
|
REVIEW: Mechanisms of AngiogenesisA. F. KaramyshevaInstitute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; fax: (495) 324-1205; E-mail: aikaram@yandex.ru |
Received December 26, 2007
Tissue activity of angiogenesis depends on the balance of many stimulating or inhibiting factors. The key signaling system that regulates proliferation and migration of endothelial cells forming the basis of any vessel are vascular endothelium growth factors (VEGF) and their receptors. The VEGF-dependent signaling system is necessary for formation of the embryonic vascular system. Neoangiogenesis during tumor growth is also associated with activation of this signaling system. The biological significance of the effect of such system on the cells depends on the content in tissue of various factors of the VEGF family and their receptors, while in the case of VEGFA it is defined by the ratio of different isoforms of this growth factor. A number of other signaling systems are also involved in regulation of the main steps of vessel formation. The signaling system Dll4/Notch regulates selection of endothelial cells for beginning of angiogenic expansion by endowing particular properties to endothelial cells leading in this process. An important step in vessel stabilization and maturation is vascular wall formation. Signaling system PDGFB/PDGFRbeta as well as angiopoietins Ang1, Ang2, and their receptor Tie2 are involved in recruiting mural cells (pericytes and smooth muscle cells). Identification of key molecules involved in the regulation of angiogenesis may provide new possibilities for development of drugs suitable for inhibition of angiogenesis or its stimulation in various pathologies.
KEY WORDS: angiogenesis, vascular endothelium growth factors (VEGF), neuropilins, PDGFB, angiopoietinsDOI: 10.1134/S0006297908070031
Abbreviations: a.a.) amino acid residues; Ang) angiopoietins; EGF) epidermal growth factor; FGF) fibroblast growth factor; PDGF) platelet-derived growth factor; PlGF) placental growth factor; RTK) receptor tyrosine kinases; TGF) transforming growth factor; VEGF) vascular endothelium growth factor; VEGFR) vascular endothelium growth factor receptor.
Blood and lymphatic vascular systems penetrate every organ and tissue to
supply cells with nutrients and oxygen, providing for circulation of
fluids and various signaling molecules.
The emergence of the blood vascular system (vasculogenesis) is one of the earliest events in embryogenesis. During early embryonic development, mesodermal cells differentiate into hemangioblasts, progenitors of both hematopoietic and endothelial cells giving rise to blood vessels. In the course of further differentiation, hemangioblasts produce angioblasts, aggregation of which results in formation of blood islands. Then fusion of blood islands results in appearance of the primary blood vascular plexus consisting of fine capillaries formed by endothelial cells. It is interesting that already at this stage capillaries acquire arterial or venous character, thus showing that the cell specificity is genetically programmed [1].
The stage of vasculogenesis is completed together with formation of primary vascular plexus, and all further transformations of the vascular net proceed during angiogenesis when new vessels are formed from already existing ones. At the stage of angiogenesis, the primary vascular plexus significantly expands due to capillary branching and is transformed into the highly organized vascular net. Angiogenesis begins from local destruction of the wall of preexisting blood vessel, activation of endothelial cell proliferation, and migration. Endothelial cells are assembled in tubular structures around which blood vessel walls are then formed. During further vascular network maturation, capillaries fuse into bigger vessels, arteries, and veins.
The walls of capillaries and fine vessels consist of a single layer of cells (pericytes), whereas walls of arteries and veins are formed by several layers of smooth muscle cells. Pericytes are cells of mesenchymal origin, the ontogeny of which is still not quite clear. They comprise a heterogeneous population of cells capable of differentiation to different types of mesenchymal cells like smooth muscle cells, fibroblasts, and osteoblasts [2]. Some characteristics of smooth muscle cells are typical of pericytes, but it is still not clear whether pericytes and smooth muscle cells are phenotypic variants of the same cell line or they originate from different progenitors.
So, vessels consist of two main cell types: endothelial cells and mural cells. Therefore, it is important for understanding mechanisms of angiogenesis to determine what processes regulate the biological activity of these cell types and to study their interaction with each other.
In adults, formation and growth of new vessels are under strict control. These processes are activated only under strictly defined conditions like wound healing. Strict regulation of this system and balanced functioning is very important for the organism, because both excessive formation of blood vessels and their insufficient development lead to serious diseases.
Activation of angiogenesis is a necessary condition for tumor development. An expanding tumor nodule, like any other tissue, must be supplied with oxygen and nutrients to maintain its vital activity. It is known that without blood supply the dimensions of a tumor nodule cannot exceed 2-3 mm3 due to hypoxia leading to death of tumor cells [3, 4]. Because of this, there exist mechanisms switching angiogenesis on in a growing tumor.
Now it becomes increasingly clear that the emergence and maturation of new vessels are extremely complex and coordinated processes requiring successive activation of a rather large series of receptors and numerous ligands and finely adjusted balance between multiple stimulating and inhibitory signals. Nevertheless, results of investigations that started in the 1990s made it possible to move forward significantly in understanding of these processes.
VASCULAR ENDOTHELIUM GROWTH FACTOR IS A KEY REGULATOR OF
ANGIOGENESIS
Although most blood vessels in an adult organism remain quiescent, endothelial cells retain the capability of rapid division in response to physiological stimuli, which may result in activation of angiogenesis. Quite a number of molecules are known that can serve as positive regulators of angiogenesis (fibroblast growth factors FGFa and FGFb, transforming growth factors TGFalpha and TGFbeta, hepatocyte growth factor HGF, tumor necrosis factor TNFalpha, angiogenin, interleukin-8, and angiopoietins). However, not all of these factors are specific for endothelial cells, and only some of them are able to influence directly endothelial cells in culture. It is now assumed that the critical event in the regulation of angiogenesis is the signaling cascade involving vascular endothelium growth factor (VEGF). This conclusion is based first of all on the biological properties of this growth factor.
Under in vitro conditions, VEGF stimulates growth of endothelial cells that originated from arteries, veins, and lymphatic vessels by direct action on them [5]. VEGF is a powerful inducer of angiogenesis in a number of experimental models in vivo [6]. It also induces the lymphangiogenic response in mice [7]. VEGF is a survival factor for endothelial cells in vivo and in vitro [8-10]. Under in vitro conditions, VEGF prevents apoptosis of endothelial cells caused by the lack of serum [9]. VEGF has been shown to induce expression of antiapoptotic proteins Bcl-2 and A1 in endothelial cells [8].
