDR1 gene encoding Pgp), and drug sensitivity of these cells was determined by comparison with the sensitivity of parental cells. Since this method has some limitations, the data cannot be considered as comprehensive [5]. For example, the ability of Pgp to transport methotrexate was not found in this kind of experiments, but was detected in those of a different type [16, 17].
Naturally, during recent years different methods have been used to determine which ABC transporters are involved in MDR. Studying the expression of 48 ABC transporters in the cell line collection of the US National Cancer Institute (NCI) using real time RT-PCR has shown the reliable correlation between expression of several ABC proteins and decreased cell sensitivity to cytostatics [18]. In this work, it was also shown that no less than 30 ABC proteins can stimulate a decrease in the drug sensitivity of tumor cells. The use of microchips to compare transcription of the ABC transporter encoding genes in cell lines resistant and sensitive to antitumor drugs (two pairs) showed that 28 transporters are able to define resistance to some drugs or to whole classes of antitumor preparations [4, 19]. So far as the functions of many ABC transporters are not involved in the protection of cells against toxins, a question arises concerning “switching” functional activity of these transport proteins from their usual physiological work to protective function. It is clear that such switching takes place. Now we shall give some examples.
As mentioned above, investigation of physiological functions of the ABC family proteins is one of presently rather intensively developing trends. Functions of ABC transporters in an organism are diverse. Thus, proteins ABCB2 and ABCB3 (TAP1 and TAP2) transport peptides. Transporters of this family also play an important role in translocation of membrane lipids, phospholipids, and cholesterol within a cell and in a whole organism. It is now known that almost half of human ABC transporters are involved in lipid transport, and quite a number of hereditary diseases are connected with mutations of genes encoding these proteins [20]. For the first time the relationship between lipid metabolism and ABC transporters was discovered during investigation of mice with knock-out of mdr2 gene (Abcb4) [21]. Bile of these mice did not contain phosphatidylcholine, which resulted in development of jaundice. Abcb4 appeared to be a flippase that transfers phosphatidylcholine from one cell membrane layer to the other. Mutations of this human gene result in development of hereditary cholestasis. Human ABCB4 (Pgp3 or PGY3) may be related to drug resistance. This protein is able, like a mouse ortholog transporting phosphatidylcholine, to determine resistance to such Pgp substrates as vinblastine and digoxin upon introduction of the encoding gene into cells in culture. Drug resistance was diminished by modifiers of the Pgp transporting activity [22]. Another member of the same subfamily--ABCB11 (SPGP, BSEP)--transports bile acids and plays a rather important role in their excretion into bile. Mutations in the ABCB11 gene also determine familial cholestasis. Transfection of this gene into cells resulted in emergence of resistance to taxol [23]. Although gene transfection into cells is quite an artificial model, results of such experiments show that there are situations in which one and the same protein is able to protect cells against drugs rather than to carry out its physiological functions. It would be very interesting to understand whether such switching really exists in nature and if so, then how it is carried out. To come closer to this understanding, we shall briefly consider data concerning how MDTs recognize their substrates.
HOW MULTIDRUG TRANSPORTERS RECOGNIZE THEIR SUBSTRATES
The capability of MDT (such as Pgp or MRP1) to bind a great number of various substrates was for a long time surprising for researchers. For example, it was shown that Pgp transports hundreds of drugs, peptides, and some lipids [1]. Classic investigations of enzymes have shown that the enzyme binds to the substrate due to specific atomic interactions between amino acid residues of the enzyme and the substrate molecule. It is not surprising that just the notion that the MDT binding site is able to interact with tens of structurally diverse molecules was perceived by many researchers as the violation of fundamental principles of biochemistry. Achievements in structural analysis of MDT and some other proteins recognizing multiple substances provided the solution to this riddle. A major contribution to solution of this problem was made by A. A. Neyfakh, Jr. [3, 6, 24]. The first results of structural analysis of Pgp, unfortunately at insufficient resolution, have been published [25]. The results of structural analysis of the bacterial Pgp homolog MsbA, involved in transmembrane transport of lipid A necessary for formation of bacterial envelope, have been published [3].
Comparison of ABC proteins of different organisms is quite reasonable because functional activity of human and bacterial ABC transporters is provided by one and the same mechanism. This is clearly shown in a remarkable work in which the LmrA gene, encoding ABC transporter of bacterium Lactococcus lactis (determining bacterial resistance to antibiotics), was introduced into human cells. In human cells the LmrA protein was inserted into plasma membrane and began to provide for drug resistance of these cells. In this case, the substrate specificity of LmrA coincided with that of Pgp [26].
The hydrophobicity of Pgp and MsbA made it difficult to obtain material for structural investigations. The study of BmrR protein gave the most complete results [27]. BmrR is a transcription regulator of Bmr, the multidrug transporter of B. subtilis. In response to binding different hydrophobic cations, it activates Bmr expression. It is important to emphasize that the structure of this protein was analyzed both in quiescent state and in their complexes with substrates [27]. Results of all these works made it possible to design a model of substrate transfer by ABC transporters [6, 24, 27].
As noted above, Pgp includes two TMD and two NBD. Structural analysis revealed within the cell membrane a large cavity (pocket) formed by transmembrane helices of the transporter molecule [27]. This pocket has two lateral holes exposed into the membrane, through which the substrates evidently can enter the pocket. Previous data suggested that substrates were discarded by transporters from the hollow space limited by the membrane inner layer, but not from the cytosol [3, 6]. According to the proposed model, after ATP binding to the intracellular NBD of the transporter (Pgp), the conformation of the protein transmembrane helices strongly changes, resulting in closing of the lateral holes [6, 27]. This process is evidently accompanied by lowering the affinity of the transporter to the substrate, which results in dissociation of the bound substrate to the extracellular environment. Following the ATP hydrolysis and ADP and phosphate dissociation, the transporter molecule restores its original conformation characterized by high affinity to substrates.
The binding mechanism is assumed to be as follows: ligands penetrating into the deep protein pocket establish Van der Waals contacts with surrounding hydrophobic residues. It is significant that the binding pocket dimensions are sufficiently big to allow different orientation of ligand molecules to interact with different sets of residues forming the walls of the pocket [6]. Since structures of human ABC transporters in their complexes with substrates have not been studied, this model for them is so far only hypothetical, and investigations in this direction should be continued [3]. Nevertheless, the main mechanisms of substrate binding by ABC transporters and similar proteins have become clear. The proposed principles of their functioning explain many riddles, including the problem of protein switching from their physiological function to the protective one. In this respect, it is important that there are works showing that ABC proteins have several sites for ligand binding [28-30]. Further investigations of this interesting problem are needed.
