Analysis of RNA Cleavage by RNA Polymerases from Escherichia coli and Deinococcus radiodurans
D. V. Pupov, N. A. Barinova, and A. V. Kulbachinskiy*
Institute of Molecular Genetics, Russian Academy of Sciences, pl. Kurchatova 2, 123182 Moscow, Russia; fax: (499) 196-0221; E-mail: akulb@img.ras.ru* To whom correspondence should be addressed.
Received February 14, 2008
RNA polymerase can both synthesize and cleave RNA. Both reactions occur at the same catalytic center containing two magnesium ions bound to three aspartic acid residues of the absolutely conserved NADFDGD motif of the RNA polymerase beta´ subunit. We have demonstrated that RNA polymerase from Deinococcus radiodurans possesses much higher rate of intrinsic RNA cleavage than RNA polymerase from Escherichia coli (the difference in the rates is about 15-fold at 20°C). However, these RNA polymerases do not differ in the rates of RNA synthesis. Comparison of the RNA polymerase sequences adjacent to the NADFDGD motif reveals the only amino acid substitution in this region (Glu751 in D. radiodurans vs. Ala455 in E. coli), which is localized in the secondary enzyme channel and can potentially affect the rate of RNA cleavage. Introduction of the corresponding substitution in the E. coli RNA polymerase leads to a slight (about 2-3-fold) increase in the cleavage rate, but does not affect RNA synthesis. Thus, the difference in the RNA cleavage rates between E. coli and D. radiodurans RNA polymerases is likely determined by multiple amino acid substitutions, which do not affect the rate of RNA synthesis and are localized in several regions of the active center.
KEY WORDS: RNA polymerase, RNA cleavage, catalysis rateDOI: 10.1134/S000629790806014X
Abbreviations: nt) nucleotides; RNAP) RNA polymerase.
RNA polymerase (RNAP) plays a key role in gene expression and is one of
the most evolutionarily conserved cell proteins. Bacterial RNAP has the
simplest structure and is a convenient model for studies on
transcription mechanisms. Transcription initiation is performed by an
RNAP holoenzyme consisting of the core enzyme (subunit composition
alpha2betabeta´omega),
which possesses catalytic activity, and the initiation factor,
sigma-subunit. RNA synthesis on the elongation stage is
performed by the RNAP core enzyme. Besides RNA synthesis, RNAP can
catalyze a hydrolytic cleavage of an RNA transcript, which can occur by
either exo- or endonuclease mechanism [1, 2]. In the latter case, RNAP moves backward along the
DNA template; the 3´-end of the RNA transcript loses connection
with the template DNA chain, and the active center accommodates the
internal part of RNA [3, 4].
The nuclease activity of RNAP plays an important role in reactivation
of the backtracked elongation complexes, as well as in correction of
transcription errors resulting from incorporation of incorrect
nucleotides [5]. Bacterial cells contain specific
protein factors (such as GreA and GreB in E. coli), which
interact with RNAP and stimulate RNA cleavage [5].
Both synthesis and cleavage of RNA are performed by the same RNAP catalytic center containing two magnesium ions bound to three aspartic acid residues of the absolutely conserved NADFDGD motif of the RNAP beta´-subunit [2, 6]. The catalytic center is localized at the junction of the main and secondary RNAP channels (Fig. 1) [7-10]. The binding of DNA template and RNA transcript occurs in the main channel. The secondary channel likely serves for nucleotide entry into the active center. Upon backtracking of the elongation complex, the 3´-end of the RNA transcript comes into the secondary channel [11, 12]. Gre-factors stimulating RNA cleavage also bind in the secondary channel area [5, 13, 14]. However, both the precise structure of the backtracked elongation complex and detailed mechanism of RNA cleavage remain unclear.
