REVIEW: Embryonic Stem Cells and the Problem of Directed Differentiation
I. A. Grivennikov
Institute of Molecular Genetics, Russian Academy of Sciences, pl. Kurchatova 2, 123182 Moscow, Russia; E-mail: igorag@img.ras.ru
Received December 29, 2007; Revision received February 18, 2008
During the last two decades molecular genetic and cell mechanisms of proliferation and differentiation of mammalian stem cells have been intensively studied in leading laboratories all over the world. Studies in this field are very important both for basic science and for the development of promising cell therapy technologies. Embryonic stem cells represent a unique experimental model for the investigation of basic principles of mammalian cell differentiation and development. Using this model, important data on similarity in genetic programs during embryonic development and embryonic stem cells differentiation have been obtained. These include basically similar consequent expression of transcription factors, cell receptors, tissue specific proteins, and ion channels. Lines of embryonic stem cells are widely used for the investigation of gene functions in ontogenesis as well as in adult organisms (using gene-knockout strategy). This review deals with different pathways of mammalian (including human) embryonic stem cells differentiation. It considers the main approaches to directed differentiation of these cells in vitro: use of feeder cells, growth factors, and other chemical compounds and also genetic modification. Some examples of application of embryonic stem cells derivatives for cell therapy of some pathological conditions are discussed.
KEY WORDS: embryonic stem cells, embryoid bodies, differentiation, pluripotency, transplantation, cell therapy, transfection, genetic modificationsDOI: 10.1134/S0006297908130051
Abbreviations: BDNF, brain derived neurotrophic factor; BMP, bone morphogenetic protein; ERK, extracellular signal-regulated kinase; ES cells/ESC, embryonic stem cells; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; GFAP, glial fibrillary acidic protein; HIF-1, hypoxia-inducible factor-1; HSC, hematopoietic stem cells; LIF, leukemia inhibitory factor; NGF, nerve growth factor; NT-3, neurotrophin-3; siRNA, small interfering RNA; SSEA, stage-specific embryonic antigen; TGF-β, transforming growth factor β; VEGF, vascular endothelial growth factor.
This review considers the main features and possible directions of
application of mammalian (including human) embryonic stem (ES) cells.
Isolation of mouse ES cells and the development of corresponding cell
lines opened new possibilities not only for studies of developmental
biology and gene functioning at the level of the whole organism, but
also for studies of directed differentiation of these cells with
subsequent development of approaches for cell therapy [1, 2].
In this review, special attention will be paid to the problem of directed differentiation of ES cells into various cell types and perspectives of their use in practical medicine.
After injection into blastocysts, ES cells can be involved in formation of all germinal tissues including generative ones; this gives rise to the development of chimeric animals [3]. Manipulations with mouse ES cells in combination with the method of homologous recombination (which may cause directed changes in ES cells genome) gives a possibility for the development of transgenic animals with particular altered genotype. This approach was originally used in 1987 by Thomas and Capecchi [4]; using mouse ES cells, they demonstrated gene inactivation (knockout) by means of homologous recombination. Subsequent studies on genetic modification of ES cells and directed changes of gene structure of living organisms and substantial contribution of M. Capecchi, M. Evans, and O. Smithies to this field were appreciated by scientific community. In 2007, these scientists were awarded the Nobel Prize in Physiology and Medicine “for their discoveries of principles for introducing specific gene modifications in mice by the use of embryonic stem cells”.
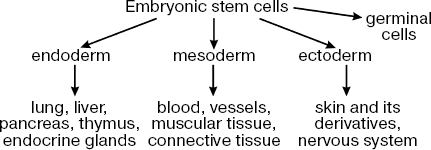
The main property of ES cells is their pluripotency, i.e. the ability for differentiation into all known types of somatic cells found in a living organism in vivo and in vitro and also unlimited proliferative potential with maintenance of initial phenotype [3, 5, 6]. Figure 1 shows the main directions of ES cell differentiation into derivatives of the three germinal layers (ectoderm, mesoderm, and endoderm).
Recently it has been demonstrated that ES cells can be differentiated not only into somatic cells but also into germ cells [7-9]. Taking into consideration this fact, I will use here the term pluripotency for ES cells rather than multipotency, which is used for characteristics of stem cells existing in most tissues of adult organisms (these cells can differentiate into a limited number of cell types).Fig. 1. Main directions of ES cell differentiation.
Three types of mammalian ES cells are recognized.
1. Cells isolated from internal cell mass of mammalian blastocyst [1, 2]. These cells usually defined as ES cells.
2. Cells of embryonic carcinomas originally obtained from teratocarcinomas (consisting of a mixture of differentiated cells from various germinal layers and undifferentiated stem cells) [10]. These cells are characterized by karyotypic instability and low activity in embryonic development during their transfer into a recipient blastocyst. These features limit their use.
3. Primary germinal embryonic cells [11, 12]. These ES cells exhibiting high proliferative activity can be maintained (under certain conditions) in cell culture in the undifferentiated state for a long period.
Maintenance of an undifferentiated phenotype in ES cells culture requires the presence of a so-called feeder layer; the latter is usually mouse primary embryonic fibroblasts or STO fibroblasts [2]. Figure 2 (see color insert) shows mouse ES cells growing at the feeder layer of mouse embryonic fibroblasts.
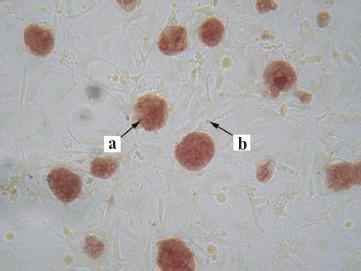
Morphological characteristics of all known ES cells are similar: these cells have a large nucleus preferentially containing euchromatin and several nucleoli. The cells are characterized by a high nucleus-to-cytoplasm ratio: the cytoplasm is a thin round band surrounding the nucleus. ES cells are also characterized by high activity of endogenous alkaline phosphatase (Fig. 3, see color insert), which is one of the commonly used tests suggesting maintenance of the pluripotent status of these cells.Fig. 2. Mouse ES cells colonies on a feeder layer of mouse embryonic fibroblasts: a) colonies formed by ES cells; b) feeder layer cells (×400).
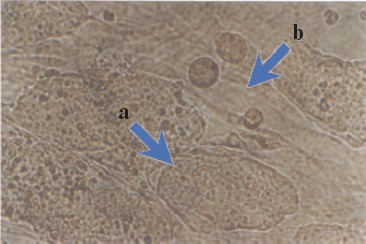
Figure 3 shows staining of colonies formed by undifferentiated ES cells. The feeder layer cells do not give positive reaction to alkaline phosphatase activity. ES cells are also characterized by high level of telomerase activity, which correlates with their undifferentiated degree.Fig. 3. Detection of alkaline phosphatase activity in mouse ES cells culture growing on a feeder layer formed by mouse embryonic fibroblasts (×200): a) stained colonies formed by ES cells; b) unstained cells of the feeder layer.
Below I consider features of some of the best-studied ES cells and the ways of their differentiation.