MULTIFACTOR MULTIPLE DRUG RESISTANCE
It is now obvious that the emergence of MDR in tumor cells is determined by various factors and often by their complexes [2]. Thus, the case is not simply MDR, but MDR determined by many factors, i.e. multifactor MDR. It is important that several ABC transporters may determine cellular MDR. We have compared five pairs of drug-sensitive and resistant tumor cells of different histogenesis (Table 3) [31]. One of the resistant sublines exhibits hyperexpression of MRP1 protein (COR-23L/R), the rest being considered as resistant due to Pgp activity. In these resistant sublines the amount of the MDR1 gene mRNA is significantly increased, and in two out of five drug-resistant sublines the increased content of mRNA of MRP1 and BCRP genes was also detected. These data were confirmed by the results of protein investigation [31]. Expression of at least three MDR genes is observed in KB8-5 cells with a low level of drug resistance. This supports the idea that multifactor MDR may emerge already at the early stages of MDR development. Since it is not observed in all resistant sublines, it is obvious that its development depends on the cell context. It is clear that the coordinated regulation of several ABC transporters upon development of drug resistance is observed often but not always. The question arises concerning the peculiarities of the cell context (elements of signal cascades) that define coordinated regulation of several ABC transporters.
Table 3. Content of ABC transporter genes
mRNA in drug-resistant and sensitive cells of different tissue lineage
[31]
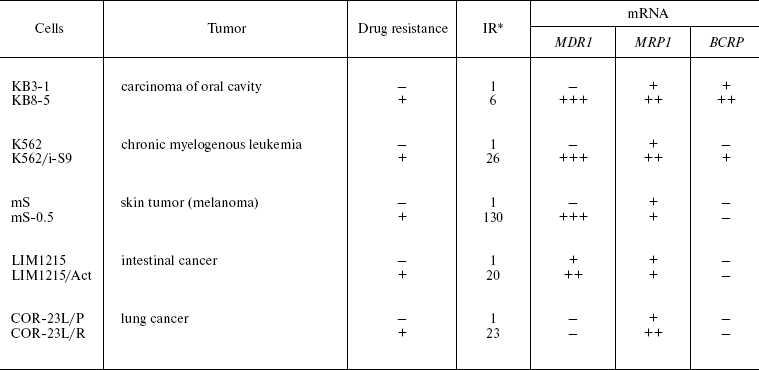
*Resistance index.
REGULATION OF ABC TRANSPORTERS
Undoubtedly, regulatory mechanisms of ABC transporters can be different. They can be regulated at different levels such as transcription and translation. So far, the main attention has been given to ABC transporter regulation at the level of transcription. This problem is considered in a review describing the structure of promoter region of the MDR1 gene and its activity regulation [32]. The promoter region of the MDR1 gene contains regulatory sequences for binding transfactors belonging to different superfamilies. Another analytical article [33] deals with transcription regulation of ABC gene promoters, and owing to this we shall not consider this problem in detail. The MDR1 gene transcription is activated by very different factors--antitumor drugs, ultraviolet radiation, inducers of differentiation, phorbol ethers, carcinogens, etc. These data indicate that various signal cascades are involved in regulation of ABC proteins. Our investigations have also shown that there are multiple signal transduction pathways involved in regulation of MDR1/Pgp expression and activity.
Signal pathway controlled by retinoids. Retinoids are involved in regulation of cell proliferation, differentiation, and programmed death. They are used for treatment of hemoblastoses and solid tumors, and they are especially successfully applied in therapy of acute promyelocytic leukemia [34]. The effect of retinoids is achieved due to their interaction with nuclear receptors of the RAR family (RARalpha, RARbeta, RARgamma) and RXR family (RXRalpha, RXRbeta, RXRgamma). There are data showing that RARalpha activity is especially important for biological effects of retinoids in some cells [35]. About 98% of cases of acute promyelocytic leukemia (AML-M3) and 10-15% of all acute myeloid leucoses are associated with chromosomal translocation t(15;17) (q22;q21), resulting in appearance of a chimeric gene PML-RARalpha. Alteration of RARalpha function makes the main contribution to malignant transformation caused by this rearrangement, though PML is also important.
In our works gene RARalpha was introduced into different cells (human melanoma, human and rat hepatoma, human hemoblastosis cell lines) and sublines constantly hyperexpressing this gene were obtained. According to our data, hyperexpression of the RARalpha gene in cells of solid tumors and some hemoblastoses enhances constitutive (basal) expression of the MDR1 gene [36, 37]. In a part of the studied cell populations, the enhancement in the RARalpha gene expression resulted in a more pronounced increase in the amount of MDR1 mRNA in response to a retinoic acid preparation (ATRA) compared to parental cells [37] (Table 4). The functional activity of Pgp in the RARalpha-transformed cells also increased to a greater extent compared to the parental cell line [37]. This shows that enhanced RARalpha expression increases both MDR1 gene inducibility and functional activity of Pgp in response to retinoic acid. This means that the signal pathway under retinoid control can be involved in Pgp regulation in different types of malignant neoplasias. Treatment of tumors by retinoid acid preparations can stimulate selection of cells expressing active Pgp.
Table 4. Effect of RARalpha
gene transduction into hemoblastosis cells on constitutive and
retinoic acid-induced (ATRA) expression of the MDR1 gene
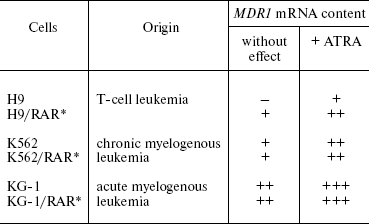
*Cells constantly expressing exogenous RARalpha
gene [37].
Sphingomyelin pathway of signal transduction. Our works also show that the sphingomyelin pathway (cascade) of signal transduction, including the secondary messenger ceramide, is involved in the regulation of MDR1/Pgp activity. The sphingolipid or sphingomyelin signal pathway is one of main signal cascades operating in regulation of cell apoptosis, differentiation, and proliferation. Ceramide is a key molecule of the sphingolipid signal pathway [38]. It is generated in response to some cytokines or different types of stress, including some antitumor drugs [38, 39]. Ceramide C2 (N-acetylsphingosine), a synthetic analog of a natural cellular ceramide, is used in experiments on investigation of the effect of intracellular ceramide accumulation on different intracellular processes. It was shown that incubation of cells with the short-chain ceramide analogs ceramide C2 or C6 imitates some types of cell response to different agents [39, 40]. We have shown in experiments with human hemoblastosis cells in culture that the treatment of cells with ceramide C2 as well as with some chemical preparations increases the amount of MDR1 gene mRNA and enhances Pgp expression and functional activity [41-43]. The c-Raf1 gene product is known to be involved in transduction of a signal whose launching depends on the accumulation of intracellular ceramide [44]. We have shown that introduction into cells of a dominantly-negative mutant of the Raf gene (Raf-C4) eliminates the stimulation of MDR1/Pgp by ceramide C2 and induction of apoptosis by the same preparation [43]. Thus, these data revealed for the first time an additional pathway of signal transduction, which is involved in regulation of activity of the MDR1 gene and Pgp.
Ras-mediated pathway. The Ras family proteins are the most important component of pathways regulated by receptor and non-receptor tyrosine kinases [45, 46]. In response to numerous external effects, Ras activates a number of effector proteins (including Raf proteins), thus regulating cell proliferation, motility, transport of macromolecules, as well as of some other functions. Certain amino acid substitutions in the Ras protein molecules result in the acquisition of the constitutively active state. Ras mutations are found in approximately 25-30% of all human tumors. Our work [43] and other investigations [47, 48] show that the Raf-mediated signal pathway may be involved in regulation of MDR1 gene transcription.
