Functional Significance of a Putative Sp1 Transcription Factor Binding Site in the Survivin Gene Promoter
M. V. Mityaev*, E. P. Kopantzev, A. A. Buzdin, T. V. Vinogradova, and E. D. Sverdlov
Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (495) 330-6538; E-mail: mityaev@humgen.siobc.ras.ru* To whom correspondence should be addressed.
Received May 5, 2008; Revision received June 26, 2008
We sequenced 1500-bp genomic DNA regions upstream from the survivin gene (BIRC5). DNA was isolated from human placenta and tumors of patients with diagnosed squamous cancer of the lung that showed high-level BIRC5 gene expression. We have revealed four new promoter allelic variants differing in single nucleotide substitutions, one variant with two nucleotide substitutions, and a variant with a TAAA tetranucleotide insertion. All promoter variants displayed low activity in cells with functionally active p53 protein and high activity in cell lines characterized by low level or absence of p53 protein function. The activity of the promoters with single nucleotide substitutions was comparable to that of the wild-type promoter, whereas two nucleotide substitutions markedly reduced the activity. We also demonstrated the functional significance of a putative Sp1 transcription factor-binding site at (-63...-54) upstream from the transcription initiation site. Mutation within this sequence led to a sharp decrease of promoter activity. The functional architecture of the survivin promoter is discussed based on results known from the literature and those obtained here.
KEY WORDS: survivin promoter, regulation of promoter activity, Sp1 binding sites, SNPDOI: 10.1134/S0006297908110035
Abbreviations: BIRC5) survivin gene; CDE) cell cycle-dependent element; SNP) single nucleotide polymorphism.
Apoptosis plays an important role in developmental processes and
maintenance of tissue homeostasis [1, 2]. As a rule, suppression of apoptosis is one of
prerequisites of tumor formation and progression [3]. An important role in apoptosis belongs to the
members of IAP (inhibitor of apoptosis proteins) family, particularly,
to survivin (BIRC5) [4].
Enhanced expression of the BIRC5 gene is observed in tumor cells in most cancer diseases, such as melanoma and cancers of lung, esophagus, stomach, intestine, pancreas, and breast, and seems to be one of important characteristics of their malignant transformation [5]. Moreover, the survivin gene is expressed in embryonal cells and in actively dividing adult human cells, such as thymocytes, CD34+ hemopoietic stem cells, basal cells of intestinal epithelium, cells of gastric mucosa and placenta, endothelial cells, and keratinocytes of skin basal layer [6]. Survivin is not expressed in differentiated cells of normal tissues [5].
Regulatory mechanisms of BIRC5 gene expression are not thoroughly explained. It is clear that the regulation largely occurs at the level of transcription.
Multiple sites involved in interaction with various regulators of transcription have been identified: CDE (cell cycle dependent element) and, possibly, CHR (cell cycle genes homology region) [7], as well as different transcription factor binding sites localized preferably in non-translated region with coordinates (-250...+70) relative to the transcription initiation point (Scheme 1). The tumor suppressors Rb [8, 9] and p53, as well as the transcription factor Egr1 [10] are involved in repression of the BIRC5 gene promotor. Alternatively, the pro-oncogenic factors c-myc [11], TCF4 (in a complex with beta-catenin), and Stat3 and transcription factors of the E2F subfamily activate expression of the BIRC5 gene [4]. The transcription factors Sp1 [12, 13], KLF5 [14], DEC1 [15], and HIF-1alpha [16] are also involved in activation of the survivin gene promotor. In regard to the listed transcription factors, functional binding sites for Sp1, KLF5, c-myc, HIF-1alpha, TCF4, Stat3, Rb, p53, and Egr1 have been found in the BIRC5 gene promoter, thus suggesting possible involvement of these factors in direct control of BIRC5 gene expression. In particular, a decrease in expression level of the survivin gene in cells in the presence of functional p53 protein was reported in a series of papers [9, 17-19].