CHARACTERISTICS OF EMBRYONIC STEM CELLS
Mouse Embryonic Stem Cells
The first mouse ES cells were obtained in the beginning of 1980s [1, 2]. Most of these cells were derived from mouse embryos of the inbred line 129/Sv at the stage of blastocyst. These include the following ESC lines: R1, W9.5, AB-1, CP-1, CCE, CC.1, PJ1-5, E14, D3 [13]. Karyotypic analysis of 35 ESC lines has shown that 25 lines are characterized by the XY karyotype whereas the others have XO and XX karyotypes. It is suggested that ES cells are preferentially formed from cells with male karyotype, because the second X chromosome may be a pluripotency destabilizer [14]. All ES cells have basically normal karyotype (2n = 40) and its changes result in the loss of pluripotency [15].
Mouse ES cells exhibit high proliferative activity; in culture, they can maintain undifferentiated state for a long time as well as normal and stable karyotype. Mouse ES cells are characterized by relatively short time for cell population doubling (about 12-15 h) with very short G1 phase. In these populations, cells of moderate size (10-12 µm) predominate; during reseeding, they exhibit adhesive and proliferating properties. Contacting the feeder layer, they form (visually) normal monolayer colonies, which can differentiate into embryoid bodies under certain conditions. In the beginning of ES cells differentiation, expression of cyclin D increases, G1 phase is prolonged, and the rate of cell divisions decreases [16].
As mentioned above, for maintenance of undifferentiated phenotype of mouse ES cells in vitro cells are cultivated on a feeder layer. The latter is usually formed by primary embryonic fibroblasts inactivated by mitomycin C or by γ-irradiation. Another method used for prevention of ES cells differentiation is addition of a specific growth factor, known as leukemia inhibitory factor (LIF), to the culture medium (lacking the feeder layer).
Some features of leukemia inhibitory factor. LIF belongs to a class of cytokines. It is a hematopoietic regulator inducing differentiation of myeloid leukemia M1 cell line. The feeder cells from primary embryonic fibroblasts produce LIF and thus maintain undifferentiated state of ES cells [17]. In the presence of bovine embryonic serum, recombinant LIF can partially function as the feeder layer and provide growth and proliferation of ESC [6].
LIF is a member of the interleukin-6 (IL-6) family, which also includes interleukin-11 (IL-11), oncostatin M, ciliary neurotrophic factor (CNTF), and cardiotrophin (CT-1).
LIF realizes its effects via interaction with a corresponding plasma membrane receptor; the latter is a heterodimer complex of proteins gp130 and LIFR. Initially LIF interacts with its receptor, LIFR, and then with gp130. This results in formation of a ternary complex [18], which activates the transcription factor STAT3 signaling pathway. This is the main factor responsible for maintenance of ES cells pluripotency [19, 20]. Besides, the signaling pathway associated with STAT3 maintenance of the undifferentiated status of ES cells can also involve the BMP-SMAD pathway. Recently, it has been demonstrated that in serum free medium LIF can block neuronal differentiation of mouse ES cells and maintain only their pluripotency to a lesser extent. However, combined effects of LIF and BMP (bone morphogenetic protein) maintain ES cell pluripotency. These authors suggest that the major contribution of BMP is attributed to induction of Id gene expression involving the SMAD pathway [21]. It should be noted that there are two other pathways involved in maintenance of the undifferentiated state of ES cells--ERK and Wnt-1 [22, 23].
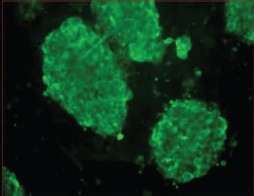
Surface antigens and maintenance of pluripotency. Mouse ES cells express the surface antigen SSEA-1 (stage-specific embryonic antigen) detected at the stage of an 8-cell mouse embryo up to the inner cell mass of the blastocyst [24].
Figure 4 (see color insert) shows immune fluorescence detection of the SSEA-1 antigen on the surface of mouse ES cells. Full staining of the cell clusters suggests that most cells are in the undifferentiated state and maintain their pluripotent properties. Earlier documentation of undifferentiated phenotype of mouse ES cells employed the ECMA-7 marker, and for differentiating cells the TROMA-1 marker was used. It has already been mentioned that mouse ES cells are characterized by high level of expression of endogenous alkaline phosphatase (Fig. 3) and telomerase, which is required for maintenance of proliferative activity [25, 26].
In joint experiments with colleagues from Sechenov Moscow Medical Academy, we were the first to find calcium channels in mouse ES cells and to characterize them [27]. It was shown that voltage-gated ion channels are basically absent on the surface of ES cells. However, our results suggested the presence of a large calcium store in ES cells; it was sensitive to thapsigargin, a specific inhibitor of calcium ATPase of intracellular calcium stores. Ryanodine-sensitive stores were weakly developed in these cells.Fig. 4. Immune fluorescence detection of SSEA-1 antigen in culture of mouse ES cells grown on a feeder layer of mouse embryonic fibroblasts (×400).
Maintenance of pluripotency of mouse ES cells required expression of some transcription factors such as Oct-4, Nanog, and Rex-1 [28-31]. Oct-4 is a member of the POU protein family; it is required for formation of the inner cell mass of the blastocyst and is expressed in early stages of the development of mouse embryos [32]. However, the increase of its expression causes differentiation of ES cells in the mesoderm and endoderm, whereas its absence results in formation of trophoectoderm [28]. The transcription factor Nanog is also involved in maintenance of pluripotency of mouse ES cells; the increase in its expression promotes maintenance of undifferentiated state of ES cells in the absence of LIF and corresponding activation of STAT3. Maintenance of pluripotency of ES cells with increased expression of Nanog but in the absence of LIF requires Oct-4 expression, because in the absence of Oct-4 cell differentiation begins [31, 33]. Gordeeva et al. [30] compared expression of the homeobox-containing transcription factors Oct-4, Pax-6, Prox-1, and Ptx-2 in mouse ES cells. This study originally demonstrated expression of Prox-1 and Ptx-2 genes at initial stages of cell differentiation. This suggests an important role of these genes in embryogenesis.
It has recently been shown that some neurotrophins (BDNF (brain derived neurotrophic factor), NT-3, and NT-4 (neurotrophin-3 and -4)) can maintain both proliferation and pluripotency of a culture of human ES cells; these neurotrophins prevent apoptosis of these cells [34]. The effects of neurotrophins are realized via a signaling pathway that includes activation of phosphatidylinositol-3-kinase.
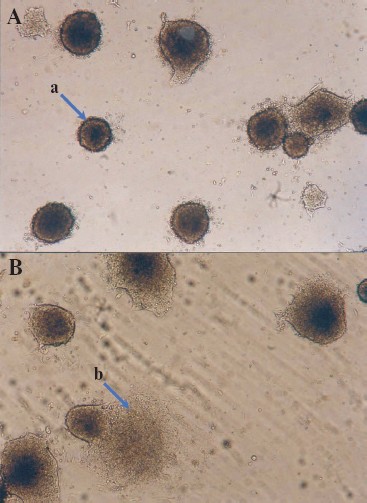
Formation of embryoid bodies. Initial stages of differentiation. In the absence of the feeder layer and LIF, ES cells form so-called embryoid bodies, which represent germs of endoderm, ectoderm, and mesoderm and resemble post-implantation development. Attachment of an embryoid body to the adhesive substrate is accompanied by disruption of tight junctions between surface cells; this results in impairment of basal membrane integrity and formation of contacts between inner cells with a substrate. After that the inner cells migrate from the embryoid body and their active proliferation followed by subsequent differentiation into various tissues (e.g. muscular, nervous, epithelial, etc.) begins [6]. Mouse ES cells can form simple and cystic embryoid bodies [5, 35].