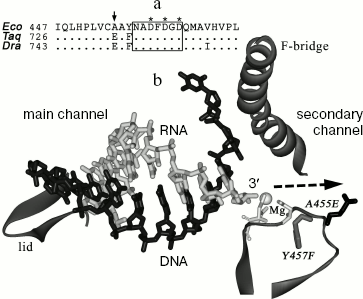
Despite high conservation of the catalytic center, RNAPs of various bacteria can significantly differ in the RNA cleavage rate. In particular, RNAP from the thermophilic bacterium T. aquaticus has significantly higher level of RNA cleavage activity (at its temperature optimum) than does RNAP from E. coli [15]. However, in this case the difference in cleavage rates combines with difference associated with temperature adaptation, which hampers the search for RNAP structural elements involved in RNA cleavage and determining species-specific differences in reaction rates.Fig. 1. Structure of the active center of RNAP. a) Alignment of RNAP beta´-subunit sequences from E. coli (Eco), T. aquaticus (Taq), and D. radiodurans (Dra) in the area of the NADFDGD-motif. Numbers correspond to the amino acid residues at the left alignment border; asterisks denote the aspartic acid residues involved in magnesium ion binding; the arrow points to the A455 residue. b) A model of nucleic acid localization in the elongation complex of T. thermophilus RNAP (from data of [10]). The figure displays the main and secondary RNAP channels, RNA is drawn in light-gray, template DNA in black. An area of the beta´-subunit containing the NADFDGD-motif (amino acids 447-472, numbering of E. coli) is in the bottom right hand corner of the figure. A gray sphere denotes the magnesium ion bound in the active center; three aspartic acid residues of the NADFDGD-motif are drawn in light gray. The amino acid substitution A455E is drawn in black, the substitution Y457F in gray. Arrow points to a pathway of the RNA 3´-end movement upon a backtracking of the elongation complex. In addition, two elements of beta´-subunit are drawn between which the RNA-DNA hybrid is positioned: the “lid” located at the rear of the hybrid and the “F-bridge” separating the main and the secondary channels.
The goal of this study was to compare the RNA cleavage activities of RNAPs from the mesophilic bacteria E. coli and D. radiodurans. Deinococcus radiodurans is the closest mesophilic relative of T. aquaticus, and, as shown earlier, many characteristics of RNAPs from these bacteria are very similar [16]. However, the D. radiodurans RNAP does not possess cold sensitivity of catalysis characteristic of thermophilic enzymes, which allows a direct comparison of its properties with those of other mesophilic bacterial RNAPs [16]. In this study, we have shown that, like the T. aquaticus RNAP, the D. radiodurans RNAP possesses increased level of RNA cleavage activity and have analyzed a possible role of non-conserved amino acids in the RNAP catalytic center area in these differences.
MATERIALS AND METHODS
Enzymes. The D. radiodurans RNAP was isolated from the strain B1422 D. radiodurans as described previously [16]. The substitution A455E was introduced into the gene rpoC encoding the beta´-subunit of E. coli RNAP using the standard methods of PCR mutagenesis. The gene carrying mutation was cloned in plasmid pET29 between the restriction sites of endonucleases NdeI and XhoI. The mutant protein was expressed in the E. coli BL21(DE3) strain, isolated, and purified as previously described [17]. The core RNAP enzymes from E. coli (wild type and with the substitution A455E) were reconstituted from individual subunits in vitro [17].