Eight possible Sp1 binding sites [12] with canonical or close to canonical sequence (G/T)(G/A)GGCG(G/T)(G/A)(G/A)(C/T) were found in the promoter region of the survivin gene [20]. It was shown using site-directed mutagenesis that at least three of these sites are functionally active [12, 13, 15]. The binding site called “Sp1-complex” (-68...-54 relative to the transcription initiation point) [12] is actually a cluster composed of two overlapping putative Sp1 binding sites. Simultaneous introduction of several mutations into the sequence of the Sp1-complex cluster affects both Sp1 binding sites and leads to a sharp decrease in BIRC5 gene promoter activity [13, 15]. However, a question still remains unanswered: which of these binding sites possesses functional significance?
Single nucleotide polymorphisms (SNP) in positions -553, -192, -169, -163, and +41 have been characterized in the BIRC5 gene promoter. The SNPs -553G>C, -192A>G, -169C>T, and -163G>A occur with equal probability in genomic DNA of both normal and tumor and cultured cancer cells and seem to have no effect on expression of the survivin gene [21, 22]. The substitution +41G>C, which leads to disruption of the consensus sequence of the regulatory CDE element, was found in genomic DNA of more than 65% of studied cancer lines (colon, prostate, and breast cancer), whereas this substitution was absent in genomic DNA of normal cells [22]. However, according to other data, the presence of substitution +41G>C does not correlate with inclination to cancer diseases [23, 24]. The results of studies on the effect of the substitution +41G>C on BIRC5 gene promoter activity are contradictory. Some data suggest decrease in the promoter activity in vitro [22], whereas other data suggest its increase [21, 24].
Besides the above-mentioned putative regulatory sites, possible regulatory elements have been described in sites of the 5´-non-transcribed region of the BIRC5 gene, which are more distant from the transcription initiation point [12]. Epigenetic regulation of the promoter activity is also described [17, 19, 25].
The given set of data demonstrates a conflicting character of information concerning the functional architecture of the survivin promoter. In this work, we have attempted to find functionally significant mutations in the promoter region of the BIRC5 gene, which might determine the enhanced level of expression of this gene in tumor cells. Nine single-nucleotide substitutions and a TAAA micro-insertion were characterized in promoter sites of the gene. Four of these substitutions and this micro-insertion were first found and described in this work. We also characterize a promoter variant carrying two single-nucleotide substitutions, whose presence led to decrease in promoter activity with a cooperative effect. Moreover, we have demonstrated a functional significance of the regulatory site, which presumably binds the Sp1 transcription factor.
MATERIALS AND METHODS
Cell cultures and tissues. Cancer cells of lines A549 (lung carcinoma), Calu1 (epidermoid lung carcinoma), NCI-H23 (lung adenocarcinoma), NCI-H358 (bronchoalveolar lung carcinoma), NCI-H460 (large-cell lung carcinoma), and HeLa (cervical adenocarcinoma) were used in the study. Fibroblasts IVL-11NS were obtained according to a standard protocol [26] from a normal lung tissue adjacent to a tumor (a specimen was obtained from a lung tumor surgery patient at the Vishnevsky Surgery Institute). The cells were grown in the DMEM/F12 (1 : 1) medium containing 10% fetal calf serum, 100 U/ml of penicillin, 100 µg/ml of streptomycin, and 0.25 µg/ml of amphotericin (Invitrogen, USA) at 37°C in an atmosphere of 5% CO2. Specimens of histologically characterized tumor tissues 290/05T and 451T were obtained from surgery of patients with non-small cell lung carcinoma. The patients were followed up at the Blokhin Cancer Research Center of the Russian Academy of Medical Sciences from May, 2004 through November, 2005. Specimens of tumor tissues contained not less than 70% cancer cells.
Isolation of genomic DNA. Genomic DNA was isolated using a Wizard Genomic DNA Purification Kit (Promega, USA) from specimens of tumor tissue, which were previously frozen and powdered in liquid nitrogen, according the manufacturer's protocol. DNA amount was determined spectrophotometrically. Genomic DNA from placenta was kindly provided by Dr. Yu. B. Lebedev (Institute of Bioorganic Chemistry, Russian Academy of Sciences).