The effect of VEGF in vivo in developing and adult organisms was found to be different: the inhibition of VEGF resulted in intensive apoptotic changes in newborn mice, whereas in mice older than four weeks the inhibition of VEGF had practically no effect [11]. This may be due to insufficient maturity of the blood-vascular system in newborn mice, because endothelial cells of newly formed but not of mature tumor vessels exhibited significant dependence on VEGF [10]. It is supposed that one of the key events resulting in the loss of dependence on VEGF is vascular wall formation with involvement of pericytes.
VEGF is also known as a factor regulating vascular permeability [12, 13]. The ability of this factor to enhance vascular permeability defines its important role in inflammation and other pathological processes. In particular, it is known that tumor vessels are characterized by enhanced permeability, and this peculiarity contributes to tumor cell penetration into vascular networks and metastasis.
The gene encoding human VEGF consists of eight exons separated by seven introns [14, 15]. The first 26 a.a. in VEGF constitute the signaling peptide showing that VEGF is a secreted protein. Alternative splicing of the VEGF gene produces four different isoforms--VEGF121, VEGF165, VEGF189, and VEGF206--containing, respectively, 121, 165, 189, and 206 a.a. after removal of the signal peptide [14, 15]. The most frequent isoform, VEGF165, is lacking amino acids encoded by the sixth exon, while isoform VEGF121 has no amino acids encoded by the sixth and seventh exons. There are data in the literature concerning the detection of less frequent spliced isoforms VEGF145 and VEGF183 [5]. In VEGF secreting cells, the most frequent isoform is VEGF165, a homodimer with molecular mass 45 kD [16].
An important characteristic of VEGF isoforms is their ability to bind heparin, because just this defines whether the secreted protein will be accumulated in extracellular matrix or will be released and thus become accessible for interaction with other cells. It appeared that isoforms VEGF189 and VEGF206 bind heparin with high affinity and are practically completely accumulated in extracellular matrix; VEGF121 does not bind heparin (it is a freely diffusible protein), and isoform VEGF165 has intermediate properties (it is a secreted molecule but the bulk of secreted VEGF165 protein remains bound to the cell surface and extracellular matrix) [17, 18].
The matrix-associated VEGF isoforms serve for the cell as a peculiar depot of this growth factor and, when necessary, they are able to be released rather quickly due to cleavage by plasmin in the C-terminal region with formation of a biologically active fragment [17]. In this case, the loss of the heparin-binding domain results in significant decrease of the mitogenic activity of VEGF [19]. Thus, the VEGF165 isoform is characterized by optimal parameters of biological activity. This is also supported by data showing that mice expressing exclusively isoform VEGF120 (VEGF in mice is one a.a. shorter) are nonviable [20].
Hypoxia is one of the most important factors inducing VEGF expression. The enhanced expression of VEGF mRNA under conditions of lowered oxygen content caused by different pathological states has been shown [21]. For example, it is known that cells of many human solid tumors express increased amounts of VEGF, thus stimulating development of new vessels in the growing tumor tissue. The expression of VEGF is also increased by epidermal growth factor (EGF), transforming growth factors (TGFalpha and TGFbeta), insulin-like growth factor 1 (IGF-1), fibroblast growth factors (FGF), platelet-derived growth factors (PDGF), etc. These data point to the possibility of autocrine or paracrine regulation of VEGF expression in the case of secretion of any of the above-mentioned factors by cells [5, 22].
In addition to VEGF, other closely related factors were later detected, which formed a family that now consists of growth factors VEGFA (VEGF), VEGFB, VEGFC, VEGFD, VEGFE, and placental growth factor PlGF.
RECEPTORS OF VEGF GROWTH FACTORS
Growth factors of the VEGF family exert their biological effect via interaction with receptors located on endothelial cell membranes. Three receptors have been identified that bind different VEGF growth factors: VEGFR1 (FLT1), VEGFR2 (Flk1/KDR), and VEGFR3 (FLT4) (initial receptor names are given in parentheses) [23-29]. These receptors belong to the superfamily of receptor tyrosine kinases (RTK) and, based on their structural peculiarities, they comprise a special class within it. Like all RTK, the VEGF receptors are transmembrane proteins with a single transmembrane domain (Scheme 1). The extracellular region of VEGFR is formed by seven immunoglobulin-like domains (IG I-VII), whereas the intracellular part exhibits tyrosine kinase activity, and the tyrosine kinase domain in these receptors is separated to two fragments (TK-1 and TK-2) by an inter-kinase insert [24, 25]. All VEGFR receptors are highly homologous [24, 30].
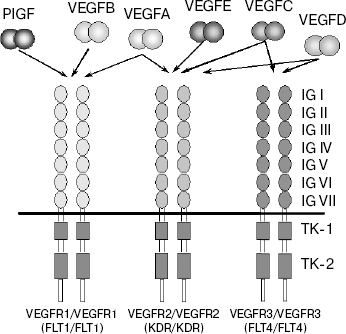
VEGFA interacts with receptors VEGFR1 and VEGFR2. In this case, the affinity binding of VEGFA to VEGFR1 exceeds that to VEGFR2 by one order of magnitude (Kd = 2-10 pM for VEGFR1 [23, 31] and 75-125 pM for VEGFR2 [32]).
VEGFR1. Although this VEGF receptor was identified first [23, 24], its functions are still not quite clear. The initial step in activation of this type of receptors in response to interaction with ligands is their dimerization followed by trans/autophosphorylation of tyrosine residues in the cytoplasmic kinase domain. However, it was shown that VEGFA stimulates only very weak autophosphorylation of VEGFR1 [33, 34]. Moreover, neither increased lethality nor any obvious distortions in the vascular network development were registered in mice expressing VEGFR1 devoid of the tyrosine kinase domain after site directed mutation of the gene [35]. It was supposed that negative regulation of the effect of VEGFA on vascular endothelial cells rather than mitotic signal transduction might be the main function of VEGFR1 [36]. This supposition is supported by such structural peculiarity of VEGFR as its soluble form produced by alternative splicing [24, 37]. This form is not a transmembrane protein, and it does not contain the tyrosine kinase domain. Owing to this, it is not able to transduce a signal. However, the soluble form of VEGFR1 retains the ability to bind VEGFA and is a negative regulator of the activity of this growth factor by prevention of its interaction with VEGFR2 receptor.