In works carried out in cooperation with members of the laboratories of B. P. Kopnin and P. M. Chumakov, we showed that introduction of N-rasAsp12 gene into human and rat cells (several lines) results in the expression of active Pgp and emergence of drug resistance in some of them [49, 50]. However, the induction of the MDR1/mdr1 gene by mutant Ras is dependent at least on the cell species affiliation. In experiments on stable cell transfection by the mutant oncogene N-rasAsp12, we found the enhancement of Pgp functional activity only in rat cells but not in human cells (cells of mS melanoma, chronic myeloleukemia K562, and colon cancer cells LIM1215) [49, 50]. Thus, the N-ras protein is able to regulate the Pgp activity. Our data and those available in the literature [32, 33] show that regulation of the MDR1 gene and its product Pgp is coordinated with different antiapoptotic cell systems providing for its survival under unfavorable conditions. Then a question arises whether in this case combined regulation of Pgp and other ABC transporters takes place. The next series of our investigations deals with this problem.
PI3K-mediated signal pathway: PTEN. One of the most important mediators in transduction of a signal for cell survival, protecting the cell against a broad spectrum of the cell death inducers, is phosphoinositol-3-kinase (PI3K) and the signal transduction pathways activated by this enzyme [45, 51-53]. PI3K can be activated due to its direct interaction with tyrosine kinases and due to binding to Ras proteins. The phosphatase PTEN is an inhibitor of this signal pathway. We have studied the role of PTEN in MDR and regulation of ABC transporter activities. The simplified scheme below shows this signal cascade. More detailed schemes of the PI3K-mediated signal pathway are shown in other reviews [51-53].
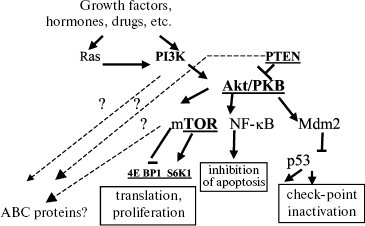
The effect of PTEN can be estimated by lowering intracellular content of phosphorylated Akt kinase. Germinal mutations of PTEN are responsible for the hereditary Kouden's syndrome including enhanced risk of development of tumor. Inactivating mutations in gene PTEN occur in many types of malignant neoplasias (cancer of uterine endometrium, prostate and mammary gland tumors, gliomas, meningiomas, melanomas, etc.). So, it is clear that PTEN is an anti-oncogene [45].
We have used the prostate cancer cell lines DU145 and PC3 with different PTEN status: in PC3 cells, PTEN is not expressed, and it is present in DU145 cells (Table 5). In DU145 cells, PTEN inhibits phosphorylation of Akt (Table 5) [54]. This is accompanied by enhanced cell sensitivity (compared to PC3 cells) to different drugs, and the extent of this enhancement is different for different preparations. These data show that functional status of PTEN phosphatase determines cell resistance to a number of chemotherapeutic preparations, i.e. it determines MDR. We have also shown that the PTEN status in prostate cancer cells correlates with expression level of genes/proteins MRP1 and BCRP (Table 5) [54]. We have found that lowering MRP1 expression and activity contributes to the alteration of cell sensitivity to drugs: probenecide (an MRP1 inhibitor) enhanced the PC3 cell sensitivity to doxorubicin. In this case, the MDR1/Pgp status in the investigated cells is independent of the Akt/PTEN status. The question arises whether the relationship between PTEN status and MRP1 expression exists only in prostate tumors. Our experiments with the introduction of the PTEN gene into cells show that PTEN is able to inhibit MRP1 in other cells as well, for example, in epidermoid carcinoma cell lines KB3-1 and KB8-5.
Table 5. Relationship of PTEN protein
activity, drug sensitivity of cultured prostate cancer cells, and
expression of ABC family genes/proteins [54]
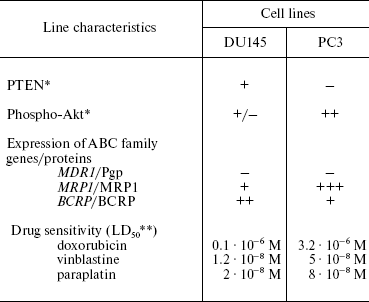
*Determined by Western-blot hybridization.
**LD50 is the drug dose causing death of 50%
cells; results of MTT test.
So, in this case it is possible to define the term “cell context” relative to regulation of a group of ABC transporters as the dependence of regulation of two of them in tumor cells on the Akt/PTEN activity and the absence of such dependence for another transporter. An additional question arises: does the activity of signal pathway PI3K/Akt/PTEN change upon MDR formation in tumor cells? We have studied the effect of transient PTEN transfection on the sensitivity of the cell line series to two different cytostatics (colchicine and doxorubicin) having different intracellular targets and structures [55]. Tumor cell lines of different histogenesis, species affiliation, and different sensitivity to antitumor preparations were used (parental variants and those obtained from these lines due to Pgp hyperexpression). The introduction of the PTEN gene into parental cell lines did not influence drug sensitivity of these cells, whereas sensitivity of some drug-resistant sublines changed. It was found that sensitivity of these cells to cytostatics could either increase or decrease. The effect of PTEN hyperexpression on alterations of cell sensitivity to drugs depends on the mechanism of action of the drug and on the cells into which the transgene was introduced, i.e. on the cell context. Our data suggests that molecular mechanisms involving the PI3K/Akt/PTEN signal pathway emerge in the studied resistant cells [55].
Thus, our data show that different pathways of signal transduction are involved in regulation of ABC transporters. Distortion of these pathways upon malignant transformation and tumor progression can seriously influence the activity of transport ABC proteins. It is clear that in order to overcome MDR caused by ABC transporters, it is necessary not only to understand that they are involved in drug resistance of one or another neoplasm, but it is also necessary to know how their activation is associated with different important processes of the cell vital activity. Searches are necessary for the multifactor MDR regulators that are activated or inhibited with participation of one or another pathway of signal transduction. These elements include the multifunctional protein YB-1, in the regulation of which the signal cascade PI3K/Akt is involved [56-58].
PARTICIPATION OF A MULTIFUNCTIONAL PROTEIN YB-1 IN REGULATION OF
ABC TRANSPORTERS
Mammalian protein YB-1 is a member of a multifunctional family of DNA/RNA-binding proteins with evolutionarily conserved cold shock domain [59]. YB-1 is an RNA-binding protein of broad specificity involved in regulation of mRNA transcription and translation, its splicing, and maintenance of stability. As a transcription factor, it regulates expression of genes having Y-boxes in promoters and enhancers (inverted CCAAT sequences). Among these genes, there is the MDR1 gene encoding Pgp. Certain contradictions exist between data on the regulation of MDR1 by YB-1 binding to the Y-box [33]. However, it has been shown in a significant number of works that YB-1 regulates expression of the MDR1 gene [60-63] and of a gene encoding the MDR protein LRP [64]. Quite a number of external factors are probably able to stimulate the functioning of YB-1 as a transcription factor: the transfer of YB-1 from the cytoplasm into the cell nuclei was observed in response to DNA-damaging substances, UV-radiation, and elevated temperature [61, 65]. The transfer of YB-1 from the cytoplasm into the nuclei of cultured intestinal cancer cells of HCT116 and HCT15 lines resulted in activation of the Pgp and MRP1 encoding genes, enhanced expression of these transporters by cells, and enhanced functional activity. However, in this case no drug resistance was developed [65]. Nuclear localization of YB-1 correlated with Pgp expression in breast cancer, osteosarcoma, and lung cancer [60, 63, 66, 67].