PCR amplification of BIRC5 gene promoter sites. The BIRC5 gene promoter from nucleotides -1456 through +42 (the transcription initiation point was taken as +1) with genomic DNA was amplified using the primers SurvF1 (5´-AGATCTAAATCTGGGTGAAGGGTATATGAGT) and SurvR1 (5´-AAGCTTCGCGATTCAAATCTGGCGGT). The length of the expected PCR product was 1498 bp. The PCR reaction mixture contained 25 ng of genomic DNA, buffer (67 mM Tris-HCl, pH 8.8, 16.6 mM (NH4)2SO4, and 0.01% Tween-20), 2.5 mM MgCl2, 0.25 mM of each dNTP, 0.2 µM of each primer, and 1 U of Taq DNA-polymerase (State Research Institute of Genetics and Selection of Industrial Microorganisms, Russia). The reaction was conducted as follows: preliminary heating at 95°C for 2 min (first cycle); followed by 30 cycles of DNA denaturation at 95°C for 45 sec, primer annealing at 64°C for 30 sec, and elongation at 72°C for 60 sec; final elongation (last cycle) - at 72°C for 5 min. After completion of the reaction, amplification products were separated electrophoretically in 1% agarose gel, and the final DNA fragments were eluted using a Wizard SV Gel and PCR Clean-Up System (Promega) and cloned in the pGEM-T vector (Promega). Plasmids carrying the insert of promoter fragments were isolated from Escherichia coli clones using a Wizard Plus SV Minipreps kit (Promega) and sequenced.
Primary structure was determined by direct sequencing of two overlapping fragments prepared by PCR of genomic DNA specimens derived from normal and tumor tissues. PCR was conducted with two primer pairs: SurvF1 plus SurvR2 (5´-CTGCACGACCTGGGTTTCC) and SurvF2 (5´-AGGACTTACTGTTGGTGGGACG) plus SurvR1. The primers SurvF2, SurvR2, SurvF3 (5´-ATTAGCCTGGTGTGGTGGTG), SurvR3 (5´-CAGTGTGTCCTCTGCTTTGG), SurvF4 (5´-TCAAGCGATTCTCCTGCCTC), and SurvR4 (5´-CTCACGCCTGTAATCCCAAC) were used for sequencing. Nucleotide sequences of promoter fragments in the pGEM-T plasmid were determined using the primers T7-promoter, SP6-promoter, SurvF2, SurvR2, SurvF3, SurvR3, SurvF4, and SurvR4.
DNA was sequenced at the Center of Collective Use “Genome” (Institute of Molecular Biology, Russian Academy of Sciences) using an ABI PRISM BigDye Terminator v.3.1 reagent kit (Applied Biosystems, USA) with following analysis of reaction products on an ABI PRISM 3100-Avant automatic DNA sequencer (Applied Biosystems). The chromatograms were analyzed using Chromas v.2.33 software (Technelysium, Australia).
Creation of gene engineering constructs. The primers SurvF1 and SurvR1, which were used for amplification of promoter sites, contained in their sequences BglII and HindIII restriction sites, respectively. Plasmids pGEM-T carrying the insert of promoter fragment were subjected to hydrolysis by BglII and HindIII restrictases. The hydrolysis products were electrophoretically separated in 1% agarose gel, and the final DNA fragments were eluted from the gel and ligated with the vector pGL3-BV (Promega) hydrolyzed by the same restrictases.
A PsurvSp1-mut fragment with the substitution -58G>A was prepared by site-directed mutagenesis of the BIRC5 gene promoter comprising the vector pGEM-T using a GeneEditor kit (Promega). The 5´-CGCGCCGCCCCACCTCTACTCC oligonucleotide was used for generation of the substitution.
The prepared plasmids were propagated in E. coli and isolated with the yield of 100-150 µg using a QIAGEN Plasmid Midi Kit (Qiagen, Germany).