Data of experiments on gene inactivation also point to the function of VEGFR1 as a negative regulator of VEGFA activity. Flt1-/- mice die before birth between 8.5 and 9.5 days of embryonic development not because of underdevelopment of the blood-vascular system, but on the contrary, due to excessive growth and disorganization of blood vessels [38].
Regulation of blood vessel permeability might be another function of receptor VEGFR1 [39]. VEGFR1 interacts with both VEGFA and other factors of this family--VEGFB [40] and PlGF [36]. The latter two factors are not ligands of VEGFR2 and can compete with VEGFA only for binding to VEGFR1. Such competitive inhibition of the receptor VEGFR1 might result in increased number of VEGFA molecules binding to VEGFR2. In fact, there are data concerning the ability of PlGF to enhance the effect of VEGFA [36].
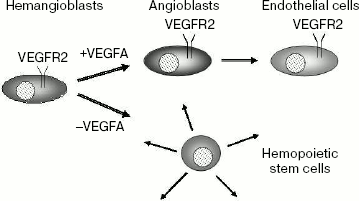
VEGFR2. Receptor VEGFR2 plays a key role both in embryonic angiogenesis and in hematopoiesis. Mice with inactive gene Flk1 are nonviable and die between 8.5 and 9.5 days of embryonic development [41]. In such mice the process of vasculogenesis is disturbed, and differentiation of endothelial cells as well as hematopoiesis are absent. These data suggest the existence of common VEGFR2 expressing progenitors for endothelial cells and hematopoietic stem cells called hemangioblasts (Scheme 2). It is of interest that in the presence of VEGFA, hemangioblasts differentiated to angioblasts and then to endothelial cells, whereas in the absence of this growth factor hemangioblasts differentiated to hematopoietic stem cells [42]. In this case, VEGFR2 expression was retained only in endothelial cells, while in hematopoietic cells expression of this receptor was inhibited.
Growth factors VEGFC, VEGFD, and VEGFE also interact with receptor VEGFR2 (Scheme 1). The latter of these growth factors is encoded by an ORF of the parapox virus genome [43].
The concept of VEGFR2 as the main mediator of VEGFA biological effect has now become generally accepted. Activation of VEGFR2 stimulates a number of signal transduction pathways that later become responsible for mitogenesis, migration, and survival of endothelial cells. This is also confirmed by data on inhibition of angiogenesis upon inactivation of VEGFR2, as well as by data showing that growth factor VEGFE, interacting exclusively with VEGFR2, caused proliferation, chemotaxis, and formation of tubular structures in endothelial cells in vitro and stimulated in vivo angiogenesis as well [27, 44-47].
Biological consequences of the interaction of the VEGFR2 receptor with a particular ligand can be different. Thus, significant increase in the number of subcutaneous blood vessels was observed in transgenic mice with VEGFE hyperexpression, but there was much lower number of such side effects as inflammation and edema compared to that upon induction of angiogenesis by growth factor VEGFA [48, 49]. Such differences might be due to peculiarities of the interaction of VEGFR2 with a particular ligand. Thus, comparison of biological effects caused by growth factors VEGFA and VEGFD in endothelial cell culture has shown that growth factor VEGFD induced tyrosine phosphorylation in VEGFR2 much more weakly and more slowly than VEGFA [50]. However, this effect lasted longer and at later stages (60 min) the efficiency of VEGFD stimulated VEGFR2 phosphorylation was not less than that of VEGFA. The lowered efficiency and retarded kinetics of VEGFD-induced VEGFR2 phosphorylation are most likely due to distinctions in the affinity of VEGFD and VEGFA binding to this receptor: the affinity of VEGFD binding to VEGFR2 is much lower. The efficiency of activation by VEGFA and VEGFD of various VEGFR2-associated signal pathways was also different, which resulted in different biological effects caused by these two growth factors. Thus, VEGFD induced endothelial cell migration but was not able to stimulate their proliferation.
Knowledge of peculiarities of the angiogenic effect of a particular growth factor of this family might be important in development on their basis of different drug preparations that can be used for stimulation of angiogenesis (as in therapy of ischemic diseases) or for inhibition of neoangiogenesis in tumors.
VEGFR3. Expression of the third VEGF receptor at the late stages of embryonic development becomes increasingly restricted to the lymphatic system endothelium. In adults, VEGFR3 is expressed mainly on endothelium of lymphatic vessels. The disturbance of the intracellular signaling cascade associated with this receptor selectively affects lymphangiogenesis [30]. In contrast, investigations of the VEGFR3-associated signaling cascade in cell culture of lymphatic vessel endothelium shows that activation of only VEGFR3 alone is enough to protect cells against apoptosis and induce their proliferation and migration [51].
Receptor VEGFR3 does not interact with VEGFA, its ligands being two other members of this family, VEGFC and VEGFD (Scheme 1) [52-54]. Similarly to the key role played by VEGFA in blood vessel growth, VEGFC is an important regulator of lymphangiogenesis. Lymphatic vessels are completely absent from Vegfc gene knockout mice, severe edema develops in them, and embryos die before birth. Even the loss of a single Vegfc allele in heterozygous mutants results in underdevelopment of lymphatic vessels and in skin lymphedema [55]. VEGFD also exhibits lymphangiogenic activity but is not critical for development of the lymphatic system [56].
Growth factors VEGFC and VEGFD differ in structure from VEGFA: in addition to the VEGFA-homologous region, they have an extended COOH region, the specific peculiarity of which is the presence of four cysteine-enriched C-X10-C-X-C-X-C repeats typical for the Balbiani ring 3 protein (BR3P). Primary VEGFC and VEGFD protein products later undergo posttranslational proteolytic processing resulting in cleavage of the NH2 region containing the signal peptide, and a significant part of the COOH region. As a result, the main part of the mature protein consists of a region homologous to VEGFA [57, 58].