Figure 5 (see color insert) shows the embryoid bodies formed by mouse ES cells grown on a gelatin substrate and also initial stages of differentiation of these cells accompanied by disruption of embryoid bodies and cell migration from the inner space along the substrate [46].
It should be noted that differentiation of ES cells can occur spontaneously, and it may also be induced by some growth factors and various chemical compounds. Differentiation of ES cells in vitro is used as a model for studies of embryogenesis and corresponding differentiation into various cell types.Fig. 5. Embryoid bodies obtained in a culture of mouse ES cells (×200). A) Embryoid bodies on the 3-4th day after attachment to the gelatin substrate; a) embryoid body. B) Onset of cell migration from the embryoid bodies; b) arrow shows cells migrating from the embryoid body after disruption of an external shell.
Human Embryonic Stem Cells
Isolation and cultivation of human ES cells was a colossal achievement of the 1990s. These cells were isolated from pre-implanted blastocysts fertilized in vitro by their extraction from the inner cell mass (which was then plated onto the feeder layer) followed by subsequent cell growth and preparation of corresponding cell lines [25, 36-39].
Figure 6 (see color insert) shows human ES cells grown on a feeder layer of mouse embryonic fibroblasts.
Injection of human ES cells under testicular capsule of immune deficient mice (SCID strain) resulted in formation of teratomas containing derivatives of germinal layers [25, 36]. Pluripotency of human ES cells is also maintained by the transcription factors Oct-4 and Nanog [33]. Similarly to mouse ES cells, human ES cells could maintain for a long time their pluripotency, high proliferative activity, and (this is especially important) normal karyotype (46XX/XY) [40]. These cells are also characterized by high activity of alkaline phosphatase and telomerase [25, 37]. However, differences between human and mouse ES cells also exist. Some of these differences are listed in Table 1.Fig. 6. Colonies of human ES cells, line hESM03, grown on a feeder layer of mouse embryonic fibroblasts (×400). This photo was kindly provided by M. A. Lagar'kova (Institute of General Genetics, Russian Academy of Sciences).
Table 1. Some properties of mouse and human
ES cells
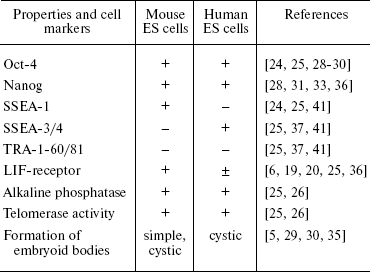
In contrast to mouse ES cells, human ES cells form only cystic embryoid bodies. Undifferentiated human ES cells express specific surface antigens SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 [25, 37, 41] and do not express SSEA-1 typical for mouse ES cells. Cell population doubling time is higher for human ES cells (30-35 h) than for mouse ES cells (12-15 h) [40]. In most cases, reseeding of mouse ES cells employs trypsin, whereas mechanical dissociation is used for reseeding of human ES cells because their enzymatic treatment often results in chromosome aberrations [35]. However, recently 17 lines of human ES cells with normal karyotype were obtained after enzymatic treatment of cells [42]. Cultivation of human ES cells in vitro also often employs the mouse embryonic fibroblast feeder, secreting necessary growth factors. However, it was demonstrated that LIF weakly inhibited differentiation of these cells in spite of activation of the LIF/STAT3 signaling pathways in human ES cells (it is possible that this activation is not sufficient for maintenance of pluripotency of these cells) [25, 36, 43]. The major difference of human ES cells from mouse ES cells is spontaneous or BMP-4-induced differentiation into trophoectoderm cells [25, 44]. It is possible that the inner cell mass of the human blastocyst maintains ability for generation of trophoectoderm. This suggestion is supported by data that a decrease in expression of Oct-4 in human ES cells results in increased expression of trophoectodermal (Cdx2) and extraembryonic endodermal markers such as GATA-6 [45]. Suppression of Nanog gene expression by means of RNA interference was also accompanied by increased expression of trophoectodermal markers in these cells [46].
Possible application of human ES cells in cell therapy requires very strict conditions of their cultivation. The possibility of human cell infection with pathogenic microorganisms, retroviruses, and other pathogens, which might contaminate embryonic serum and feeder layer cells, should be absolutely ruled out. In this connection, much attention is now paid to preparation of new lines of human ES cells that can grow on a feeder-free substrate using synthetic media with serum substitutions; such approach can significantly standardize the cultivation procedure [48].
Other Embryonic Stem Cells
Pluripotent stem cells have also been obtained from other animals including hen [48], hamster [49], rabbit [50], rat [51, 52], mink [53], pig [54], sheep [55], cattle [56-58], and some primates [59-63].
However, only mouse and chicken ES cells can differentiate into germinal pathway cells. Development of studies on human ES cells also required ES cell lines from other primates. Monkey ES cells are characterized by typical markers of human ES cells; they also maintain normal karyotype and high proliferative activity in vitro [60]. These cells can be a convenient model for studies of human ES cells. In addition, the monkey ES cell line Cyno-1 isolated from a blastocyst obtained by parthenogenesis in vivo exhibited many features typical for human ES cells: high activity of telomerase and alkaline phosphatase, expression of corresponding cell markers (Oct-4, SSEA-3, SSEA-4, TRA-1-60, TRA-1-81), and ability to differentiate into various cell types. This gives hope that parthenogenetically derived cells might serve as a source for autologous therapy; this would exclude the necessity of embryo development. However, use of cells lines obtained using such an approach has certain limitations because of impairments in expression of imprinting genes in these cells [65].
Application of ES Cells
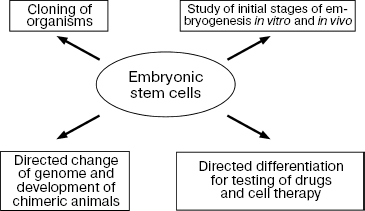
ES cells are now used both in basic scientific studies and in practice (Fig. 7).
Since their discovery, ES cells have been used for studies of initial stages of embryonic development of various species, especially mammals, and recently for their cloning as well. Some experiments have shown that in vitro ES cells undergo all stages of initial development that normally occur in embryos in vivo.Fig. 7. Directions for use of ES cells in biology and medicine.
ES cells have been successfully used to create chimeric animals. It was already mentioned that mouse ES cells were used as a model for the development of methods of homologous recombination; use of these methods allowed the introduction of extremely accurate changes in genomes of living cells and the monitoring of functions of these altered genes at the level of the whole organism [4, 13].
The most recent and intensively developing directions of application of ES cells include their directed differentiation into certain cell types. This is especially important for human cells, which may be used for cell transplantation and also for the development of cell models for testing of drugs (Fig. 7).
DIRECTED DIFFERENTIATION OF EMBRYONIC STEM CELLS AND FACTORS
INFLUENCING THIS PROCESS
Studies of differentiation of ES cells and especially their directed differentiation into certain types of cells represent not only theoretical interest, but they give hope to develop perspective methods of cell or tissue therapy of various serious human diseases.
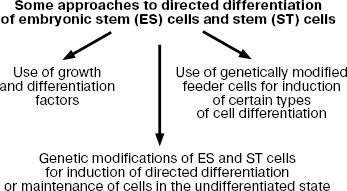
Here I consider some approaches to regulation of differentiation of ES cells, which are summarized in Fig. 8.