Cell transfection. Cells were transfected in 24-well plates with Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Plasmid DNA (0.88 µg) containing a reporter plasmid with inserted firefly luciferase gene and a normalization plasmid pRL-TK (Promega) in the ratio 10 : 1 was used for transfection. Cells were grown in antibiotic-free DMEM/F12 (1 : 1) medium containing 10% fetal calf serum for 48 h, and activities of firefly (Photinus pyralis) and sea pansy (Renilla reniformis) luciferases were measured in cell extracts using the Dual-Luciferase Reporter Assay System (Promega) on a GENios Pro luminometer (Tecan, Switzerland). In parallel experiments, the cells were transfected with the promoterless plasmid pGL3-BV and plasmid pGL3-PV (positive control) containing the firefly P. pyralis luciferase gene under the control of SV40 virus early promoter. The plasmid pRL-TK providing a constitutive expression of R. reniformis luciferase was used as an internal control for reduction of error associated with different efficiency of transfection in a series of independent experiments. The firefly luciferase activity values were normalized to the values of R. reniformis luciferase activity. Three to five independent transfections were performed for each experimental construct.
Bioinformational analysis. The data on SNP distribution in the BIRC5 gene promoter region were acquired from the dbSNP built 126 database (http://www.ncbi.nlm.nih.gov/SNP/index.html).
Statistical analysis. Mean and standard deviation values were calculated using Excel software (Microsoft, USA). Significance of difference between the values of promoter activity was estimated with Student's t-test assuming that the value distribution is normal. The values were assumed to be statistically different at p < 0.05.
RESULTS AND DISCUSSION
Identification of single-nucleotide substitutions in the BIRC5 gene promoter sequence. To compare nucleotide sequences, we amplified the BIRC5 gene promoter fragments from specimens of genomic DNA isolated from placenta and tumor tissues of two patients suffering from squamous-cell lung carcinoma, whose cells were characterized by elevated BIRC5 gene expression level. These fragments with coordinates -1456 through +42 according to the data of [12] possess the highest promoter activity.
Following the cloning of three promoter fragments obtained from PCR amplification of promoter fragments into the vector pGEM-T and subsequent transformation of E. coli cells with recombinant plasmids, six clones from each independent cloning were chosen, and nucleotide sequences of corresponding inserts were determined. The sequences of cloned fragments were compared with the sequence adjacent to the BIRC5 gene obtained from GenBank (U75285).
Ten of 18 analyzed fragments contained various single-nucleotide substitutions and one insertion of TAAA tetranucleotide (Scheme 1a). The character of distribution of single-nucleotide substitutions in sequences amplified from different specimens of genomic DNA is presented on Scheme 2a. The same substitutions were found in several fragments amplified from one specimen of genomic DNA, thus suggesting that they do not result from errors of PCR amplification. This conclusion is also supported by direct sequencing of the amplification products of promoter DNA obtained from three tested specimens of genomic DNA. Double peaks are observed on chromatograms in expected positions (data not shown).
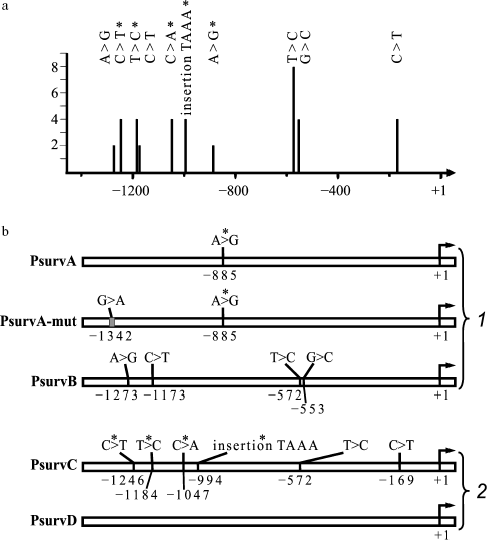
Thus, we have found nine single-nucleotide sequence variations in the primary structure of the BIRC5 gene promoter (Scheme 2a); five of them are SNPs, which were previously reported in the database dbSNP built 126 (identification numbers in dbSNP are given in round brackets): -1273A>G (rs3764382), -1173C>T (rs3764381), -572T>C (rs8073903), -553G>C (rs8073069), and -169C>T (rs17878467), and other four: -1246C>T, -1184T>C, -1047C>A, and -885A>G are first found, as well as the TAAA tetranucleotide insertion between nucleotides -994 and -993.