Both VEGFC and VEGFD are ligands for receptors VEGFR3 and VEGFR2, and the affinity of their binding to a particular receptor changes during proteolytic maturation. Immature forms of VEGFC and VEGFD, generated during partial processing, bind only to VEGFR3, whereas mature forms of these proteins retain their ability to interact with VEGFR3 and simultaneously activate VEGFR2 as well. Due to this peculiarity, the biological effect of growth factors VEGFC and VEGFD can be ambiguous: they are able to activate both lymphangiogenesis via interaction with VEGFR3 and angiogenesis by stimulation of VEGFR2. Moreover, it was shown that VEGFR3 was able to form heterodimers with VEGFR2 [59]. Thus, the biological consequences of VEGFC and VEGFD activation can be quite ambiguous and depend on many factors, including quantitative ratios of receptors VEGFR3 and VEGFR2 in the tissue.
Changes in the ratio of VEGFR receptors can be observed during tumor progression. In the course of investigation of VEGFR3 gene receptor expression in benign and malignant human thyroid tumors, we have found increased amounts of the corresponding mRNA in adenomas compared to that in normal thyroid tissue, whereas expression of this gene in adenocarcinomas was much more heterogeneous and on the whole it was decreased [60]. We obtained similar data during investigation of VEGFR3 and VEGFR2 gene expression in samples of human bladder cancer. Expression of both genes at early stages of tumor progression was rather high. At later stages, expression of VEGFR2 remained practically at the same level, whereas expression of VEGFR3, as in the case of thyroid tumors, became heterogeneous, and the total level of expression of this gene was decreased.
NEUROPILINS
A different class of receptors interacting with VEGF was found later on the surface of some tumor and endothelial cells. These receptors differ from VEGFR by both binding affinity and molecular mass [61]. It turned out that for the interaction of VEGFA with these receptors the presence of a fragment encoded by the seventh exon is of fundamental significance because isoform VEGF121 devoid of this fragment did not bind the newly found receptors. It became clear later that transmembrane neuropilin receptors (NRP1 and NRP2), whose ligands are members of the semaphorine family involved in nerve cell regulation, correspond to these newly found receptors [62].
Although neuropilins can interact with VEGF, there are no data on their signal transduction after binding to VEGF [63]. However, the expression of these receptors is necessary for angiogenesis: mice with damaged Nrp1 gene die during embryonic development [64]. Neuropilins are supposed to be co-receptors for VEGF, the role of which is the presentation of VEGF growth factor to VEGFR2 receptor, which enhances binding efficiency. It is possible that just this explains the higher mitotic activity of VEGF165 isoform compared to VEGF121.
Interestingly, neuropilins exhibit precise specificity towards arterial or venous vessels. While NRP1 is found in arteries, expression of NRP2 is restricted to veins and lymphatic vessels [65-67]. NRP2 interacts with VEGFR3 and binds VEGFC and VEGFD. Expression of this receptor was shown to be important for lymphangiogenesis [65, 68].
Experimental results make clear that the cell response to the action of VEGF growth factors is strictly regulated by a number of mechanisms including expression of various members of this family and their binding to different receptors. Expression of different VEGFA isoforms produced by alternative splicing plays an important role in the regulation of cell response as well. In turn, the accessibility of different VEGFA isoforms for interaction with cells largely depends on proteolytic release of isoforms attached to the extracellular matrix and thus on the activity of appropriate proteases [69]. It has been found quite recently that alternative splicing can also produce VEGFA isoforms with antiangiogenic properties. These are so-called b-isoforms differing by the last six C-terminal amino acid residues [70]. So, even change in the balance of different isoforms of only a single member of this family, VEGFA, can significantly influence the development of the blood-vascular system.
The elucidation of mechanisms regulating angiogenesis, and first of all of endothelial cell activity, opened up the elaboration of new therapeutic approaches. As already noted, activation of angiogenesis is a necessary condition for tumor growth and progression; development of new vascular
networks in tumors and their increased permeability contribute to metastasis. Because of this, it was supposed that drugs aimed at suppression of tumor neoangiogenesis would block the process of tumor growth. It was also supposed that one of advantages of antiangiogenic therapy over traditional chemotherapy, aimed directly at tumor cells, might be the genetic stability of endothelial cells. It is known that tumor cells are genetically unstable, and one of consequences of chemotherapy is the development of drug resistance.
The drug preparation Avastin (Genentech) (antibodies to VEGF) is already used in clinical practice. Drug preparations based on compounds that in one way or another block the signaling cascade associated with receptor VEGFR2 are now in the stage of development. The first results of the application of Avastin have shown that it contributes to the prolongation of life in patients with rectal, breast, and lung cancers, but only in combination with traditional chemotherapy. Limitation of the antitumor effect of Avastin shows that for inhibition of tumor growth it may be necessary to consider, in addition to VEGF, different angiogenic factors that also contribute to regulation of tumor angiogenesis. It is also necessary to take into account factors influencing other cell types, like mural cells.
SELECTION OF ENDOTHELIAL CELLS FOR CREATION OF NEW
CAPILLARIES
If all endothelial cells reacted equally to angiogenic stimulus, then the part of the vascular network that underwent this affect would have to be disintegrated and the blood supply to tissue in this region would be disturbed. To prevent this, there is a mechanism that enables selection of just some endothelial cells inside the capillary to initiate angiogenic expansion. These cells called “tip-cells” occupy the leading position while new vessels grow: they react to the VEGFA gradient that specifies the direction of their migration and move forward of the growing capillary (Scheme 3). Angiogenic stimulus causes major change in the tip-cell phenotype. They acquire such properties as invasiveness and the ability to
migrate. They also activate secreted or cell surface proteases for partial destruction of adjacent basement membrane.
Cell contacts between tip-cells and surrounding endothelial cells must change.
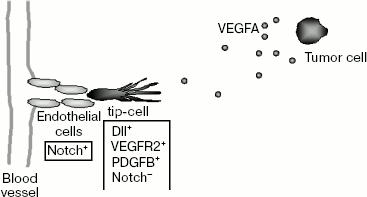
During embryonic development of mice, selection of tip-cells is monitored by Notch family receptors and their transmembrane ligands Dll4 (Delta like ligand 4) [71, 72]. In mammals, four Notch receptors and five ligands (Jagged1, Jagged2, Dll1, Dll3, and Dll4) are found [73]. In vascular endothelial cells, Notch1, Notch4, Jagged1, Jagged2, Dll1, and Dll4 are expressed. Both Notch receptors and their ligands are expressed on the surface of cell membranes. Activation of an intracellular signaling cascade connected with Notch receptors is due to the interactions of the receptors with their ligands upon making intercellular contacts.