It should be noted that at early stages of the development of these approaches the major attention has been paid to search and employment of various growth and differentiation factors; however, now genetic modifications of ES cells for induction of certain types of cell differentiation attracts much attention. During recent years, much attention is paid to genetic modifications of ES cells for induction of certain types of cell differentiation.Fig. 8. Modes of induction of directed differentiation of ES cells in vitro.
Below is a list of conditions that should be used during the development of approaches for directed differentiation of ES cells for their subsequent use for transplantation into a particular recipient (particularly a human).
1. Initial conditions should favor elaboration of a large number of undifferentiated ES cells.
2. During the next stage, it is necessary to differentiate cells and to obtain maximally possible number of cells of a certain type (ideally it should be a homologous population) suitable for transplantation.
3. Solution of all these tasks requires the development of standardized conditions for elaboration of ES cells and for their subsequent differentiation as well; these are important preconditions to get reproducible results.
In vivo studies of directed differentiation of ES cells and the effects of cell environment on this process use cell transplantation into various organs of adult animals (brain, liver, heart, etc.) followed by subsequent monitoring of the transplanted cells. Such studies employ ES cells marked with fluorescent proteins. Cells expressing such proteins are easily detected in sections by means of fluorescence microscopy of sections of organs and tissues of interest.
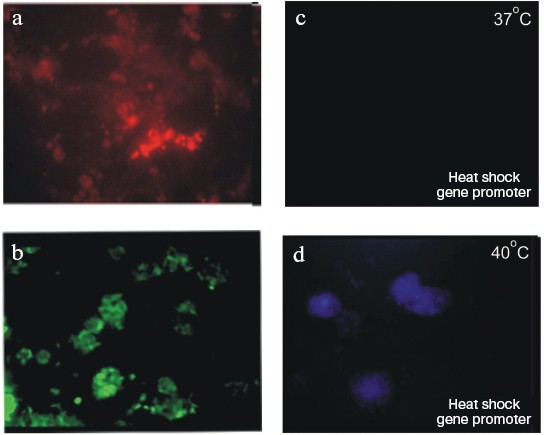
Figure 9 (see color insert) shows ES cells of R1 line transformed with plasmids carrying genes encoding various fluorescent proteins: “green”, “red”, and “blue”. The use of genes under controllable promoters is a very promising approach. For example, Fig. 9 (c and d) shows expression of a “blue” protein under control of promoter of heat shock protein gene. The figure clearly shows lack of staining of transfected cells incubated at 37°C and appearance of blue fluorescence of cells incubated at 40°C [66].
Use of Feeder Layer CellsFig. 9. Mouse ES cells, line R1, transfected with vectors carrying “colored” protein genes. (Data obtained in collaboration with L. I. Korochkin and his colleagues at the Institute of Gene Biology, Russian Academy of Sciences.) a) ES cells transfected with a plasmid carrying “red” protein gene under control of the cytomegalovirus promoter. b) ES cells transfected with a plasmid carrying “green” protein gene under control of the cytomegalovirus promoter. c, d) ES cells transfected with a plasmid carrying “blue” protein gene under control of the heat shock protein promoter: c) cells grown at 37°C; d) the same cells grown at 40°C (×200).
Cells of the feeder layer can be used for induction of differentiation of ES cells; this has recently been demonstrated by Schmitt et al. [67].
Mouse ES cells were cultivated on a substrate of OP9 stromal cells expressing the delta-1 ligand required for activation of corresponding Notch receptors on ES cells. During experiments, the authors achieved differentiation of the ES cells into T-lymphocytes, which expressed surface markers specific for this type of cells. Moreover, functional competence of resultant T-lymphocytes was demonstrated during their transplantation into immune deficient mouse recipients lacking these cells. Thus, this is a rather perspective approach for elaboration of a basically unlimited number of cells required for particular experiments and also for their directed differentiation with high yield of desired cells.
Use of Growth and Differentiation Factors
Various chemical compounds are now widely used for induction of differentiation of ES cells in certain direction. Retinoic acid, a vitamin A derivative, was one of the first compounds used for this purpose. Interestingly, various concentrations of retinoic acid determine preferential differentiation of ES cells into either neuronal or myogenic directions.
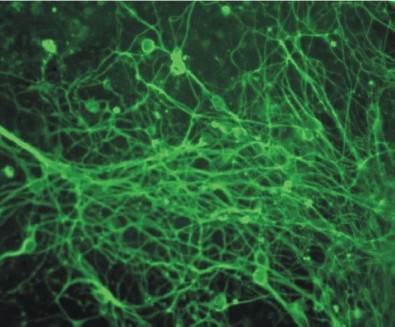
Figure 10 (see color insert) shows differentiation of mouse ES cells in the neuronal direction; this was caused by 1 µM retinoic acid added to cell cultures at the stage of embryoid bodies.
Studies of genetic control of myogenic and epithelial differentiation of ES cells have shown that sequential expression of tissue-specific genes in these cells is the same as during the process of normal development [68, 69].Fig. 10. Induction of neuronal differentiation in a culture of mouse ES cells treated with 1 µM retinoic acid. Immunofluorescent detection of cells by means of antibodies against β-III-tubulin (×200).
Ways and modes of differentiation of these cells into some cell types induced by factors of growth and differentiation are considered below.
Differentiation into cardiomyocytes. Cardiovascular diseases are the leading cause of mortality in such countries as USA, Russia, China, and India. In our country up to one million people die every year from these diseases. So, in vitro cardiomyocyte preparation is one of the important directions of induction of ES cell differentiation. This is related to the possibility of elaboration of large quantities of cells for replacement therapy. ES cell differentiation into cardiomyocytes can be easily observed in vitro by appearance of spontaneously contracting cell clusters. In most modern protocols describing differentiation of ES cells into cardiomyocytes, initial stages of manipulations with cells include the stage of embryoid body formation in the absence of LIF [70].
There are several factors influencing differentiation of ES cells into cardiomyocytes: type of ES cell line, initial number of cells in the embryoid body, medium composition, and the presence of serum, growth factors, and other additions as well as time of embryoid body seeding. Among conditions promoting differentiation of mouse and human ES cells in the cardiogenic direction one should mention: retinoic acid (10 nM) [71], dimethyl sulfoxide (0.5-1.5%) [72], 5-azo-deoxycytidine [73], and ascorbic acid [74]. Recently a stimulating effect of nitric oxide on differentiation of mouse ES cells into cardiomyocytes has also been found [75]. Initial stages are characterized by the increase of regions with spontaneous contractions followed by subsequent increase in the contraction rate. Aggregation of cells and changes in the rate of spontaneous contractions may last from several days up to one month depending on cell number at the initial stages. It is important to understand that totally differentiated cardiomyocytes often stop spontaneous contractions, but they can be maintained in culture for a long time. Thus, three stages of differentiation can be arbitrarily recognized depending on duration of cell cultivation: early (appearance of pacemaker-like cells and primary cardiomyocytes), intermediate, and terminally (differentiated atrial and ventricular cells). Methods of differentiation of ES cells into cardiomyocytes as well as modes of enrichment of differentiating cells with cardiomyocytes have been developed [76].