Determination of activities of survivin gene promoter site variants. Activity of survivin gene promoter sites was determined for five promoter variants; three of them were amplified from normal tissue DNA (PsurvA, PsurvA-mut, and PsurvB) and two from DNA of tumor tissue of patients with squamous cell carcinoma (PsurvC and PsurvD). All listed promoter sites contained various substitutions (Scheme 2b). The effect of substitutions on the transcription activity of promoters in cell lines of various genesis was determined from the expression level of the reporter luciferase gene. To do this, the promoter sites were cloned in the reporter vector pGL3-BV containing the firefly luciferase gene. Both normal human fibroblasts (IVL-11NS) and cells of lines A549, Calu1, NCI-H23, NCI-H358, NCI-H460, and HeLa were transfected with the plasmid constructs pGL3-PsurvA, pGL3-PsurvA-mut, pGL3-PsurvB, pGL3-PsurvC, and pGL3-PsurvD. Activities of the BIRC5 gene promoter sites in the transfected cells were compared with activity of the SV40 virus early promoter comprising the reporter vector pGL3-PV (positive control). Promoter activity was determined as the ratio between the luciferase activity in extracts of cells transfected with a plasmid containing the insert of tested promoter and the luciferase activity in the cells transfected with a plasmid containing the promoterless vector pGL3-BV. A minimum of three transfections were carried out for each experimental construct.
The data are presented in Fig. 1. All cloned fragments possessed promoter activity in all tested cell lines. The spread of activity values determined in independent experiments did not exceed 15% within the data for each of the promoter variants.
Activities of allelic variants PsurvA, PsurvB, PsurvC, and PsurvD were nearly equal within each of the tested cell lines, but differed from line to line.Fig. 1. Activity of the survivin gene (BIRC5) promoter sites carrying single-nucleotide substitutions in transformed cells falling into the “p53+” (a) and “p53-” (b) groups and in normal lung fibroblasts (c). The cells were transfected with reporter plasmids containing the insert of corresponding promoter sites of the BIRC5 gene, pGL3-PV with the SV40 virus promoter, and promoterless plasmid pGL3-BV. The luciferase activity in extracts of cells transfected with the pGL3-BV plasmid was taken to be unity. The column height reflects the mean value of luciferase activity from at least three transfections, and error bars indicate the value of standard error of the mean.
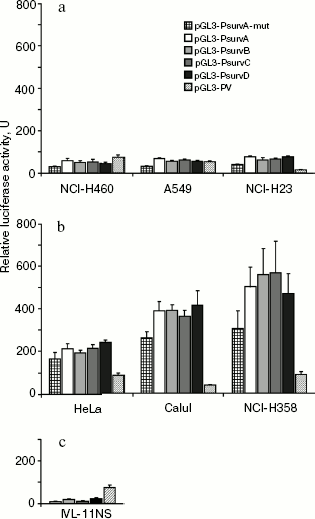
Activities of all promoter variants were low in the cell lines we referred to the group “p53+” with either wild-type p53 (A549 and NCI-H460) [27, 28] or mutant, but active, p53 protein (NCI-H23) [27] (Fig. 1a). The cells of lines Calu1 and NCI-H358, in which the p53 gene is homozygously deleted [28], and HeLa, in which the level of the p53 protein is dramatically decreased due to its accelerated ubiquitin-dependent degradation [29] were referred to the group “p53-”; activity of all promoter variants in this group at least three-fold exceeded that in “p53+” cells (Fig. 1b). Activity of all allelic variants in lung fibroblasts IVL-11NS obtained from normal human tissue was decreased in relation to tumor cells (Fig. 1c). Its value varied insignificantly for all allelic variants. It is worth noting that the p53 protein of NCI-H23 cells carries a missense mutation M246I in the DNA-binding domain [27]. It was shown that efficiency of p53-M246I binding depended on the specific nucleotide sequence of the p53 binding site [30]. In the case of the BIRC5 gene promoter, one can suppose - based on our results - that this mutation has no significant effect on efficiency of p53 binding, because the mutant p53-M246I protein (NCI-H23) suppressed the promoter activity to the same degree as the wild-type p53 (A549 and NCI-H460). However, this supposition undoubtedly requires additional verification.