Notch receptors are heterodimeric proteins; their ligand-binding extracellular region contains 29-36 repeats structurally similar to epidermal growth factor EGF (EGF-like repeats). Interaction of a Notch receptor with its ligand results in proteolytic cleavage of the receptor [74]. The first site in which the cleavage takes place is located in the extracellular region of the receptor near the transmembrane domain. The extracellular domain separated from the receptor then undergoes “trans-endocytosis” by a ligand-expressing neighboring cell. The second receptor cleavage site is located in the transmembrane domain. The second cleavage results in translocation of the cytoplasmic domain of the Notch receptor into the cell nucleus, where it binds transcription factor CBF1 (C promoter binding factor 1) and activates transcription [75].
Dll4 was identified rather recently [76] and it appears that this ligand of Notch1 and Notch4 receptors plays a key role in the regulation of angiogenesis [77]. This follows from data on the preferable expression of Dll4 in vascular endothelium [76-79]. Moreover, in most mouse lines the deletion of one of the Dll4 alleles causes significant disturbances in the development of the vascular system during early embryogenesis resulting in death of the embryo [78, 80, 81]. Among the great number of genes involved in regulation of vasculogenesis and angiogenesis, such significant defects of the development of the vascular system and death of embryos caused by deletion of a single allele were described only for VEGFA and Dll4 [82, 83].
The mechanism of the Notch/Dll4-associated signaling cascade that determines the differing behavior of endothelial cells that experience angiogenic stimuli is evidently connected with the intracellular signal transduction stimulated by VEGFA. When VEGFA affects endothelial cells it activates expression of Dll4 and its Notch receptors [84]. In this case, the Dll4 and Notch1 expression is tessellated among endothelial cells in the vessel area, where activation of angiogenesis takes place, and the tip-cell specific characteristics are preferably acquired by endothelial cells devoid of Notch1 expression. In endothelial cells expressing Notch1 receptor, activation by Dll4 ligand of the signaling cascade associated with this receptor prevents their transition to active state and thus restricts emergence of an excessive number of tip-cells [85-87]. Inhibition of the active state of endothelial cells due to stimulation of Dll4/Notch-associated transduction of intracellular signal is evidently caused by lowering of their sensitivity to VEGFA. It was shown that in Dll4-hyperexpressing endothelial cells expression of VEGFR2 and its co-receptor neuropilin-1 was significantly inhibited [88].
Lowered levels of Dll4 expression or blocking Notch-dependent signaling cascade enhances tip-cell formation, resulting in significant enhancement of activity of formation, branching, and fusion of newly formed endothelial tubules [71, 85, 86, 89-91]. Such excessive enhancement of angiogenesis results in the disturbance of correct vascularization. Thus, studying tumor progression on experimental animal models has shown that destruction of Dll4-Notch interaction causes intensive growth and branching of blood vessels. However, such vessels appear to be of low functionality--resulting in increased hypoxia, insufficient tissue perfusion, and finally, in inhibition of tumor growth. These data show that the signal pathway regulated by the Notch receptor along with the VEGF-dependent signaling cascade might be a useful target for antiangiogenic therapy [89, 90].
In response to the action of VEGFA, tip-cells sprout phyllopodii towards the VEGFA gradient [92]. Thus, the direction of tip-cell migration and accordingly the direction of capillary growth are regulated by the spatial distribution of this growth factor in the tissue. This effect is caused by the interaction of VEGFA with VEGFR2 receptor, the concentration of which is especially high in tip-cells. Evidently, in this case tip-cells themselves do not proliferate in response to VEGFA. Thus, only endothelial cells localized in the growing capillary branch rather than tip-cells proliferate in the growing mouse retina [92].
Once tip-cells are selected and begin to move forward, formation of new capillaries should begin because of the proliferation and migration of other endothelial cells. Proliferation of cells present in the growing capillary branch is stimulated due to the effect of VEGFA on the same VEGFR2 receptor. This means that tip-cells and endothelial cells forming the growing capillary response differently to the activation of this receptor. The data show that VEGFA independently controls migration of tip-cells and proliferation of endothelial cells forming a new capillary [92]. It is still not clear why one and the same effect is so differently interpreted by tip- and other endothelial cells that form the capillary. This problem requires further investigation.
Differences in biological response to VEGFR2 activation and higher expression levels of Dll4, Vegfr2, and Pdgfb transcripts in tip-cells show that tip- and endothelial cells of the main part of a growing capillary are two different subpopulations of endothelial cells. It is not clear whether differences between these two endothelial cell types are genetically controlled, or this is the result of temporary adaptation due to peculiarities of the cell arrangement inside the vessel.
PROCESSES THAT REGULATE MATURATION OF NEWLY FORMED VESSELS
The process of vessel maturation includes a step-by-step transition from actively growing vessel bed to the quiescent fully formed and functional network. Inhibition of endothelium proliferation and emergence of new capillaries take place in this case along with stabilization of already existing newly formed vascular tubules and incorporation of mural cells [93, 94].
The initial step of maturation is fusion of the newly formed capillaries with others. In this case the behavior of the tip-cells should also change: making contact with other tip-cells or with already existing capillaries, tip-cells should stop moving and highly adhesive intercellular interactions should be established at the place of contact.
Simultaneously with making contacts, the vessel lumen should be formed, and this can happen both before and after making contacts with other capillaries. The emerging blood flow contributes to stabilization of the newly formed blood vessel, while oxygen supply by the blood flow lowers the local expression level of VEGFA and other angiogenic signals induced earlier by hypoxia.
An important step of maturation is recruiting mural cells--pericytes and smooth muscle cells of blood vessels (vSMCs). Pericytes are in direct intercellular contact with endothelial cells and form walls of capillaries and immature blood vessels, whereas walls of mature blood vessels and those of large diameter like arteries and veins are formed by several layers of smooth muscle cells separated from endothelium by a layer of basement membrane.