Analysis of RNA cleavage. An artificial elongation complex was prepared as described previously [18, 19]. The following oligonucleotides were used for the complex assembly: (1) 5´-AGGATACTTAGAGCCTACGAGAGGGACACGGCGAATAGCGAT (non-template DNA); (2) 5´-ATCGCTATTCGCCGTGTCCCTCTCGTAGGCTCTAAGTATCCT (template DNA); (3) 5´-AUCGAGAGGGACA (RNA). The RNA-oligonucleotide was labeled at 5´-end using the T4 polynucleotide kinase and [gamma-32P]ATP. Assembly was carried out in buffer containing 40 mM of Tris-HCl, pH 7.9, and 40 mM of KCl. The oligonucleotide (2) (final concentration 50 nM) was incubated with the oligonucleotide (3) (20 nM) for 3 min at 45°C followed by addition of the core RNAP enzyme (30 nM) and incubation for 10 min at 25°C. The oligonucleotide (1) (200 nM) was added to the samples followed by incubation for 10 min at 37°C and cooling at 20°C. The cleavage reaction was initiated by addition of MgCl2 solution (final concentration 10 mM). After certain intervals (20, 60, 180, 600, 1800, 3600, and 7200 sec), aliquots were sampled, and the reaction was terminated by addition of equal volume of the stop-buffer containing 8 M urea and 2× TBE buffer. RNA products were separated electrophoretically in denaturing 23% polyacrylamide gel and analyzed using a phosphorimager (GE Healthcare, USA). Two to three independent experiments were performed for each RNAP. The RNA cleavage reaction rate was calculated from the equation:
C = Cmax(1 - exp(-kobst)),
where C is the level of RNA cleavage in the given time, Cmax is the maximum cleavage level, and kobs is the apparent first-order reaction rate constant. The data processing and calculation of constants were performed using GraFit software (Erithacus).
Measurement of elongation rate. A promoter DNA fragment used in experiments was synthesized by PCR with a pIA226 plasmid containing a lambda promoter PR and hisT terminator (localized at the distance of 156 nt from the promoter) and primers localized at the distance of -81 and +387 nt from the transcription starting point. Transcription was carried out in buffer containing 40 mM Tris-HCl, pH 7.9, 40 mM KCl, and 10 mM MgCl2. The RNAP holoenzyme (100 nM core enzyme and 500 nM sigma-subunit) was incubated with the DNA fragment (10 nM) for 10 min at 37°C for the promoter complex formation. The mixture of three nucleotides (12 µM of ATP, GTP, and UTP and 10 µCi of [alpha-32P]UTP) and RNA-primer corresponding to the first two nucleotides in the point of transcription initiation (20 µM ApU) were added to the samples. After 7 min, the samples were placed on ice (0°C), and the mixture of all four nucleotides at concentration of 0.5 mM was added. The reaction was terminated by addition of equal volume of the stop buffer, and RNA products were analyzed by electrophoresis in 15% polyacrylamide gel.
RESULTS
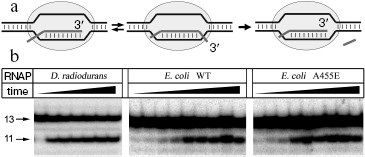
Reaction of RNA cleavage by RNAPs from E. coli and D. radiodurans was studied using an artificial elongation complex containing RNA of fixed length. This complex was prepared by incubation of the RNAP core enzyme with two complementary DNA-oligonucleotides corresponding to template and non-template DNA strands and an RNA-oligonucleotide corresponding to the synthesized RNA (Fig. 2a). It was shown earlier that such complex possesses catalytic activity and can both synthesize and cleave RNA [18]. The used RNA-oligonucleotide (13 nt in length) was complementary to the central part of the template DNA-oligonucleotide throughout the length except for two 5´-terminal nucleotides. In the complex containing RNA of this structure, RNAP can backtrack by two nucleotides and cleave two nucleotides from the 3´-end of the RNA (during this process the length of RNA-DNA hybrid remains constant and is 9 bp) [19].