Activity of the promoter variant PsurvA-mut carrying two single-nucleotide substitutions -1342G>A and -885A>G in the cell lines HeLa, Calu1, NCI-H358, and NCI-H460 was on an average 25-30% lower (p < 0.05) than the activity of allelic variants PsurvA, PsurvB, PsurvC, and PsurvD of the survivin gene promoter. A 45% decrease (p < 0.01) in activity of the PsurvA-mut promoter in comparison with the specified allelic variants was observed in cell lines A549 and NCI-H23 (Fig. 1, a and b). A promoter variant PsurvA-mut2 carrying only the substitution -1342G>A insignificantly differed from the allelic variant PsurvA carrying only the substitution -885A>G (data not shown).
Functional tests performed with the reporter luciferase system have shown that the found single-nucleotide substitutions and the TAAA micro-insertion have no significant effect on the BIRC5 gene promoter activity both in cancer and normal cells. However, all promoter fragments expressed significantly higher activity in cancer cells compared with normal ones. Moreover, activity of promoter fragments depended on the status of p53 in cells: it was lowered in cells with functionally active p53 (including the p53-M246I mutant form) and several times elevated in cells in which the p53 protein is absent or functionally inactive.
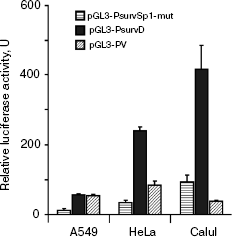
Analysis of the effect of mutation in a putative Sp1 binding site on BIRC5 gene promoter activity. The survivin gene promoter has eight putative Sp1 protein-binding sites including the site called “Sp1-complex” with coordinates (-68...-54) relative to the transcription initiation point. Analysis of nucleotide sequence of the promoter revealed two overlapping putative Sp1 protein binding sites comprising the Sp1-complex. The site with coordinates (-63...-54) fully corresponded to the consensus sequence, whereas the site with coordinates (-68...-59) differed from the consensus sequence at position -66 (Scheme 1b). We have introduced the substitution -58G>A into the sequence corresponding to the consensus site of Sp1 binding, so that it has disrupted the invariant CGCC sequence in a putative Sp1 binding site. Activities of initial PsurvD promoter and PsurvSp1-mut promoter containing this substitution were compared - like in the preceding series of experiments - in ability to provide expression of the reporter gene. Transfection of HeLa, Calu1, and A549 cells with the plasmid constructs pGL3-PsurvD and pGL3-PsurvSp1-mut resulted in decrease of relative luciferase activity for modified promoter (Fig. 2). Luciferase activity decreased on average 7-fold in HeLa cells and 4.5-fold in A549 and Calu1 cells (p < 0.01). This allows mapping of a new functional regulatory site, which presumably binds with the Sp1 transcription factor.
Functional architecture of the survivin gene promoter. Scheme 1a represents the data from the literature and our data on localization of functionally significant and possibly significant sites and polymorphisms in the survivin gene promoter. Most of the polymorphisms we have found are localized in a distal part of the promoter and do not touch functional sites. Such localization agrees with the data we found on independence of promoter activities on allelic variant in this area.Fig. 2. Activity of the survivin gene promoter site PsurvSp1-mut with mutation -58G>A in the Sp1 binding site and the PsurvD promoter in various cell lines. Luciferase activity in extracts of cells transfected with the promoterless plasmid pGL3-BV was taken to be unity. Columns denote the mean value of luciferase activity from three independent experiments, and error bars indicate the standard error of the mean.
At the same time, the majority of binding sites for transcription factors is clustered in a proximal area of the promoter (-250...+70 from the transcription initiation point). This corresponds to a position of proximal promoters in a majority of known genes. An extremely high density of cis-regulatory modules is in this area, which is in agreement with very complex regulation of the BIRC5 gene. Many modules repeat themselves. Their alternative use is possible in different cell types, at different stages of cell cycle, depending on distinct contents of transcription factors. Their alternative use is also evident from overlapping of binding sites for the transcription factors c-myc, Sp1, and CDE element on the locus (-104...-92), factors KLF5, Sp1, and CDE element on the locus (-33...-20), as well as factors Sp1, HIF-1alpha, and p53 on the locus (-17...+7). For instance, the c-myc binding in cells expressing this factor might prevent the binding of Sp1 or proteins interacting with CDE and, contrariwise, high contents of the factors may eliminate the effect of c-myc on the promoter activity.