FORMATION OF BLOOD VESSEL WALLS
Currently, due to the discovery of angiogenic factors and especially VEGF, the main attention of researchers is attracted to the regulation of endothelial cells. However, mural cells are also functionally significant, because disturbance of the correct formation of the wall causes an increase in vessel wall permeability, blood vessel dilatation that later results in edemas and even in embryonic lethality. The stage of maturation, associated with formation of the newly formed vessel walls, is often distorted in various pathological situations. In particular, the tumor vascular system consists of poorly organized immature hemorrhagic “leaky” vessels creating favorable conditions for tumor cell invasion and spreading of metastasis [95-98].
Several different factors are involved in recruiting pericytes to form walls of newly formed vessels, but evidently PDGFB plays the key role [99]. In mice deficient in receptor PDGFRbeta or its ligand PDGFB, the number of pericytes is sharply decreased. Blood vessels of such mice are characterized by enhanced dilatation, due to which edemas emerge during embryonic development and result in the death of the embryo [100].
The PDGF family consists of four different PDGF strands (A-D) establishing functional homodimers (PDGF-AA, PDGF-BB, PDGF-CC, and PDGF-DD) or a heterodimer PDGF-AB [101]. PDGF and VEGF families have much in common in their structure. Analysis of genomic sequences encoding VEGF and PDGF shows that these families originated from a common progenitor [102].
Receptors PDGF (PDGFRalpha and PDGFRbeta) like receptors VEGFR belong to the superfamily of receptor tyrosine kinases and these two receptor classes are structurally very similar. Like VEGFR, receptors PDGFR are transmembrane proteins whose intracellular region contains the tyrosine kinase domain separated to two fragments by an interkinase insert, but unlike VEGFR, the extracellular region of PDGFR is formed not by seven, but by five immunoglobulin-like domains [103].
During interaction with their own ligands, PDGFR receptors form homo- or heterodimers [99, 101]. In this case, PDGF-AA binds only PDGFRalpha, while PDGF-BB exhibits higher affinity upon interaction with receptor PDGFRbeta but is also able to bind PDGFRalpha and PDGFR heterodimers [99]. Less abundant forms PDGF-CC and PDGF-DD bind, respectively, homodimers PDGFRalpha and PDGFRbeta as well as heterodimer PDGFRalphabeta. It is interesting that these two forms exhibit higher structural similarity with VEGF compared to other PDGF variants [101, 104].
During angiogenesis, blood vessel endothelium cells express PDGFB, and expression of the mRNA of this factor is especially high in tip-cells. The increased levels of PDGFB expression in tip-cells generate a gradient of the concentration of this factor that stimulates both recruiting pericytes with PDGFRbeta receptor expressed on their surface and creation of the wall of the newly formed capillary [92, 105].
No expression of PDGFRbeta receptor was found in endothelial cells and because of this PDGFB has no effect on them [100, 106, 107]. Thus, PDGFB provides for paracrine regulation between endothelial cells secreting this factor and the PDGFRbeta receptor-expressing cells forming blood vessel walls--pericytes and vascular smooth muscle cells (VSMCs) [108, 109]. PDGFB exhibits a mitogenic effect on pericytes/VSMCs causing their proliferation, directed migration, and incorporation into the vessel wall [110]. In this case, the expression of PDGFB only by endothelial cells is important for the appropriate formation of vessel wall because in mice with endothelial cells deficient in expression of this growth factor defects in the capillary wall formation due to insufficient content of pericytes were found [111].
Pericytes, in turn, exert a stabilizing effect on newly formed vessels and arrest their growth [2, 112, 113]. TGF-beta1 (transforming growth factor beta1) plays an important role in this process. Data from in vitro experiments have shown that TGF-beta1 is activated upon making contact between endothelial cells and pericyte progenitors. The activation of TGF-beta1 resulted in inhibition of endothelial cell proliferation and migration [114, 115], inhibition of receptor VEGFR2 expression in these cells [116], and induced differentiation of progenitor cells to pericytes [112, 117]. Recruitment of pericytes and accumulation of extracellular matrix proteins in the adjacent basement membrane contributes to vessel maturation and its transition to the quiescent state.
ANGIOPOIETINS AND Tie RECEPTORS
An additional signaling system is involved in regulation of complex interactions between endothelium and surrounding cells, namely, the tyrosine kinase receptor Tie2 (Tek) and its ligands--angiopoietins (Ang) [118, 119]. Like the signaling cascade associated with VEGF and its receptors, the Tie/Ang signaling system is necessary for vascular system development during embryogenesis. Transgenic mice with inactive gene Tie2 die between 9.5 and 10.5 days of embryonic development. In this case no noticeable deviations from normal embryos are observed at the stage of vasculogenesis and primary vascular plexus formation, but during further development processes of capillary maturation and stabilization are significantly disturbed, and the primary capillary plexus is not transformed to the more complex branched vascular network [120-122].
A similar phenotype is observed in mice with knockout of the gene encoding the Tie2 receptor ligand angiopoietin Ang1, or hyperexpressing another ligand of the same receptor Ang2 [123, 124]. These data show that, despite structural similarity, angiopoietins Ang1 and Ang2 exhibit differently directed action on the Tie2-associated signaling cascade. In fact, though both these ligands bind Tie2, consequences of their interaction with the receptor are different. While Ang1 stimulates Tie2 phosphorylation, interaction with Ang2 does not result in activation of the receptor. So, Ang2 is a competitive inhibitor of Ang1 [123].
Under in vitro conditions, Ang1 causes chemotaxis of endothelial cells but does not induce their proliferation [124, 125]. At the same time, Ang1 stimulates angiogenesis in vivo [126].
Experimental data show that the Tie2/Ang1-dependent signaling cascade promotes the association of pericytes and endothelium, lowers vascular permeability, and exhibits anti-inflammatory activity [110, 119, 127]. Angiopoietin Ang1 in a developing organism is expressed by mesenchymal cells, including pericytes, and by smooth muscle cells [117]. It is supposed that Ang1 binds receptor Tie2 expressed on the surface of endothelial cells and thus promotes interaction between endothelial cells and pericytes and in this way stabilizes the maturing vascular system [128].