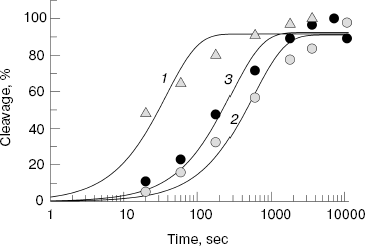
Assembling of E. coli and D. radiodurans RNAP elongation complexes was carried out in absence of magnesium ions; since RNAP is inactive under these conditions, this prevented premature RNA cleavage and allowed preparation of the complex in homogeneous state. After the preparation of the complex, Mg2+ was added to the mixture, and the levels of RNA cleavage were measured at increasing time intervals (Fig. 2b). It was shown that both RNAPs could efficiently cleave RNA under these conditions; in each case, the maximum cleavage level likely corresponds to the amount of catalytically active complex. The data on kinetics of RNA cleavage were used for calculation of apparent first order reaction rate constant (kobs) (Fig. 3). Analysis of the data showed that the D. radiodurans RNAP has much greater RNA cleavage rate (kobs = 0.027 ± 0.007 sec-1) than E. coli RNAP (kobs = 0.0016 ± 0.0003 sec-1).Fig. 2. Analysis of RNA cleavage by D. radiodurans and E. coli RNAPs. a) The scheme of RNA cleavage in an artificial elongation complex. RNA is drawn in dark-gray and the RNAP core enzyme as light-gray oval. Upon transition from the active (left) to the backtracked state (middle) a cleavage of two 3´-terminal RNA nucleotides occurs (right). b) Kinetics of RNA cleavage by RNAPs from D. radiodurans and E. coli (wild type and with substitution A455E). The cleavage reaction was conducted at 20°C and terminated after increasing time intervals. Arrows indicate the initial RNA (13 nt in length) and the reaction product (11 nt in length).
One can suppose that the difference in RNA cleavage rates between E. coli and D. radiodurans RNAPs results from amino acid substitutions in the area of the NADFDGD-motif involved in binding of catalytic magnesium ions. The protein loops of E. coli and D. radiodurans RNAPs, which contain this motif (see Fig. 1b), differ from each other in three positions: A455E, Y457F, and V468I (E. coli numbering) (Fig. 1a). The first two substitutions are also present in the T. aquaticus RNAP, whereas the third one is only found in the D. radiodurans RNAP. Since the T. aquaticus RNAP also possesses an increased level of RNA cleavage activity, one can conclude that the substitution V468I has no apparent effect on the rate of RNA cleavage. The substitution Y457F involves the amino acid, which is buried inside the enzyme and cannot be directly involved in catalysis. The remaining substitution A455E is localized not far from the catalytic center, in the area of the secondary channel, in which the 3´-end of RNA is going upon backtracking of the elongation complex. To examine the role of this substitution in RNA cleavage, we prepared and tested E. coli RNAP carrying the corresponding mutation in the beta´-subunit.Fig. 3. Quantitative analysis of kinetics of RNA cleavage by D. radiodurans and E. coli RNAPs. The plot displays the level of RNA cleavage versus the reaction time (for experiment presented in Fig. 2). For each RNAP, the cleavage efficiency is given as a percentage of maximum cleavage: 1) D. radiodurans RNAP (Dra); 2) wild-type E. coli RNAP (WT); 3) E. coli RNAP with substitution A455E. The curves correspond to first-order kinetics and are the best approximations of experimental data.
Control experiments showed that the substitution A455E has no effect on both the interaction of E. coli RNAP with promoters and initiation of transcription (data not shown). The RNA cleavage activity of mutant RNAP was examined in the same test as was the activity of the wild-type RNAP (Figs. 2 and 3). The mutation was found to result in some increase in the rate of RNA cleavage by the E. coli RNAP (kobs = 0.0041 ± 0.0009 sec-1), which, however, does not achieve the rate of RNA cleavage by the D. radiodurans RNAP.