Antagonistic relationships are also possible between regulation exercised by p53 and HIF-1alpha protein, which binds to the HRE (hypoxia response element) site. Under normoxia conditions, p53 negatively controls the BIRC5 gene promoter activity. This fact allows an explanation that all allelic promoter variants we tested had lower activity in the cells with functionally active p53. It is well known that hypoxia leads to induced apoptosis with involvement of p53. As a rule, the level of HIF-1 dramatically increases under hypoxic conditions [16], which leads to activation of transcription of multiple genes, thus making the cells capable of surviving and avoidance of apoptosis under hypoxic conditions. An important function of p53 under these conditions may be a prevention of activation of the antiapoptotic survivin protein expression due to prevention of HIF binding. This can be achieved via competition for binding with overlapping sites. Moreover, p53 may counteract the binding of Sp1 factor, which is a transcription activator, thereby suppressing the BIRC5 gene promoter activity.
Several elements of the promoter sequence might be tissue-specific. They include the binding sites for TCF4 [31] and KLF5 [32]. On the other hand, the putative functional site of Sp1 binding, which we have found, appears to be used in various cell types: in our experiments these are lung adenocarcinoma and squamous-cell cervical cancer.
The mutation we have analyzed in this work implicates a putative Sp1 transcription factor-binding site, which overlaps another putative binding site for this factor. The role of this overlap also requires special analysis. It might be that these sites are differently used under different conditions. It is also possible that a combination of two sites in one module leads to strengthening of the factor binding and augments its activating role. Thus, the promoter structure combines both the regulatory sites, which are common for many cell types, and tissue-specific ones, thus providing - in accordance with functional multiplicity - wide combinative capabilities for flexible regulation of survivin expression [4].
The authors are indebted to V. K. Potapov and N. V. Skaptsova for synthesis of oligonucleotides used in this work, to Yu. B. Lebedev for kindly provided specimen of genomic DNA, and to fellow workers of the Center of Collective Use “Genome” (Institute of Molecular Biology, Russian Academy of Sciences).
The study was supported by Federal Special-Purpose Program “Studies and developments for priority trends in development of the scientific and technological basis of Russia in 2007-2012” (grant 2007-02-2.2-05-01-006) and by State Support of Leading Scientific Schools (grant 2395.2008.4).
REFERENCES
1.Henson, P. M., and Hume, D. A. (2006) Trends
Immunol., 27, 244-250.
2.Jacobson, M. D., Weil, M., and Raff, M. C. (1997)
Cell, 88, 347-354.
3.Vermeulen, K., van Bockstaele, D. R., and Berneman,
Z. N. (2005) Ann. Hematol., 84, 627-639.
4.Altieri, D. C. (2008) Nat. Rev. Cancer,
8, 61-70.
5.Ambrosini, G., Adida, C., and Altieri, D. C. (1997)
Nat. Med., 3, 917-921.
6.Fukuda, S., and Pelus, L. M. (2006) Mol. Cancer
Ther., 5, 1087-1098.
7.Li, F., Ambrosini, G., Chu, E. Y., Plescia, J.,
Tognin, S., Marchisio, P. C., and Altieri, D. C. (1998) Nature,
396, 580-584.
8.Jiang, Y., Saavedra, H. I., Holloway, M. P., Leone,
G., and Altura, R. A. (2004) J. Biol. Chem., 279,
40511-40520.
9.Raj, D., Liu, T., Samadashwily, G., Li, F., and
Grossman, D. (2008) Carcinogenesis, 29, 194-201.
10.Wagner, M., Schmelz, K., Dorken, B., and Tamm, I.
(2008) Int. J. Cancer, 122, 1278-1287.
11.Cosgrave, N., Hill, A. D., and Young, L. S.
(2006) J. Mol. Endocrinol., 37, 377-390.
12.Li, F., and Altieri, D. C. (1999) Biochem.
J., 344, Pt. 2, 305-311.