As already mentioned, Ang2 blocks the stabilizing action of Ang1. However, consequences of the effect of Ang2 on angiogenesis depend on the presence of VEGF: in the absence of VEGF Ang2 contributes to vascular regression, but in the presence of this growth factor Ang2 stimulates angiogenesis [123]. Such differences in action of one and the same factor are explained by the fact that inhibition of the cascade of signal transduction via Tie2 receptor inhibits the supply of mural cells. This results in disturbance of stabilization of capillaries newly formed by endothelial cells. However, at the same time distortion of interaction between endothelial cells and mural cells makes vessels hypersensitive to the stimulating effect of VEGF [123, 128]. The coordinated action of VEGFA and Ang2 was shown during investigation of tumor angiogenesis: in the tumor regions with initiated vascular growth, expression of these angiogenic factors was activated [129, 130].
Thus, regulation of angiogenesis largely depends on the balance of factors stimulating and inhibiting different steps of the vascular network formation. Shift of the balance towards angiogenesis stimulation is characteristic of tumor. In conditions of hypoxia, a number of angiogenic factors, first of all VEGF, are activated. Elaborations of the first antiangiogenic antitumor drug preparations were first of all aimed at blocking just this factor. However, the use of anti-VEGF preparation Avastin (Genentech) was efficient only in combination with traditional chemotherapy [131]. The use of anti-VEGF preparation in an experimental model with spontaneous tumor development in mice was also not sufficiently efficient: after prolonged use of the preparation drug resistance developed in the mice, showing that tumor cells are able to maintain angiogenesis by switching to different angiogenic factors [132].
Inefficiency of the first anti-VEGF preparations can be explained by simultaneous contribution of different factors to the switching on of tumor angiogenesis, especially at late stages of tumor progression. In addition, in order to inhibit tumor angiogenesis, it is possible to influence not only endothelial cells, but other cell types like those forming vascular walls or stromal cells.
Nevertheless, the knowledge accumulated to date on the mechanisms of regulation of angiogenesis and peculiarities of the activation of this process in tumors have already made possible the elaboration of new approaches to therapy of malignancy. Further improvement of angiogenesis-inhibiting antitumor preparations could lead the way to development of drugs for combined effect using different angiogenic molecules as targets.
REFERENCES
1.Coultas, L., Chawengsaksophak, K., and Rossant, J.
(2005) Nature, 438, 937-945.
2.Gerhardt, H., and Betsholtz, C. (2003) Cell
Tissue Res., 314, 15-23.
3.Folkman, J. (1990) J. Natl. Cancer
Inst., 82, 4.
4.Folkman, J. (1995) N. Engl. J. Med.,
333, 1757-1763.
5.Ferrara, N., and Davis-Smyth, T. (1997) Endocr.
Rev., 18, 4-25.
6.Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel,
D. V., and Ferrara, N. (1989) Science, 246,
1306-1309.
7.Nagy, J. A., Vasile, E., Feng, D., Sundberg, C.,
Brown, L. F., Detmar, M. J., Lawitts, J. A., Benjamin, L., Tan, X.,
Manseau, E. J., Dvorak, A. M., and Dvorak, H. F. (2002) J. Exp.
Med., 196, 1497-1506.
8.Gerber, H. P., Dixit, V., and Ferrara, N. (1998)
J. Biol. Chem., 273, 13313-13316.
9.Gerber, H. P., McMurtrey, A., Kowalski, J., Yan,
M., Keyt, B. A., Dixit, V., and Ferrara, N. (1998) J. Biol.
Chem., 273, 30366-30343.
10.Benjamin, L. E., Golijanin, D., Itin, A., Pode,
D., and Keshet, E. (1999) J. Clin. Invest., 103,
159-165.
11.Gerber, H. P., Hillan, K. J., Ryan, A. M.,
Kowalski, J., Keller, G. A., Rangell, L., Wright, B. D., Radtke, F.,
Aguet, M., and Ferrara, N. (1999) Development, 126,
1149-1159.
12.Senger, D. R., Galli, S. J., Dvorak, A. M.,
Perruzzi, C. A., Harvey, V. S., and Dvorak, H. F. (1983)
Science, 219, 983-985.
13.Dvorak, H. F., Brown, L. F., Detmar, L., and
Dvorak, A. M. (1995) Am. J. Pathol., 146, 1029-1039.
14.Houck, K. A., Ferrara, N., Winer, J., Cachianes,
G., Li, B., and Leung, D. W. (1991) Mol. Endocrinol., 5,
1806-1814.
15.Tischer, E., Mitchell, R., Hartman, T., Silva,
M., Gospodarowicz, D., Fiddes, J. C., and Abraham, J. A. (1991) J.
Biol. Chem., 266, 11947-11954.
16.Ferrara, N., and Henzel, W. J. (1989) Biochem.
Biophys. Res. Commun., 161, 851-858.
17.Houck, K. A., Leung, D. W., Rowland, A. M.,
Winer, J., and Ferrara, N. (1992) J. Biol. Chem., 267,
26031-26037.
18.Park, J. E., Keller, H.-A., and Ferrara, N.
(1993) Mol. Biol. Cell, 4, 1317-1326.
19.Keyt, B. A., Berleau, L. T., Nguyen, H. V., Chen,
H., Heinsohn, H., Vandlen, R., and Ferrara, N. (1996) J. Biol.
Chem., 271, 7788-7795.
20.Carmeliet, P., Ng, Y.-S., Nuyens, D., Theilmeier,
G., Brusselmans, K., Cornelissen, I., Ehler, E., Kakkar, V. V.,
Stalmans, I., Mattot, V., Perriard, J.-C., Dewerchin, M., Flameng, W.,
Nagy, A., Lupu, F., Moons, L., Collen, D., D'Amore, P. A., and Shima,
D. T. (1999) Nat. Med., 5, 495-502.
21.Dor, Y., Porat, R., and Keshet, E. (2001) Am.
J. Physiol. Cell Physiol., 280, C1367-C1374.
22.Neufeld, G., Cohen, T., Gengrinovitch, S., and
Poltorak, Z. (1999) FASEB J., 13, 9-22.
23.De Vries, C., Escobedo, J. A., Ueno, H., Houck,
K., Ferrara, N., and Williams, L. T. (1992) Science, 255,
989-991.
24.Shibuya, M., Yamaguchi, S., Yamane, A., Ikeda,
T., Tojo, A., Matsushime, H., and Sato, M. (1990) Oncogene,
5, 519-524.