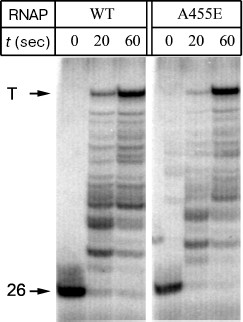
To analyze the effect of substitution A455E on the rate of RNA synthesis, we compared the mean rates of transcription elongation by E. coli wild-type RNAP and RNAP with the mutation. A DNA fragment containing the phage lambda promoter PR and hisT-terminator was used as the template. The 5´-terminal region of RNA synthesized on this template contains only A, G, and U residues at first 26 positions and C residue at position 27. As a result, when only three substrates (ATP, GTP, and UTP) are added, the formed elongation complexes are arrested at position 26 of the template. Addition of all-nucleotide mixture at saturating concentration to the arrested elongation complexes allowed RNAP to synthesize the full-size RNA. It was found that both RNAPs possess equal elongation rates, and the major part of RNAP molecules synthesizes the full-length RNA at 1 min after the beginning of reaction (Fig. 4). Thus, the substitution A455E does not affect the rate of RNA synthesis.
Fig. 4. A comparison of elongation rates between the E. coli wild-type RNAP and RNAP with the substitution A455E. The mixture of all four nucleotides was added to elongation complexes containing 26-nt RNA, and the reaction was carried out for 20 and 60 sec at 0°C. Arrows show positions of the initial transcript and full-length RNA formed after transcription termination (T).
DISCUSSION
Results of our study indicate that the RNAP from the mesophilic bacterium D. radiodurans possesses significantly higher level of RNA cleavage activity than the RNAP from E. coli. However, as found earlier, these RNAPs do not differ in the RNA synthesis rates and have equal temperature optima of activity [16]. The data suggest that reaction of RNA cleavage involves somewhat different sites of active center than reaction of RNA synthesis. A comparison of RNAPs from E. coli and D. radiodurans makes possible the search for structural elements of RNAP involved in reaction of RNA cleavage, which may give us important information on mechanisms of functioning of the enzyme active center.
Earlier, mutations in E. coli RNAP were described affecting the rate of RNA cleavage. In particular, it was shown that substitutions of residues E813 and D814 in the beta-subunit by alanine disrupt stimulation of RNA cleavage upon increase of pH, and the substitution R1106A leads to significant stimulation of the cleavage at all pH values [2]. It was supposed that residue D814 may simulate the binding of one of the catalytic magnesium ions, and the residue R1106 interacts with D814 and influences its position in the active center. It is worth noting that stabilization of magnesium ion binding in the active center of RNAP is also the main mechanism of action of Gre-factors stimulating RNA cleavage [5, 13]. However, the amino acids referred to above are identical in all RNAPs. Moreover, mutations in these positions affect not only cleavage, but also synthesis of RNA [2]. It is obvious that difference in RNA cleavage rate between RNAPs from E. coli and D. radiodurans should be determined by differences in other, non-conserved, sites of the active center.
As the first step to identification of these sites, we have shown that the substitution A455E in the area of the NADFDGD-motif in the beta´-subunit of E. coli RNAP leads to about 2-3-fold increase in the rate of RNA cleavage. This amino acid residue is the only residue in the site of magnesium binding, which is different in E. coli and D. radiodurans RNAPS and localized in the secondary channel of the enzyme. The effect of this substitution may possibly be in stabilization of the RNA 3´-end binding in backtracked elongation complex in optimal position for the cleavage reaction. Moreover, this substitution may allosterically influence the active center structure, particularly by altering conformation of residues E813 and D814 of the RNAP beta-subunit and thus influencing the magnesium ion binding. An important task of further studies is the search for other RNAP sites determining the difference in RNA cleavage rates among the enzymes of various bacteria. In particular, analysis of differences in the area of the RNAP secondary channel is of great interest.
The functional role of high RNA cleavage activity of RNAP from D. radiodurans remains unknown. It was shown earlier that the RNAP of the related thermophilic bacterium T. aquaticus also exhibits high level of RNA cleavage activity [15]. It was supposed that this feature of T. aquaticus RNAP is a result of temperature adaptation, because it is known that increase of the temperature stimulates formation of backtracked elongation complexes [15]. Our data have shown that this feature of RNAPs from D. radiodurans and T. aquaticus may be either a phylogenetic characteristic of the given group of bacteria or be a consequence of adaptation to various extreme habitats: to high level of radiation in the case of D. radiodurans RNAP and high temperature in the case of T. aquaticus RNAP. Further experiments (including analysis of the role of RNA cleavage in vivo) are required for understanding the possible adaptive role of this feature.