Differentiation into central nervous system cells. Directed differentiation of ES cells via the ectodermal pathway, which includes differentiation of cells of the nervous system, is now well documented. Most studies are focused on preparation of nervous tissue from ES cells. In addition, other ectoderm elements have also been obtained; these include cells expressing keratinocyte markers [69]. Using these cells, structures similar to embryonic skin have been obtained; this suggests that they may normally function in tissues [77]. Formation of skin tissue cells is particularly regulated by BMP4 [78]. Interestingly, addition of BMP4 to a culture of cells differentiating via the neuroectodermal pathway results in inhibition of the development of nervous tissue and cell differentiation into keratinocytes.
It is very difficult to investigate differentiation of ES cells via the neuroectodermal pathway because of very complex structure of the mammalian nervous system. Taking into consideration the practical impossibility of investigation of initial stages of cells differentiating via the neuroectodermal pathway in vivo (especially in humans), ES cells should be considered as a unique model of these processes, because during differentiation in the neuroglial direction in vitro they pass all the stages as during in vivo development [79, 80].
It should be noted that differentiation in this direction results in formation of various cell types: neurons and glial cells. In addition, a “common pool” of glial cell precursors gives rise to various differentiated cells such as astrocytes (also subdivided into several types), oligodendrocytes, and Schwann cells. The population of neurons formed during differentiation is also heterogeneous; it consists of neurons of various ergicity including dopaminergic, cholinergic, GABA-ergic, glutamatergic neurons, etc. Compared with this, differentiation of ES cells into cardiomyocytes is a rather “simple” task.
If directed differentiation of ES cells (especially human ES cells) is considered as the source for subsequent transplantation into brain, strict requirements for methodological regulations become quite understandable.
Differentiation of human ES cells in the neuroglial direction was achieved using a system of artificial three-dimensional carriers of polyhydroxy esters [81]. In this system, co-treatment of ES cells with NGF (nerve growth factor) and NT-3 resulted in significant increase in number of both nestin-positive (neuronal precursors) and β-III-tubulin positive (neurons) cells. In the differentiating culture, structures resembling vessels were also observed. Gerrard et al. [82] developed effective differentiation of human ES cells in the neuronal direction. The method is based on blockade of BMP signaling in cells by means of its antagonist noggin used during early stages of differentiation. Use of such approach during long-term cultivation yielded up to 90% of cells expressing such markers of neuron precursors as nestin and PSA-NCAM (nerve cell adhesion molecule). Subsequent cultivation resulted in appearance of mature nerve cells expressing MAP2 and β-III-tubulin. It is important to indicate that on the developed system appearance of glial cells was not observed during long time of cultivation. Only after 80 days of cultivation, appearance of GFAP (glial fibrillary acidic protein)-positive cells was observed.
Use of this method resulted in preferential differentiation of cells into GABA-ergic neurons. There was small number of TH (tyrosine hydroxylase)-positive cells (presumably dopaminergic neurons) and lack of any glutamatergic neurons. However, if cells were treated at a certain stage of differentiation (cultivation for about 40 days) with some growth factors such as FGF8 (fibroblast growth factor), GDNF (glial derived neurotrophic factor), BDNF, and also with ascorbic acid (in various combinations), the number of TH-positive cells significantly increased. Thus it seems that using various combinations of growth factors it is possible to obtain neuronal enriched cell populations.
Lecithin-modified BDNF also increased neuronal differentiation of ES cells both in vitro and in vivo [83]. Cell differentiation into neuronal direction is accompanied by intracellular activation of ERK 1/2 (extracellular signal regulated protein kinase) [84]. Corresponding inhibition of these kinases by a specific inhibitor (UO126) resulted in significant decrease in neuronal differentiation. In addition, the inhibitor caused activation of caspase 3; this suggests that the ERK signaling pathway is involved not only in differentiation, but also in maintenance of cell viability.
Study of neuronal differentiation of ES cells lacking JNK1, JNK2, and JNK3 protein kinases (belonging to the family of stress-activated protein kinases) revealed interesting features [85]. ES cells lacking JNK2 and JNK3 kinases exhibited normal ability to differentiate in the neuronal direction as normal cells, whereas ES cells lacking JNK1 did not exhibit such feature. However, these cells demonstrated preferential epithelial differentiation. Neuronal differentiation of mammalian ES cells could also be induced by factors synthesized by avian neurons. Addition of conditioned medium obtained from cultures of chicken dorsal root ganglion neurons significantly stimulated such differentiation [86].
Peptide regulators of neuroglial differentiation. In contrast to growth and differentiation factors, which are rather large (and therefore expensive) protein molecules, regulatory peptides are potentially more attractive and perspective factors influencing processes of directed differentiation of ES cells.
Cazillis et al. [87] used two peptides for differentiation of mouse ES cells: pituitary adenylate cyclase activating polypeptide (PACAP) and vasoactive intestinal peptide (VIP). Both peptides act at VPAC1 and VPAC2 receptors coupled to GTP-binding proteins. Incubation of ES cells with each peptide (final concentration 100 nM) for eight consecutive days after seeding and subsequent formation of embryoid bodies resulted in induction of directed differentiation of these cells in the neuronal direction. In control cultures the proportion of cells with neuronal phenotype (positive for staining on neuron specific enolase) varies from 10 to 25%; after treatment with PACAP and VIP it increase to 80 and 91%, respectively. PACAP and VIP did not influence the number of O4-positive cells (an oligodendrocyte marker). In cultures treated with these peptides, there were no GFAP-positive cells (an astrocyte marker) found also. In differentiated cultures, TH- and GABA-positive cells represented 47 and 52% and 54 and 42% after induction of differentiation by PACAP and VIP, respectively. Both peptides stimulated neuron formation and increased their length (in the treated cultures). These peptides are no exception. For example, Kim et al. [88] demonstrated that agonists of ì- and ê-opioid receptors (also transducing signals by means of a system of GTP-binding proteins) stimulated differentiation of ES cells into neuronal precursors. It is suggested that their action is realized via the system of corresponding (ERK) protein kinases. Thus, use of short peptide may represent a perspective approach to directed differentiation of ES and other stem cells, which might then be used in cell therapy.
Hematopoietic differentiation pathway. Hematopoiesis is formation of blood cells from their stem cell origin. Bone marrow stem cells give rise to two lineages: lymphoid (B- and T-lymphocytes) and myeloid (erythrocytes, macrophages, segment nuclear cells, and megakaryocytes). During embryo development, hematopoiesis occurs via two independent (primary and secondary) pathways. Primary hematopoiesis required only for embryonic development involves yolk-sac cells, which produce embryonic erythrocytes and macrophages, but cannot form hematopoietic stem cells (HSC) and lymphoid lineage cells. Secondary hematopoiesis gives rise to HSC and then to all blood cells typical of the adult organism.
During cultivation in a medium containing embryonic serum, ES cells undergo preferential differentiation via the hematopoietic pathway (with high reproducibility of such experiments) [89]. In the forming embryoid bodies more that 50% of cells express Flk-1 receptor (for VEGF), the characteristic marker of hematopoietic cells and blood vessel cells [90]; more than 5% of cells can give stably dividing line of hematopoietic cells. Detailed study of differentiation kinetics, set of working genes, the level of their expression, and comparison with data on yolk-sac cells has shown that ES cells develop via the primary hematopoietic pathway [91-93].
The direction of ES cells into the secondary hematopoiesis pathway required changes in conditions of cell growth. During common cultivation of ES cells with OP9 stromal cells, the former underwent lymphoid cytokine-induced differentiation into B-lymphocytes [94, 95].