13.Xu, R., Zhang, P., Huang, J., Ge, S., Lu, J., and
Qian, G. (2007) Biochem. Biophys. Res. Commun., 356,
286-292.
14.Zhu, N., Gu, L., Findley, H. W., Chen, C., Dong,
J. T., Yang, L., and Zhou, M. (2006) J. Biol. Chem., 281,
14711-14718.
15.Li, Y., Xie, M., Yang, J., Yang, D., Deng, R.,
Wan, Y., and Yan, B. (2006) Oncogene, 25, 3296-3306.
16.Peng, X. H., Karna, P., Cao, Z., Jiang, B. H.,
Zhou, M., and Yang, L. (2006) J. Biol. Chem., 281,
25903-25914.
17.Esteve, P. O., Chin, H. G., and Pradhan, S.
(2007) J. Biol. Chem., 282, 2615-2625.
18.Grossman, D., Kim, P. J., Blanc-Brude, O. P.,
Brash, D. E., Tognin, S., Marchisio, P. C., and Altieri, D. C. (2001)
J. Clin. Invest., 108, 991-999.
19.Mirza, A., McGuirk, M., Hockenberry, T. N., Wu,
Q., Ashar, H., Black, S., Wen, S. F., Wang, L., Kirschmeier, P.,
Bishop, W. R., Nielsen, L. L., Pickett, C. B., and Liu, S. (2002)
Oncogene, 21, 2613-2622.
20.Briggs, M. R., Kadonaga, J. T., Bell, S. P., and
Tjian, R. (1986) Science, 234, 47-52.
21.Jang, J. S., Kim, K. M., Kang, K. H., Choi, J.
E., Lee, W. K., Kim, C. H., Kang, Y. M., Kam, S., Kim, I. S., Jun, J.
E., Jung, T. H., and Park, J. Y. (2008) Lung Cancer, 60,
31-39.
22.Xu, Y., Fang, F., Ludewig, G., Jones, G., and
Jones, D. (2004) DNA Cell Biol., 23, 527-537.
23.Borbely, A. A., Murvai, M., Szarka, K., Konya,
J., Gergely, L., Hernadi, Z., and Veress, G. (2007) J. Clin.
Pathol., 60, 303-306.
24.Wagner, M., Schmelz, K., Dorken, B., and Tamm, I.
(2008) Leuk. Res., 32, 1054-1060.
25.Hattori, M., Sakamoto, H., Satoh, K., and
Yamamoto, T. (2001) Cancer Lett., 169, 155-164.
26.Adams, R. L. P. (1980) Cell Culture for
Biochemists, Elsevier/North-Holland Biomedical Press, Amsterdam-New
York.
27.Mashima, T., Oh-hara, T., Sato, S., Mochizuki,
M., Sugimoto, Y., Yamazaki, K., Hamada, J., Tada, M., Moriuchi, T.,
Ishikawa, Y., Kato, Y., Tomoda, H., Yamori, T., and Tsuruo, T. (2005)
J. Natl. Cancer Inst., 97, 765-777.
28.Nicholson, S. A., Okby, N. T., Khan, M. A.,
Welsh, J. A., McMenamin, M. G., Travis, W. D., Jett, J. R., Tazelaar,
H. D., Trastek, V., Pairolero, P. C., Corn, P. G., Herman, J. G.,
Liotta, L. A., Caporaso, N. E., and Harris, C. C. (2001) Cancer
Res., 61, 5636-5643.
29.Scheffner, M., Munger, K., Byrne, J. C., and
Howley, P. M. (1991) Proc. Natl. Acad. Sci. USA, 88,
5523-5527.
30.Chen, J. Y., Funk, W. D., Wright, W. E., Shay, J.
W., and Minna, J. D. (1993) Oncogene, 8, 2159-2166.
31.Kim, P. J., Plescia, J., Clevers, H., Fearon, E.
R., and Altieri, D. C. (2003) Lancet, 362, 205-209.
32.Ghaleb, A. M., Nandan, M. O., Chanchevalap, S.,
Dalton, W. B., Hisamuddin, I. M., and Yang, V. W. (2005) Cell
Res., 15, 92-96.