Nevertheless, there are results showing that the high intracellular level of YB-1 may be insufficient for the activation of the MDR1 gene in gene-toxic stress conditions [68]. There are also data showing that the relationship between YB-1 and MDR depends on the cell context, i.e. on peculiarities of the signal pathways activities in these cells and tissues [69]. Thus, data on the relationship between YB-1 and MDR are ambiguous, and additional investigations are necessary. Now we shall show our data. All investigations of the role of YB-1 in MDR were carried out in cooperation with our colleagues from the Institute of Protein Research, Russian Academy of Sciences.
The first question that we wanted to answer was about changes in YB-1 expression and intracellular localization in drug-resistant cells. There had been no systemic study of this problem. We studied five pairs of human tumor cell lines sensitive (parental) and resistant to cytostatics and the sixth resistant cell line (Table 6) [31, 70, 71]. Expression of genes responsible for the tumor cell drug resistance, namely MDR1, MRP1, BCRP, and gene YB-1 was studied by a semiquantitative RT-PCR technique. In half of resistant variants, the enhanced expression of the MDR genes/proteins was accompanied by enhanced expression of the YB-1 gene/protein, whereas in half of cases no such correlation was observed. The extent of increase in the YB-1 mRNA amount did not correlate with the drug resistance level in resistant cells. It is clear that the increase in the YB-1 mRNA amount in resistant cells is not an obligatory feature of drug resistance, although it is detected rather often. We have found a simultaneous increase in the content of mRNA of several MDR genes in resistant cells with the most pronounced expression of the YB-1 gene (Table 6) [31].
Table 6. Changes in the YB-1 mRNA
content and intracellular localization of the protein in drug-resistant
cells compared to parental variants
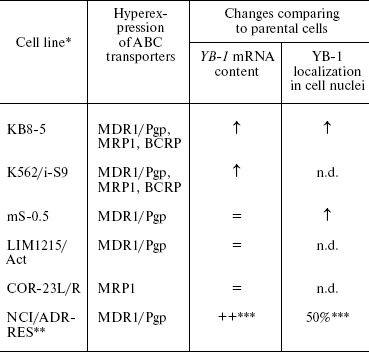
Note: n.d., not determined.
*See Table 3.
**NCI/ADR-RES is the new name of the line [70].
Since this line was not compared with the parental one, the estimation of parameters under study is given.
We have used indirect immunofluorescence microscopy to study the YB-1 localization in the nuclei and cytoplasm of sensitive and resistant cells. It appeared that in two of three studied line pairs, the number of cells with nuclear localization of YB-1 was increased in resistant cell populations compared to the original sensitive cells (Table 6) [31]. In the adriablastin-resistant cell population NCI/ADR-RES, the number of cells with YB-1 in the nuclei is high (50%). Thus, changes in YB-1 localization and increased expression of the protein encoding gene often accompany MDR.
Another unsolved problem is the effect of YB-1 on early steps of MDR emergence. We have found that the increase in intracellular YB-1 expression provides a selective advantage for these cells upon proliferation in the presence of cytostatics ([72] and unpublished data). These experiments were carried out using transient cell transfection of HCT116 and HEK293 cells with plasmids containing the full-sized YB-1 gene cross-linked to the gene of fluorescent protein GFP. This made it possible to follow the survival of the transgene containing cells after cultivation in the presence of vinblastine or cisplatin. Cells transfected with the plasmid containing only the gene of GFP protein served as control. Analysis was carried out on a flow cytofluorimeter. The experiments showed that in five days after transfection the fraction of cells with the YB-1/GFP exceeded by 1.5 times that with GFP gene, which is indicative of selective advantage of cells transfected with the YB-1 gene upon proliferation in the presence of cytostatic. It can prove indirectly that the enhanced YB-1 activity stimulates the emergence of drug resistance in the tumor cell populations. Our data suggest that the YB-1 activity is necessary first of all for formation of new resistant cell populations characterized by multifactor MDR rather than for maintenance of already existing drug resistance.
Results of our experiments, in which the amount of YB-1 mRNA was increased due to the cell transfection with YB-1 gene or decreased by introduction of YB-1 siRNA, show that the YB-1 activity and multifactor MDR are interrelated [72]. It was shown by RT-PCR on HCT116, NCI/ADR-RES, and mS-0.5 cells that the decrease of the YB-1 gene expression caused by YB-1 siRNA is followed by the decrease in expression of different MDR genes [72, 73]. The alteration in the YB-1 gene expression (increase due to gene transduction or decrease in response to siRNA) affects cell proliferation. The HCT116 cells with transgene YB-1/GFP proliferated at a higher rate compared to control cells with transgene GFP. Cells KB3-1 treated with YB-1 siRNA proliferated at a rate lower than that of control cells. It can be assumed that the increase in YB-1 expression in response to the stress effect, including that of chemotherapeutic preparations, may cause an increase in expression of some MDR proteins. In this case there emerges a pool of cells with selective advantages upon action of cytostatics and characterized by enhanced proliferation. A population of resistant cells emerges in response to the repeated drug effect (the above-mentioned data show that after transient transfection of YB-1/GFP gene into HCT116 and HEK293 cells during the first 2-5 days these cells survived better in the presence of vinblastine and cisplatin compared to cells with GFP).
Thus all described results concerning cells with MDR, the effect of cell treatment by cytotoxic drugs on YB-1 expression and localization, transient transfection of YB-1 gene, and introduction into cells of YB-1 siRNA show that YB-1 most often influences simultaneously expression of several ABC transporters. The set of these proteins varies depended on the cell context and drugs. YB-1 is able to regulate MDR proteins at transcription level, as a translation factor, and, probably, using other functions, i.e. the YB-1 effect on the ABC transporter activities may be associated with different activities of this protein.
ALTERATIONS IN ABC TRANSPORTER ACTIVITIES IN HUMAN NEOPLASMS AND
DRUG RESISTANCE OF TUMORS
The role of different ABC transporters in human neoplasms has been studied in many works. However, comparison of the results is not a simple task [4]. Despite necessary caution in estimation of results of different investigations, one should not doubt that ABC transporters may be responsible for clinical MDR, and their expression may be a sign of poor prognosis.