The authors are indebted to I. A. Bass and G. V. Ershova for their help in producing RNAP mutations, to I. Artsimovitch for kindly provided pIA226 plasmid, to S. A. Moshkovski (from the Institute of Biomedical Chemistry of Russian Academy of Medical Sciences) for kindly provided possibility to use phosphorimager.
This work is partially supported by the Russian Foundation for Basic Research (grant No. 07-04-00247).
REFERENCES
1.Orlova, M., Newlands, J., Das, A., Goldfarb, A.,
and Borukhov, S. (1995) Proc. Natl. Acad. Sci. USA, 92,
4596-4600.
2.Sosunov, V., Sosunova, E., Mustaev, A., Bass, I.,
Nikiforov, V., and Goldfarb, A. (2003) EMBO J., 22,
2234-2244.
3.Komissarova, N., and Kashlev, M. (1997) J. Biol.
Chem., 272, 15329-15338.
4.Komissarova, N., and Kashlev, M. (1997) Proc.
Natl. Acad. Sci. USA, 94, 1755-1760.
5.Laptenko, O., Lee, J., Lomakin, I., and Borukhov,
S. (2003) EMBO J., 22, 6322-6334.
6.Steitz, T. A. (1998) Nature, 391,
231-232.
7.Zhang, G., Campbell, E. A., Minakhin, L., Richter,
C., Severinov, K., and Darst, S. A. (1999) Cell, 98,
811-824.
8.Vassylyev, D. G., Sekine, S., Laptenko, O., Lee,
J., Vassylyeva, M. N., Borukhov, S., and Yokoyama, S. (2002)
Nature, 417, 712-719.
9.Murakami, K. S., Masuda, S., and Darst, S. A.
(2002) Science, 296, 1280-1284.
10.Vassylyev, D. G., Vassylyeva, M. N., Perederina,
A., Tahirov, T. H., and Artsimovitch, I. (2007) Nature,
448, 157-162.
11.Korzheva, N., Mustaev, A., Kozlov, M., Malhotra,
A., Nikiforov, V., Goldfarb, A., and Darst, S. A. (2000)
Science, 289, 619-625.
12.Epshtein, V., Mustaev, A., Markovtsov, V.,
Bereshchenko, O., Nikiforov, V., and Goldfarb, A. (2002) Mol.
Cell, 10, 623-634.
13.Sosunova, E., Sosunov, V., Kozlov, M., Nikiforov,
V., Goldfarb, A., and Mustaev, A. (2003) Proc. Natl. Acad. Sci.
USA, 100, 15469-15474.
14.Opalka, N., Chlenov, M., Chacon, P., Rice, W. J.,
Wriggers, W., and Darst, S. A. (2003) Cell, 114,
335-345.
15.Minakhin, L., Nechaev, S., Campbell, E. A., and
Severinov, K. (2001) J. Bacteriol., 183, 71-76.
16.Kulbachinskiy, A., Bass, I., Bogdanova, E.,
Goldfarb, A., and Nikiforov, V. (2004) J. Bacteriol.,
186, 7818-7820.
17.Borukhov, S., and Goldfarb, A. (1993) Protein.
Expr. Purif., 4, 503-511.
18.Komissarova, N., Kireeva, M. L., Becker, J.,
Sidorenkov, I., and Kashlev, M. (2003) Meth. Enzymol.,
371, 233-251.
19.Sosunov, V., Zorov, S., Sosunova, E., Nikolaev,
A., Zakeyeva, I., Bass, I., Goldfarb, A., Nikiforov, V., Severinov, K.,
and Mustaev, A. (2005) Nucleic Acids Res., 33,
4202-4211.