Cells exhibiting HSC properties were obtained during cultivation of ES cells in medium containing IL-3, IL-6, and SCF (stem cell factor). Cells possessing CD45 and c-kit markers were isolated and injected into thighbone of mice subjected to irradiation induced bone marrow cell killing. Some time after injection, cells derived from lymphoid and myeloid lineages formed from corresponding ES cells were found in peripheral blood of these mice [96]. It was also demonstrated that increased expression of HoxB4 resulted in formation of HSC, capable for functioning in vivo [97]. ES cells represent a convenient model for studies of early stages of the hematopoietic pathway because in embryos this system is hardly available for investigation.
Differentiation of ES cells into insulin-producing cells. About 4-5% of the human population suffers from diabetes mellitus, and the total number of patients may exceed 350 million by 2010. So development of directed differentiation of ES cells into β-cells of islets of Langerhans and subsequent transplantation of such cells are very important for the development of new methods for treatment of this disease. Using H9 human ES cells, Assady et al. [98] demonstrated that long-term cultivation of these cells is accompanied by formation of embryoid cells and results in formation of a rather large number of cells producing insulin. After 19 days of differentiation, up to 60% of the embryoid cells gave positive immune staining for insulin. The first cells synthesizing insulin appeared on the 14th day of cultivation. Among the cells giving positive staining for insulin, about 1-3% of cells exhibited high level of insulin secretion. However, the authors failed to detect induction of insulin secretion by glucose. Lumelsky et al. [99] developed a five-stage method for differentiation of ES cells into insulin-producing cells. Subsequently Shi et al. [100] improved the procedure of ES cell differentiation and shortened the time required for β-cell production to two weeks. The major approach used by these was sequential treatment of mouse R1 ES cells with various growth factors: activin A, retinoic acid, and bFGF. The resulting cells regulated insulin secretion in response to addition of glucose. This is an important achievement of this group. These cells expressed specific markers presented at the β-cells of Langerhans islets. Administration of such cells to mice with experimental diabetes normalized blood glucose. However (as in other cases), transplantation of such insulin-producing cells frequently caused the development of malignant tumors in the recipient mice.
Differentiation of ES cells into other cell types. A combination of various factors may give preferential differentiation of ES cells into other cell types. For example, hepatocyte growth factor (HGF) in combination with NGF caused a significant shift of differentiation of mouse ES cells to formation of hepatocytes: after cultivation with these factors for 15 days, there was appearance of a large number of functionally active hepatocytes. This was confirmed by detection of specific markers in these cells: α-1-antitrypsin, α-fetoprotein, albumin, and hepatocytes nuclear factor [101]. Inhibition of TGF-β (transforming growth factor-β) and signaling associated with the factor resulted in inhibition of differentiation of ES cells into smooth muscle cells.
Use of Genetic Modifications in ES Cells for Their Directed Differentiation
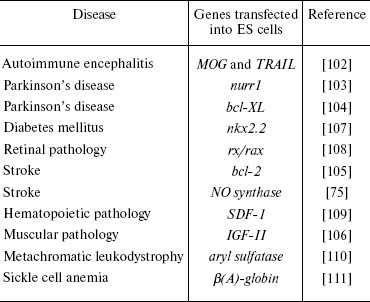
Insertion of certain genes into ES cells of various origin is used not only for studies of functions of these genes, but also for solution of problems of directed differentiation and elaboration of a large number of cells with certain phenotype that would be suitable for cell therapy. Some examples of the use of the genetic modification of ES cells are summarized in Table 2.
Table 2. Some examples illustrating use of
genetically modified ES cells for cell therapy
For preparation of dopaminergic neurons, Kim et al. [103] used genetic modification of ES cells with the transcription factor Nurr1 followed by subsequent cell differentiation in the presence of PA6 stromal cells. Cell lines overexpressing Nurr1 formed twice as many neurons as control cultures and the number of TH-positive cells in these cultures exceeded 50% of total cell number. The resultant TH-positive cells not only synthesized and secreted dopamine into the medium, but also exhibited electrophysiological characteristics typical for midbrain dopaminergic neurons. Transplantation of such cells into mouse striatum resulted in their effective integration into the brain tissue.
Another approach was realized by Shim et al. [104]. They obtained ES cells transfected with bcl-xl gene; these cells expressed antiapoptotic protein of the Bcl-2 family. Differentiation of the transfected cells resulted in appearance of a larger number of cells expressing markers of dopaminergic neurons compared with control cells. Moreover, such cells were less sensitive to the cytotoxic effect of 1-methyl-1,4-phenylpyridine, a known cytotoxic agent for dopaminergic neurons. Transplantation of cells expressing the bcl-xl gene to the brain of rats with symptoms of Parkinson's disease caused clearer normalization of behavioral symptoms than in recipient animals receiving control cells.
In attempt to correct brain impairments after experimental stroke, Wei et al. [105] also used mouse ES cells transfected with the bcl-2 gene. Treatment of the transfected cells with retinoic acid (for induction of neuronal differentiation), followed by their transplantation into the cavity formed after stroke, in 1-8 weeks after transplantation resulted in filling of the cavity with cells expressing markers of neurons, astrocytes, and oligodendrocytes. Moreover, the transplanted cells exhibited higher viability and ability for neuronal differentiation and the recipient animals were characterized by accelerated restoration of impaired brain functions. Thus, genetically modified ES cells might be used as a very valuable tool for therapy of severe strokes.
Using genetic modifications of ES cells, it is possible to achieve their differentiation into other cell types. Kanno et al. [75] used an adenovirus vector carrying the gene of the inducible form of NO synthase for induction of mouse ES cell differentiation into cardiomyocytes. The transfected cells formed four times more clusters of contracting cardiomyocytes than control cultures. At the stage of embryoid bodies, the treatment of ES cells with compounds generating NO caused similar effect. The cardiomyocytes formed in vitro expressed antigenic markers of mature cardiomyocytes (troponin I and myosin light chains). Interestingly, NO not only induced differentiation of ES cells into cardiomyocytes, but also stimulated apoptosis in undifferentiated cells.
Induction of myogenic differentiation was obtained after transfection of mouse ES cells with the gene encoding IGF-II (insulin-like growth factor-2) [106]. After transfection and corresponding selection, the cells exhibited expression of both early (myogenin, myoD) and later (dystrophin) markers of skeletal musculature. Transplantation of such cells into mice with injured anterior tibial muscle significantly improved their motor functions. This was accompanied by formation of new myofibrils. The authors concluded that genetic modification of ES cells with IGF-II might be perspective for cell therapy of various types of muscle diseases and injuries.
Genetic modifications of ES cells by means of the Nkx2.2 gene resulted in appearance of pancreatic β cells in vitro [107]. These cells were able to synthesize and secrete insulin into the medium and also to express some cell markers of β cells (proinsulin 1, proinsulin 2, and PDX1). However, the authors failed to demonstrate insulin secretion induced by glucose.
Modification of ES cells by transcription factor Rx/rax resulted in differentiation of some of the cells into retinal neurons in vitro [108]. According to electrophysiological data, these cells exhibited functional activity.
Some factors produced by stromal cells can influence differentiation of ES cells towards hematopoietic cells. For example, transfection of mouse ES cells with the stromal cell-derived factor SDF-1/CXCL12 stimulated formation of erythroid, macrophage, and multipotent precursors of hematopoietic cells in vitro [109].