The increased number of cases with hyperexpression of Pgp or another ABC transporter, following courses of drug therapy, are usually used as arguments in favor of the role of ABC transporters in tumor MDR, and the correlation between the gene or MDR protein expression and inefficiency of used therapy is considered as well [2, 74]. The best studied is the clinical significance of MDR determined by the Pgp activity. Data in favor of the significance of Pgp-MDR in therapy were obtained in such investigations for adenocarcinomas of lungs and ovaries, breast cancer, and sarcomas (including osteosarcomas). The role of Pgp-MDR in hemoblastoses was best studied for acute myelogenous leukemia. According to different authors, from 30 to 50% of cases are considered as Pgp-positive (Pgp+), and Pgp is more often detected after chemotherapy in incurable patients [2].
In many neoplasms, the role of ABC transporters in therapeutic outcome is still not clear. However, analysis of their expression can give curious results. Below are some examples based on the results of our investigations of prognostic significance of expression of one of the main MDR proteins, Pgp, in chronic myelogenous leukemia (CML).
It is known that the main molecular event responsible for CML and resulting in the CML cell resistance to chemotherapy is formation of a chimeric protein Bcr-Abl [75]. We had to determine the contribution of Pgp to MDR in CML (in the case of traditional chemotherapy) and the ability of Pgp to serve as a prognostic factor in CML therapy. Peripheral blood samples of 121 CML patients in chronic phase (CP) and blast crisis (BC) were investigated in cooperation with the Hematological Research Center of the Russian Academy of Medical Sciences [76-78]. Repeated examinations of patient groups were a feature of this work. Our results allowed us to draw several conclusions. First, along with the disease progression (transition from CP to BC), an increased number of patients whose peripheral blood cells express functionally active Pgp (28% in CP and 55% in BC) appears. Second, although the number of patients with the Pgp expressing cells increases during disease, these cells do not acquire selective advantage in the course of chemotherapy. So, expression of this protein by blood cells cannot be considered as a factor influencing the response of CML patients to chemotherapy. Third, the comparison of the duration of BC with the Pgp expression and functional activity has shown that in some patients with the Pgp-negative cell phenotype (Pgp-), and especially in those characterized by the absence of Rh123 ejection, the blast crisis duration exceeds that in patients whose cells efflux Rh123 [77]. Fourth, the number of patients whose cells efflux Rh123 increases along with the disease progression. Detection of positive results of the test using Rh123 is ahead of registered increase in Pgp expression. These results can be indicative of involvement in MDR of other ABC transporters together with Pgp. Studying evolution of the ABC transporter expression during CML therapy with a new target drug imatinib revealed similar regularities.
We have studied changes in the Pgp expression and functional activity during therapy by imatinib (imatinib mesylate, STI-571, Gleevec) of CML patients in acceleration phase (AP). AP is the start of rapid CML progression. Until recently this stage of CML was practically incurable. Imatinib is a specific inhibitor of chimeric protein kinase Bcr-Abl [79]. Our work in cooperation with the Hematological Research Center has shown that in a significant number of cases of CML AP, imatinib is efficient.
Repeated examinations of patients during therapy with imatinib have shown that in most cases the amount of Pgp-positive cells either increases or remains at the initial level, i.e. that most often in vivo selection of Pgp+ cells by imatinib takes place. After therapy for 6-12 months, the fraction of patients with Pgp+ blood cell phenotype reaches 80% [80]. Probably at this time in most patients, therapy with imatinib results in formation of certain cell clones expressing Pgp. We have found that in the case of prolonged therapy with imatinib active efflux of Rh123 can be often observed in the absence of Pgp expression [80].
Such clones of leukemic cells express both Pgp and other proteins able to determine MDR. Our data show that in all patients examined after therapy with imatinib for six months, peripheral blood cells express several MDR proteins, at least MRP1, BCRP, and LRP. The K562/i-S9 cells selected by enhanced Pgp expression [64] also expressed increased amounts of these proteins [31].
Probably selection of Pgp+ cells by different drugs is simultaneously selection of cells with hyperexpression of other ABC family transporters. Stem leukemic cells may be among them.
STEM CELLS AND ABC TRANSPORTERS
An interesting fact concerning ABC transporters is hyperexpression of several proteins of this family by stem cells [15]. Recent data show that tumor emergence and progression are associated with proliferation of tumor stem cells whose properties resemble to a high extent those of normal stem cells of these tissues [81, 82]. The best studied are leukemic stem cells [83-85]. The problem of stem cells is an extensive one and has been discussed in many reviews. For our subject it is significant that stem cells (both normal and leukemic) make up the so-called side population of hemopoietic cells. One of first discovered properties characterizing this population was their capability for accelerated efflux from the cells of fluorescent dyes Hoechst 33342 and Rhodamine 123 (Rh123), which are transported by the ABC family proteins [15]. The side population is the bone marrow fraction that remains dark upon staining with these fluorescent dyes.
Markers of stem hemopoietic cells are well known: they are characterized by the CD34+CD38- phenotype. It was shown more than 15 years ago that CD34+ cells hyperexpress Pgp [86]. The ABC transporter BCRP (ABCG2) is also expressed by stem hemopoietic cells [87] and is considered as a stem cell marker. The Pgp and BCRP activities explain the existence of the side population of the bone marrow cells. There are data showing that Rh123 is removed from stem cells by Pgp, and Hoechst 33342 is removed by BCRP [88]. Recently obtained data show that stem hemopoietic cells express a significant number of different transporters of the ABC family [89]. Thus, other proteins of this family are probably able to efflux dyes and other substances from stem cells.
Functions of ABC transporters in stem cells remain unclear. Most often it is supposed that transport proteins of the ABC family protect stem cells (both normal and tumor) against damaging substances [15, 88]. By now this supposition is justified for BCRP. Investigations of mice with knockout of the ABCG2 (BCRP) gene have shown that the bone marrow cells acquired enhanced sensitivity to toxic agents [90]. It is also supposed that ABC transporters may be involved in regulation of key features of the stem cells like their capability of self-renewal and differentiation. It was shown that the Rh123 transporter (RhT) activity in amoeba Dictyostelium discoideum prevents its differentiation. RhT resembles by its function ABC transporters. Its effect on differentiation was associated with efflux of differentiation factors from the cells [91]. For mammalian hemopoietic stem cells, the role of ABC transporters in regulation of their properties has not been determined, although works in this direction are under way.
Stem cells can also be detected in solid tumors [92]. The existence of tumor stem cells is substantiated by the fact that tumors (like normal tissues) emerge from cells capable of self-reproduction and are able to produce progeny in the form of differentiating cells. Authors who for the first time isolated stem cells from human mammary gland tumor have shown that these cells form tumors in mice with immunodeficiency [78]. It was shown that cells able to form tumors have the CD44+CD24- phenotype. A fraction characterized by the side population features was detected in a population of breast cancer cultured cell line MCF7 [93]. The cell side population expressed increased amounts of BCRP (ABCG2) mRNA compared to the main cell population, whereas cells of tumor xenotransplants expressed BCRP in a small cell fraction (0.5-3%).
So, it is clear that investigations of the association of stem cells with ABC transporters are important, but there are still only a few of them. Many questions arise during consideration of this problem, including regulation of groups of the ABC family transport proteins and the significance of expression of the ABC transporter group for evolution of the malignant cell populations.