25.Terman, B. I., Carrion, M. E., Kovacs, E.,
Rasmussen, B. A., Eddy, R., and Shows, T. B. (1991) Oncogene,
6, 1677-1683.
26.Matthews, W., Jordan, C. T., Gavin, M., Jenkins,
N. A., Copeland, N. G., and Lemischka, I. R. (1991) Proc. Natl.
Acad. Sci. USA, 88, 9026-9030.
27.Millauer, B., Wizigmann-Voos, S., Schnurch, H.,
Martinez, R., Moller, N. P. H., Risau, W., and Ullrich, A. (1993)
Cell, 72, 835-846.
28.Galland, F., Karamysheva, A., Pebusque, M. J.,
Borg, J. P., Rottapel, R., Dubreuil, P., Rosnet, O., and Birnbaum, D.
(1993) Oncogene, 8, 1233-1240.
29.Pajusola, K., Aprelikova, O., Kohronen, J.,
Kaipainen, A., Petrovaara, L., Alitalo, R., and Alitalo, K. (1992)
Cancer Res., 52, 5738-5743.
30.Alitalo, K., and Carmeliet, P. (2002) Cancer
Cell, 1, 219-227.
31.Sawano, A., Takahashi, T., Yamaguchi, S., Aonuma,
M., and Shibuya, M. (1996) Cell Growth Differ., 7,
213-221.
32.Terman, B. I., Dougher-Vermazen, M., Carrion, M.
E., Dimitrov, D., Armaltno, D. C., Gospodarowicz, D., and Bohlen, P.
(1992) Biochem. Biophys. Res. Commun., 187,
1579-1586.
33.Waltenberger, J., Claesson-Welsh, L., Siegbahn,
A., Shibuya, M., and Heldin, C. H. (1994) J. Biol. Chem.,
269, 26988-26995.
34.Seetharam, L., Gotoh, N., Maru, Y., Neufeld, G.,
Yamaguchi, S., and Shibuya, M. (1995) Oncogene, 10,
135-147.
35.Hiratsuka, S., Minowa, O., Kuno, J., Noda, T.,
and Shibuya, M. (1998) Proc. Natl. Acad. Sci. USA, 95,
9349-9354.
36.Park, J. E., Chen, H. H., Winer, J., Houck, K.
A., and Ferrara, N. (1994) J. Biol. Chem., 269,
25646-25654.
37.Kendall, R. L., and Thomas, K. A. (1993) Proc.
Natl. Acad. Sci. USA, 90, 10705-10709.
38.Fong, G. H., Rossant, J., Gertsentein, M., and
Breitman, M. L. (1995) Nature, 376, 66-70.
39.Stacker, S. A., Vitali, A., Caesar, C., Domagala,
T., Groenen, L., Nice, E., Achen, M. A., and Wilks, A. F. (1999) J.
Biol. Chem., 274, 34884-34892.
40.Olofsson, B., Korpelainen, E., Pepper, M. S.,
Mandriota, S. J., Aase, K., Kumar, V., Gunji, Y., Jeltsch, M. M.,
Shibuya, M., Alitalo, K., and Eriksson, U. (1998) Proc. Natl. Acad.
Sci. USA, 95, 11709-11714.
41.Shalaby, F., Rossant, J., Yamaguchi, T. P.,
Gertsenstein, M., Wu, X. F., Breitman, M. L., and Schuh, A. C. (1995)
Nature, 376, 62-66.
42.Eichmann, A., Corbel, C., Nataf, V., Vaigot, P.,
Breant, C., and le Douarin, N. M. (1997) Proc. Natl. Acad. Sci.
USA, 94, 5141-5146.
43.Lyttle, D. J., Fraser, K. M., Fleming, S. B.,
Mercer, A. A., and Robinson, A. J. (1994) J. Virol., 68,
84-92.
44.Millauer, B., Shawver, L. K., Plate, K. H.,
Risau, W., and Ullrich, A. (1994) Nature, 367,
567-579.
45.Scobe, M., Rockwell, P., Goldstein, N., Vosseler,
S., and Fusenig, N. E. (1997) Nat. Med., 3,
1222-1227.
46.Meyer, M., Clauss, M., Lepple-Wienhues, A.,
Waltenberger, J., Augustin, H. G., Ziche, M., Lanz, C., Buttner, M.,
Rziha, H. J., and Dehio, C. (1999) EMBO J., 18,
363-374.
47.Wise, L. M., Veikkola, T., Mercer, A. A., Savory,
L. J., Fleming, S. B., Caesar, C., Vitali, A., Makinen, T., Alitalo,
K., and Stacker, S. A. (1999) Proc. Natl. Acad. Sci. USA,
96, 3071-3076.
48.Larcher, F., Murillas, R., Bolontrade, M., Conti,
C. J., and Jorcano, J. L. (1998) Oncogene, 17,
303-311.
49.Shibuya, M. (2006) J. Biochem. Mol. Biol.,
39, 469-478.
50.Jia, H., Bagherzadeh, A., Bicknell, R., Duchen,
M. R., Liu, D., and Zachary, I. (2004) J. Biol. Chem.,
279, 36148-36157.
51.Makinen, T., Veikkola, T., Mustjoki, S.,
Karpanen, T., Catimel, B., Nice, E. C., Wise, L., Mercer, A., Kowalski,
H., Kerjaschki, D., Stacker, S. A., Achen, M. G., and Alitalo, K.
(2001) EMBO J., 20, 4762-4773.
52.Lee, J., Gray, A., Yuan, J., Luoh, S.-M.,
Avraham, H., and Wood, W. I. (1996) Proc. Natl. Acad. Sci. USA,
93, 1988-1992.
53.Yamada, Y., Nezu, J., Shimane, M., and Hirata, Y.
(1997) Genomics, 42, 483-488.
54.Achen, M. G., Jeltsch, M., Kukk, E., Makinen, T.,
Vitali, A., Wilks, A. F., Alitalo, K., and Stacker, S. A. (1998)
Proc. Natl. Acad. Sci. USA, 95, 548-553.
55.Karkkainen, M. J., Haiko, P., Sainio K.,
Partanen, J., Taipale, J., Petrova, T. V., Jeltsch, M., Jackson, D. G.,
Talikka, M., Rauvala, H., Betsholtz, C., and Alitalo, K.