Metachromatic leukodystrophy is a very severe disease characterized by impairments in lipid metabolism and progressive demyelinization in the central nervous system. It is caused by deficiency of aryl sulfatase (AS), and AS-deficient mice have been obtained to model this disease. ES cells transfected with the AS gene were used for correction of this disease in these mice. After transfection, cells were differentiated into glial cells, which were then transplanted into the brain of AS-deficient mice. The transplanted cells maintained their viability and integrated into various brain regions. Four weeks after transplantation, there was significant decrease (by ~50%) in sulfatide deposits in brain regions of these animals; this suggests success of the cell therapy [110].
Wu et al. reported about successful correction of sickle cell anemia modeled in mice by means of expression of a human mutant gene of β(S)-globin [111]. The authors replaced the gene of β(S)-globin for the normal gene of β(A)-globin in ES cells obtained from such animals, and then they obtained transgenic mice lacking pathological symptoms of this disease. Recently Hanna et al. [112] reported similar results, but these authors used autologous induced pluripotent cells corrected by means of homologous recombination with the human β-globin gene.
Modifications of ES cells with various viral genes are interesting not only for studies of functioning of these genes and their effects on early embryonic development and cell differentiation; they can be used for the development of cell models for pharmacological studies.
During studies of functions of various immunodeficiency virus genes, mouse ES cells transfected with the regulatory genes tat and nef of this virus were obtained [113]. Earlier it was demonstrated that the tat gene exhibits oncogenic potential and it activates transcription of some genes and increases cell proliferation. The nef gene suppresses expression of some genes and decreases growth of cells, but it cooperates with the tat gene during formation of transformation foci [114]. Transfection of mouse ES cells with these genes revealed some interesting facts [115]. The cells transfected with tat gene demonstrated higher proliferative activity compared with control cells as well as compared with cells transfected with the nef gene. The time required for formation of embryoid bodies by all cell lines insignificantly differ from control cells. However, in cultures transfected with the nef gene and the tat gene, the number of embryoid bodies formed was two times higher and two times less than in control, respectively. In cells transfected with the nef gene the percent of the embryoid bodies with contracting cardiomyocytes was 1.5-fold higher than in the control, whereas in ES cells transfected with the tat gene this parameter was lower than in the control. Thus, there was a reverse correlation between the effects of HIV regulatory genes on proliferation of ES cells and differentiation of these cells [115].
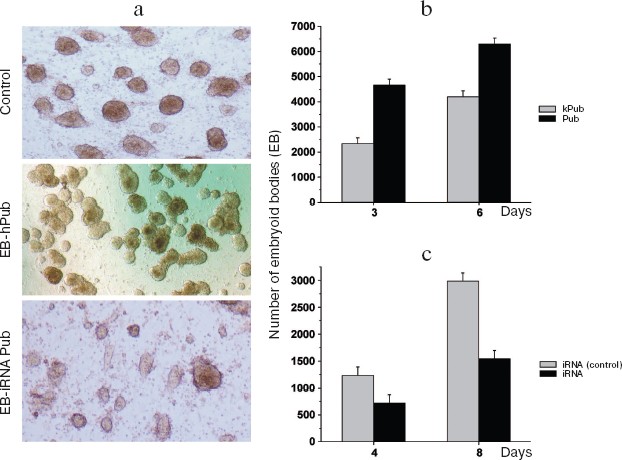
Genetic modification of ES cells influences the earliest stages of their differentiation. The effects of the pub gene, a specific inhibitor of the transcription factor PU.1 (involved in regulation of hematopoiesis), was investigated on proliferation and initial stages of differentiation of ES cells in vitro. Two approaches were employed: increased expression of human pub gene (hpub) and suppression of endogenous mouse pub expression by means of RNA interference [116, 117]. Genetically modified polyclonal ES cells transfected by plasmids expressing hpub or with plasmids generating small interference RNA (siRNA) for this gene under control of cytomegalovirus promoter did not influence proliferative activity compared with control cells. Suppression of endogenous pub expression in ES cells by means of siRNA resulted in a twofold decrease in formation of embryoid cells, whereas additional expression of exogenous hpub resulted in a twofold increase in formation of embryoid bodies compared with control. Figure 11 (see color insert) shows results of these experiments.
These results suggest that the pub gene is involved into very early stages of differentiation of ES cells resulting in formation of embryoid bodies.Fig. 11. Effect of increased hpub gene expression and decreased pub gene expression on formation of embryoid bodies of mouse ES cells. a) Photo of embryoid bodies in a culture 3-4 days after cell seeding (×200). Control: ES-DNA3 cell line. Cells were transfected by control vector without the hPub gene. A similar pattern was observed in the case of the control ES-Ineo cell line. EB-hPub: ES-Pub cell line. Cells were transfected by the vector carrying hPub. EB-iRNA Pub: ES-iRNA cell line. Cells were transfected by the vector carrying a sequences generating interfering RNA for the Pub gene. b) Histogram of embryoid bodies in transfected ES cell cultures after cultivation for 3 and 6 days. Grey columns, kPub (control ES-DNA3 cell line); black columns, Pub (ES-Pub cell line). c) Histogram of embryoid bodies in transfected ES cells cultures after cultivation for 4 and 8 days. Grey columns, iRNA (control) (control ES-Ineo cell line); black columns, iRNA (ES-iRNA cell line). Ordinate, number of embryoid cells; abscissa, cultivation time (days); *p < 0.05.
Subsequent estimation of the effect of increased and decreased expression of this gene on in vitro spontaneous differentiation of transfected cells into cardiomyocytes and neurons is shown in Figs. 12 (see color insert) and 13, respectively.
Fig. 12. Effect of increased hpub gene expression and decreased pub gene expression on differentiation of mouse ES cells into cardiomyocytes. The table shows statistical data on the effect of increased (ES-hPub line) and decreased (ES-iRNA line) expression of formation of clusters of contracting cardiomyocytes. Controls were ES-Ineo and ES-DNA3 cell lines. Immunocytochemical staining of cardiomyocytes with antibodies to troponin I is shown on the microphotograph on the right (×200).
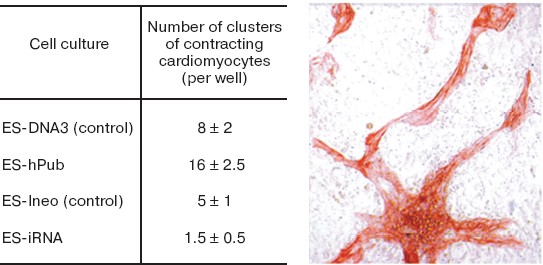
The data of Fig. 12 suggest that hpub overexpression caused a 2-fold increase in formation of clusters of contracting cardiomyocytes in cell cultures, whereas the decrease in pub gene expression caused a 3-4-fold decrease in formation of such clusters in corresponding cell cultures. Interestingly, under conditions of neuronal differentiation the increase and the decrease of expression of these genes caused completely different results (Fig. 13).Fig. 13. Effect of increased hpub gene expression (a) and decreased pub gene expression (b) on neuronal differentiation of mouse ES cells in vitro. Histogram of neuron number in the transfected ES cell cultures on the 20th day after seeding. a) Ordinate shows number of neurons per well (in percent) (n = 10). Gray column, ES-DNA3 cell line (control); black column, ES-hPub cell line. *p < 0.05. b) Ordinate shows number of neurons per well (in percent) (n = 10). Gray column, ES-Ineo cell line; black column, ES-iRNA cell line. *p < 0.05.