In conclusion, we should emphasize that in this article we have analyzed only some problems of investigations of the ABC transporters proteins. These proteins function in all living cells. They are highly conserved. This is indicative of an important role of ABC proteins in the vital activity of cells. Therefore, it is natural that the interest of researchers in these proteins is high and there are numerous works dealing with ABC transporters. Simultaneously, there are many problems associated with their investigation. We have analyzed some problems connected with peculiarities of regulation of ABC transporters in tumor cells, first of all of the best studied Pgp. Regulation of ABC proteins is closely connected with the signal transduction pathways, elements of which often change upon malignant transformation. Therefore it is clear that problems of tumor MDR, caused by ABC transporters, are inseparably associated with problems of carcinogenesis as a whole as well as with searches for new targets for therapy. In this paper, we did not touch numerous investigations dealing with the overcoming of MDR. We considered these problems in part in our previous reviews and there are many special reviews concerning this problem [2, 4, 94-96]. Another important problem, not analyzed in this paper but very important for understanding the role of ABC transporters in individual human drug sensitivity, is the problem of polymorphism of ABC transporter encoding genes [94]. This problem is also intensively developed and analyzed in a number of reviews [97-99].
Investigations of the role of ABC transporter expression and activity for clinical oncology are still necessary. First of all, it should be accounted that many ABC transporters may be involved in the multifactor MDR of tumors. At the same time, for most ABC proteins the character of their expression in different tissues and changes in expression and activity upon different neoplasms are not studied. Works using expression microchips, methods of proteomics, as well as those enabling investigation of intracellular protein localization and functional activity are ahead. Considering MDR, first of all it is necessary to understand what drugs are transported by each ABC protein and to study its physiological functions. It is also important to find all possible specific inhibitors of different ABC transporters.
Our works and those of other authors show that different signal transduction pathways, different transcription factors, and different molecular events (not only transcription regulation and regulation of these proteins at the level of translation) are involved in regulation of ABC transporters. What factors activating various pathways of signal transduction activate ABC transporters? Are antitumor drugs such factors? In most cases, such questions still require answers. What elements of signal cascades can be identified as markers of multifactor MDR (by analogy with YB-1)?
Evidently, expression of ABC transporters by solid tumor or leukemic stem cells is able to stimulate selection of tumor stem cells under conditions of various therapies. It is necessary to understand mechanisms of this process in order to choose further strategy of the therapy of neoplasms. These and many other problems require further investigations.
This work was supported by the Russian Foundation for Basic Research (grants 05-04-48283 and 07-04-00720).
REFERENCES
1.Ambudkar, S. V., Dey, S., Hrycyna, C. A.,
Ramachandra, M., Pastan, I., and Gottesman, M. M. (1999) Annu. Rev.
Pharmacol. Toxicol., 39, 361-398.
2.Stavrovskaya, A. A. (2000) Biochemistry
(Moscow), 65, 95-106.
3.Higgins, C. F. (2007) Nature, 446,
749-757.
4.Szakacs, G., Paterson, J. K., Ludwig, J. A.,
Booth-Genthe, C., and Gottesman, M. M. (2006) Nat. Rev. Drug
Discov., 5, 219-234.
5.Dean, M., Rzhetsky, A., and Allikmets, R. (2001)
Genome Res., 11, 1156-1166.
6.Neyfakh, A. A. (2003) Biol. Membr. (Moscow),
20, 206-212.
7.Website: http://nutrigene.4t.com/humanabc.htm
8.Sarkadi, B., Homolya, L., Szakacs G., and Varadi,
A. (2006) Physiol. Rev., 86, 1179-1236.
9.Kruh, G. D., and Belinsky, M. G. (2003)
Oncogene, 22, 7537-7552.
10.Deeley, R. G., Westlake, C., and Cole, S. P. C.
(2006) Physiol. Rev., 86, 849-899.
11.Klein, I., Sarkadi, B., and Varadi, A. (1999)
Biochim. Biophys. Acta, 1461, 237-262.
12.Rocchi, E., Khodyakov, A., Volk, E. L., Yang, C.
H., Litman, T., Bates, S. E., and Schneider, E. (2000) Biochem.
Biophys. Res. Commun., 271, 42-46.
13.Gadsby, D. C., Nagel, G., and Hwang, T. C. (1995)
Annu. Rev. Physiol., 57, 387-416.
14.Kimura, Y., Morita, S., Matsuo, M., and Ueda, K.
(2007) Cancer Sci., 98, 1303-1310.
15.Dean, M., Fojo, T., and Bates, S. (2005)
Nature Rev. Cancer, 5, 275-284.
16.De Graaf, D., Sharma, R. C., Mechetner, E. B.,
Schimke, R. T., and Roninson, I. B. (1996) Proc. Natl. Acad. Sci.
USA, 93, 1238-1242.
17.Norris, M. D., de Graaf, D., Haber, M.,
Kavallaris, M., Madafiglio, J., Gilbert, J., Kwan, E., Stewart, B. W.,
Mechetner, E. B., Gudkov, A. V., and Roninson, I. B. (1996) Int. J.
Cancer, 65, 613-619.
18.Szakacs, G., Annereau, J. P., Lababidi, S.,
Shankavaram, U., Arciello, A., Bussey, K. J., Reinhold, W., Guo, Y.,
Kruh, G. D., Reimers, M., Weinstein, J. N., and Gottesman, M. M. (2004)
Cancer Cell, 6, 129-137.
19.Annereau, J. P., Szakacs, G., Tucker, C. J.,
Arciello, A., Cardarelli, C., Collins, J., Grissom, S., Zeeberg, B. R.,
Reinhold, W., Weinstein, J. N., Pommier, Y., Paules, R. S., and
Gottesman, M. M. (2004) Mol. Pharmacol., 66,
1397-1405.
20.Van Meer, G., Halter, D., Sprong, H., Somerharju,
P., and Egmond, M. R. (2006) FEBS Lett., 580,
1171-1177.
21.Smit, J. J., Schinkel, A. H., Oude Elferink, R.
P., Groen, A. K., Wagenaar, E., van Deemter, L., Mol, C. A., Ottenhoff,
R., van der Lugt, N. M., van Roon, M. A., van der Valk, M. A.,
Offerhaus, G. J. A., Berns, A. J. M., and Borst, P. (1993) Cell,
75, 451-462.
22.Smith, A. J., van Helvoort, A., van Meer, G.,
Szabo, K., Welker, E., Szakacs, G., Varadi, A., Sarkadi, B., and Borst,
P. (2000) J. Biol. Chem., 275, 23530-23539.
23.Childs, S., Yeh, R. L., Hui, D., and Ling, V.
(1998) Cancer Res., 58, 4160-4167.
24.Neyfakh, A. A. (2002) Mol. Microbiol.,
44, 1123-1130.
25.Rosenberg, M. F., Velarde, G., Ford, R. C.,
Martin, C., Berridge, G., Kerr, I. D., Callaghan, R., Schmidlin, A.,
Wooding, C., Linton, K. J., and Higgins, C. F. (2001) EMBO J.,
20, 5615-5625.
26.Van Veen, H. W., Callaghan, R., Soceneantu, L.,
Sardini, A., Konings, W. N., and Higgins, C. F. (1998) Nature,
391, 291-295.
27.Zheleznova, E. E., Markham, P. N., Neyfakh, A.
A., and Brennan, R. G. (1999) Cell, 96, 353-362.
28.Martin, C., Berridge, G., Higgins, C. F., Mistry,
P., Charlton, P., and Callaghan, R. (2000) Mol. Pharmacol.,
58, 624-632.
29.Shapiro, A. B., and Ling, V. (1997) Eur. J.
Biochem., 250, 130-137.
30.Zelcer, N., Huisman, M. T., Reid, G., Wielinga,
P., Breedveld, P., Kuil, A., Knipscheer, P., Schellens, J. H.,
Schinkel, A. H., and Borst, P. (2003) J. Biol. Chem.,
278, 23538-23544.
31.Vaiman, A. V., Stromskaya, T. P., Rybalkina, E.
Yu., Sorokin, A. V., Guryanov, S. G., Zabotina, T. N., Mechetner, E.
B., Ovchinnikov, L. P., and Stavrovskaya, A. A. (2006) Biochemistry
(Moscow), 71, 146-154.
32.Scotto, K. W., and Johnson, R. A. (2001) Mol.
Interv., 1, 117-125.
33.Scotto, K. W. (2003) Oncogene, 22,
7496-7511.
34.Altucci, L., and Gronemeyer, H. (2001) Nature
Rev. Cancer, 1, 181-193.
35.Schneider, S. M., Offterdinger, M., Huber, H.,
and Grunt, T. W. (2000) Cancer Res., 60, 5479-5487.
36.Stromskaya, T., Rybalkina, E., Shtil, A.,
Zabotina, T., Filippova, N., and Stavrovskaya, A. (1998) Brit. J.
Cancer, 77, 1718-1725.
37.Stromskaya, T., Rybalkina, E., Zabotina, T.,
Shishkin, A., and Stavrovskaya, A. (2005) Cancer Cell. Int.,
24, 5-15.
38.Obeid, L., and Hannun, Y. A. (1995) J. Cell.
Biochem., 58, 191-198.
39.Hannun, Y. A., and Luberto, C. (2000) Trends
Cell Biol., 10, 73-80.
40.Ogretmen, B., Pettus, B. J., Rossi, M. J., Wood,
R., Usta, J., Szulc, Z., Bielawska, A., Obeid, L. M., and Hannun, Y. A.
(2002) J. Biol. Chem., 277, 12960-12969.
41.Shtil, A. A., Ktitorova, O. V., Kakpakova, E. S.,
and Holian, O. (2000) Leukemia and Lymphoma, 31, 1-5.
42.Ktitorova, O. V., Kakpakova, E. S., Vinogradova,
M. M., Il'ina, E. N., Govorun, V. M., Ivanov, P. K., Zabotina, T. N.,
Stavrovskaya, A. A., and Shtil, A. A. (2001) Ontogenez,
32, 295-301.
43.Kakpakova, E. S., Ktitorova, O. V., Rybalkina, E.
Yu., Mechetner, E. B., and Stavrovskaya, A. A. (2004) Biol. Membr.
(Moscow), 21, 458-465.
44.Huwiler, A., Brunner, J., Hummel, R.,
Vervoordeldonk, M., Stabel, S., van den Bosch, H., and Pfeilschifter,
J. (1996) Proc. Natl. Acad. Sci. USA, 93, 6959-6963.
45.Kopnin, B. P. (2004) in Encyclopedia of
Clinical Oncology (Davydov, M. I., ed.) [in Russian], RLS-Press,
Moscow, pp. 34-53.
46.Schubbert, S., Shannon, K., and Bollag, G. (2007)
Nat. Rev. Cancer, 4, 295-308.
47.Miltenberger, R. J., Farnham, P. J., Smith, D.
E., Stommel, J. M., and Cornwell, M. M. (1995) Cell Growth
Differ., 6, 549-556.
48.Kim, S. H., Lee, S. H., Kwak, N. H., Kang, C. D.,
and Chung, B. S. (1996) Cancer Lett., 98, 199-205.
49.Stromskaya, T. P., Grigorian, I. A., Ossovskaya,
V. S., Rybalkina, E. Yu., Chumakov, P. M., and Kopnin, B. P. (1995)
FEBS Lett., 368, 373-376.
50.Kopnin, B., Stromskaya, T., Kondratov, R.,
Ossovskaya, V., Pugacheva, E., Rybalkina, E., Khokhlova, O., and
Chumakov, P. (1995) Onc. Res., 7, 299-306.
51.Krasilnikov, M. A. (2000) Biochemistry
(Moscow), 65, 59-67.
52.Vogt, P. K., Bader, A. G., and Kang, S. (2006)
Cell Cycle, 5, 946-949.
53.McCubrey, J. A., Steelman, L. S., Chappell, W.
H., Abrams, S. L., Wong, E. W., Chang, F., Lehmann, B., Terrian, D. M.,
Milella, M., Tafuri, A., Stivala, F., Libra, M., Basecke, J.,
Evangelisti, C., Martelli, A. M., and Franklin, R. A. (2007)
Biochim. Biophys. Acta, 1773, 1263-1284.
54.Shcherbakova, E. A., Stromskaya, T. P.,
Rybalkina, E. Yu., Kalita, O. V., and Stavrovskaya, A. A. (2008)
Mol. Biol. (Moscow), accepted for publication.
55.Shcherbakova, E. A., Stromskaya, T. P.,
Rybalkina, E. Yu., and Stavrovskaya, A. A. (2007) Biol. Membr.
(Moscow), 24, 143-150.
56.Sutherland, B. W., Kucab, J., Wu, J., Lee, C.,
Cheang, M. C., Yorida, E., Turbin, D., Dedhar, S., Nelson, C., Pollak,
M., Grimes, L. H., Miller, K., Badve, S., Huntsman, D., Blake-Gilks,
C., Chen, M., Pallen, C. J., and Dunn, S. E. (2005) Oncogene,
24, 4281-4292.
57.Evdokimova, V., Ruzanov, P., Anglesio, M. S.,
Sorokin, A. V., Ovchinnikov, L. P., Buckley, J., Triche, T. J.,
Sonenberg, N., and Sorensen, P. H. (2006) Mol. Cell. Biol.,
26, 277-292.
58.Evdokimova, V., Ovchinnikov, L. P., and Sorensen,
P. H. (2006) Cell Cycle, 5, 1143-1147.
59.Skabkin, M. A., Skabkina, O. A., and Ovchinnikov,
L. P. (2004) Uspekhi Biol. Khim., 44, 3-52.
60.Bargou, R. C., Jurchott, K., Wagener, C.,
Bergmann, S., Metzner, S., Bommert, K., Mapara, M. Y., Winzer, K. J.,
Dietel, M., Dorken, B., and Royer, H. D. (1997) Nat. Med.,
3, 447-450.
61.Ohga, T., Uchiumi, T., Makino, Y., Koike, K.,
Wada, M., Kuwano, M., and Kohno, K. (1998) J. Biol.
Chem., 273, 5997-6000.
62.Oda, Y., Ohishi, Y., Saito, T., Hinoshita, E.,
Uchiumi, T., Kinukawa, N., Iwamoto, Y., Kohno, K., Kuwano, M., and
Tsuneyoshi, M. (2003) J. Pathol., 199, 251-258.
63.Saji, H., Toi, M., Saji, S., Koike, M., Kohno,
K., and Kuwano, M. (2003) Cancer Lett., 190, 191-197.
64.Stein, U., Bergmann, S., Scheffer, G. L.,
Scheper, R. J., Royer, H. D., Schlag, P. M., and Walther, W. (2005)
Oncogene, 24, 3606-3618.
65.Stein, U., Jurchott, K., Walther, W., Bergmann,
S., Schlag, P. M., and Royer, H. D. (2001) J. Biol. Chem.,
276, 28562-28569.
66.Oda, Y., Sakamoto, A., Shinohara, N., Ohga, T.,
Uchiumi, T., Kohno, K., Tsuneyoshi, M., Kuwano, M., and Iwamoto, Y.
(1998) Clin. Cancer Res., 4, 2273-2277.
67.Shibahara, K., Sugio, K., Osaki, T., Uchiumi, T.,
Maehara, Y., Kohno, K., Yasumoto, K., Sugimachi, K., and Kuwano, M.
(2001) Clin. Cancer Res., 7, 3151-3155.
68.Hu, Z., Jin, S., and Scotto, K. W. (2000) J.
Biol. Chem., 275, 2979-2985.
69.Shibao, K., Takano, H., Nakayama, Y., Okazaki,
K., Nagata, N., Izumi, H., Uchiumi, T., Kuwano, M., Kohno, K., and
Itoh, H. (1999) Int. J. Cancer, 83, 732-737.
70.Liscovitch, M., and Ravid, D. (2007) Cancer
Lett., 245, 350-352.
71.Twentyman, P. R., Fox, N. E., Wright, K. A., and
Bleehen, N. M. (1986) Br. J. Cancer, 53, 529-537.
72.Weiman, A. V., Stromskaya, T. P., Rybalkina, E.
Yu., Sorokin, A. V., Ovchinnikov, L. P., and Stavrovskaya, A. A. (2007)
Byul. Eksp. Biol. Med., 43, 442-446.
73.Weiman, A. V., Gens, G. P., Stromskaya, T. P.,
Rybalkina, E. Yu., Sorokin, A. V., Gur'yanov, S. G., Ovchinnikov, L.
P., and Stavrovskaya, A. A. (2007) Mol. Med. (Moscow), 1,
31-37.
74.Leonard, G. D., Fojo, T., and Bates, S. E. (2003)
Oncologist, 8, 411-424.
75.Gordon, M. Y. (1996) Brit. J. Haematol.,
95, 10-20.
76.Turkina, A. G., Baryshnikov, A. Yu., Sedyakhina,
N. P., Folomeshkina, S. V., Sokolova, M. A., Choroshko, N. D., and
Stavrovskaya, A. A. (1996) Brit. J. Haematol., 92,
88-96.
77.Stavrovskaya, A., Turkina, A., Sedyakhina, N.,
Stromskaya, T., Zabotina, T., Khoroshko, N., and Baryshnikov, A. (1998)
Leukemia and Lymphoma, 28, 469-472.
78.Turkina, A. G., Logacheva, N. P., Stromskaya, T.
P., Zabotina, T. N., Kuznetzov, S. V., Sachizdaeva, K. K., Tagiev, A.,
Juravlev, V. S., Khoroshko, N. D., Baryshnikov, A. Y., and
Stavrovskaya, A. A. (1999) Adv. Exp. Med. Biol., 457,
477-488.
79.Deininger, M., Buchdunger, E., and Druker, B. J.
(2005) Blood, 105, 2640-2653.
80.Stromskaya, T. P., Rybalkina, E. Yu., Kruglov, S.
S., Zabotina, T. P., Mechetner, E. B., Turkina, A. G., and
Stavrovskaya, A. A. (2008) Biochemistry (Moscow), 73,
29-37.
81.Tan, B. T., Park, C. Y., Ailles, L. E., and
Weissman, I. L. (2006) Lab. Invest., 86, 1203-1207.
82.Wicha, M. S., Liu, S., and Dontu, G. (2006)
Cancer Res., 66, 1883-1890.
83.Chertkov, I. L., and Gurevich, O. A. (1984) in
Stem Hemopoietic Cell and Its Microenvironment [in Russian],
Meditsina, Moscow.
84.Chertkov, I. L., and Drize, N. I. (2005)
Vestnik RAMN, 10, 5-10.
85.Drize, N. I. (2006) Onkogematologiya,
1/2, 5-9.
86.Chaudhary, P. M., and Roninson, I. B. (1991)
Cell, 66, 85-94.
87.Zhou, S., Schuetz, J. D., Bunting, K. D.,
Colapietro, A. M., Sampath, J., Morris, J. J., Lagutina, I., Grosveld,
G. C., Osawa, M., Nakauchi, H., and Sorrentino, B. P. (2001) Nat.
Med., 7, 1028-1034.
88.Raaijmakers, M. H. (2007) Leukemia,
21, 2094-2102.
89.De Grouw, E. P., Raaijmakers, M. H., Boezeman, J.
B., van der Reijden, B. A., van de Locht, L. T., de Witte, T. J.,
Jansen, J. H., and Raymakers, R. A. (2006) Leukemia, 20,
750-754.
90.Zhou, S., Morris, J. J., Barnes, Y., Lan, L.,
Schuetz, J. D., and Sorrentino, B. P. (2002) Proc. Natl. Acad. Sci.
USA, 99, 12339-12344.
91.Good, J. R., and Kuspa, A. (2000) Dev.
Biol., 220, 53-61.
92.Blanpain, C., Horsley, V., and Fuchs, E. (2007)
Cell, 128, 445-458.
93.Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A.,
Morrison, S. J., and Clarke, M. F. (2003) Proc. Natl. Acad. Sci.
USA, 100, 3983-3988.
94.Stavrovskaya, A. A. (2003) Biol. Membr.
(Moscow), 20, 196-205.
95.Kellen, J. A. (2003) J. Exp. Ther. Oncol.,
3, 5-13.
96.Modok, S., Mellor, H. R., and Callaghan, R.
(2006) Curr. Opin. Pharmacol., 6, 350-354.
97.Marzolini, C., Paus, E., Buclin, T., and Kim, R.
B. (2004) Clin. Pharmacol. Ther., 75, 13-33.
98.Sakaeda, T., Nakamura, T., and Okumura, K. (2004)
Curr. Top. Med. Chem., 4, 1385-1398.
99.Sakaeda, T. (2005) Drug Metab.
Pharmacokinet., 20, 391-414.