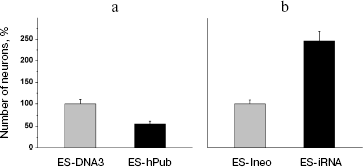
Overexpression of hpub caused a 2-fold decrease in neuron number in cultures 20 days after cell seeding (Fig. 13a). At the same time, the decrease in pub gene expression by means of RNA interference caused a 2.5-fold increase in neuron number (Fig. 13b). Thus, change of pub gene expression in ES cells cause oppositely directed effect on the differentiation of these cells into cardiomyocytes (mesoderm derivatives) and neurons (ectoderm derivatives).
Small interfering RNAs (siRNAs) are widely used for studies of gene functioning in ES cells during their differentiation into various cell types [118-120]. For example, transfection of mouse ES cells with siRNA for genes Oct-4 and Sox-2 resulted in significant decrease of their neuronal differentiation [121]. In control ES cells the number of colonies detected by means of antibodies to neuronal specific β-III-tubulin was about 90%, whereas suppression of Sox-2 and Oct-4 gene expression decreased this value to 65 and 45%, respectively. Using models of ES cells and human embryonic carcinoma cells, it was demonstrated that suppression of Oct-4 and Sox-2 gene expression by means of RNA interference resulted in induction of mesodermal differentiation [122].
Maintenance of undifferentiated state of these two cell types ultimately required normal functioning of the FGF-FGFR (fibroblast growth factor and its receptor) system.
At the same time, suppression of Oct-3/4 in mouse ES cells by means of siRNA or antisense sequences resulted in inhibition of their mesodermal and (subsequent) cardiomyocyte differentiation [123].
Suppression of L3/Lhx8 gene expression in mouse ES cells revealed its specific role in processes of neuronal differentiation [124]. Transfection of ES cells with a vector carrying sequences generating L3/Lhx8 siRNA under control of H1.2 promoter significantly influenced their directed differentiation into nervous system cells induced by retinoic acid; there was basically total inhibition of cholinergic neuron formation (detected by choline acetyltransferase staining). However, no significant changes in the number of GABA-ergic neurons were found in these cultures. Induction of L3/Lhx8 gene expression normalized formation of cholinergic neurons, and the authors concluded that this gene plays a key role in differentiation of ES cells into cholinergic neurons and that it is involved in the development of forebrain basal nuclei.
Opposite effects on gene expression (consisting in suppression or stimulation of gene expression) may demonstrate involvement of their products in regulation of cell proliferation, viability, and differentiation. For example, stimulation of expression of well-known antiapoptotic gene Bcl2 in mouse ES cells caused increased formation of hematopoietic embryoid cells with their subsequent differentiation into colonies during their seeding on methylcellulose [125]. At the same time, ES cells transfected with a plasmid carrying sequences of siRNAs for this genes were characterized by decreased ability for differentiation into hematopoietic lineages.
During embryogenesis, mammalian embryos are developed (especially at early stages) under oxygen deficit. It was demonstrated that hypoxia induced factor (HIF-1) can inhibit signal transduction through the LIF-STAT3 system of mouse ES cells; this effect is associated with direct inhibition of LIFR expression via binding to a regulatory element present in the promoter of this gene [126]. HIF-1 overexpression and suppression of its expression by means of RNA interference confirmed its effect on LIFR expression. Thus, HIF-1 can stimulate differentiation of ES cells by inhibiting LIF-STAT3 signaling responsible for maintenance of pluripotency of ES cells.
There are developments showing genetic vectors carrying RNA interference sequences and their expression can be regulated by various factors. For example, Wang et al. used tetracycline-regulated plasmid generating siRNA, which stably integrated into mouse ES cells [127]. Using this model, they demonstrated inducible and reversible repression of the Npm1 gene in these cells by specific siRNA (this resulted in suppression of ES cell proliferation).
The possibility of suppression of expression of certain genes (especially key genes for certain types of cell differentiation) opens perspectives for preparation of large homogenous cell populations with certain type of cell differentiation. This is ultimately important for their use in cell therapy.
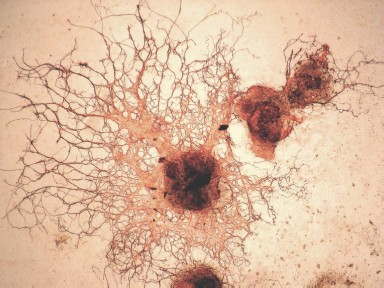
Differentiation of ES cells towards neuronal lineages can be induced using genes encoding neurotrophins, trophic factors influencing proliferation, cell differentiation into neurons of certain ergicity, and their viability [34]. Figure 14 (see color insert) shows results on induction of neuronal differentiation in ES cells by means of overexpression of the ngf gene encoding nerve growth factor. Mouse ES cells were transfected with a plasmid carrying the ngf gene fused with GFP gene under control of the cytomegalovirus promoter.
One can easily see long neurite outgrowths of forming neurons and associates of these cells morphologically similar to nerve nodes. Cells were immunocytochemically detected after 24 days of their cultivation in vitro. No such structures were found in the control.Fig. 14. Immunocytochemical staining of NGF-GFP ES cells with antibodies to β-III-tubulin (cultivation for 24 days) (×200).
Thus, different approaches to genetic modification of ES cells, which include both suppression of expression of some genes and also their overexpression, are perspective directions in studies of differentiation of these cells.
However, genetically modified ES cells with certain direction of cell differentiation have limited application in cell therapy of severe human diseases due to poor knowledge of delayed effects of such modifications on the recipient's body, particularly risk of development of malignant tumors.
Preparation of ES cells and the development of methods for manipulations with them represent one of the most remarkable and important scientific achievements at the beginning of the third millennium. Since mammalian ES cells are basically unlimited sources of undifferentiated and nontransformed cells with normal diploid karyotype, they will remain as major objects for studies on pathways of embryonic development, formation, and functioning of particular tissues under normal and pathological conditions. In addition, human ES cells (including genetically modified ones) will be employed in cell therapy of severe human diseases.
However, results of recent studies open principally new possibilities in cells therapy. It is possible to reprogram human somatic cells into pluripotent stem cells for their subsequent differentiation into various cell types and their subsequent transplantation into patients suffering from various incurable diseases.
In 2006, Japanese scientists Takahashi and Yamanaka [128] reprogrammed mouse adult and embryonic fibroblasts into pluripotent stem cells. They defined these cells as induced pluripotent stem (iPS) cells. Initially they selected 24 candidate genes that would be involved in induction of pluripotency in somatic cells and maintenance for such pluripotency in ES cells. Fibroblasts were reprogrammed in vitro by transfection with retroviral vectors carrying genes encoding four transcription factors, Oct3/4, Sox2, c-Myc, and Klf4. Resultant iPS cells exhibited morphological and growth properties similar to those of ES cells; they also expressed specific markers typical for ES cells. Transplantation of such cells into immunodeficient mice resulted in formation of tumors containing cells of various tissues derived from three germinal layers (ectoderm, mesoderm, and endoderm). However, administration of iPS into mouse blastocyst did not reveal chimeric animals among offspring (27 animals). Thus, these experiments demonstrated the possibility of in vitro preparation of pluripotent mammalian cells by their treatment with a limited number of particular regulatory factors. Later, in 2007 this group obtained chimeric animals using iPS cells [129, 130].
In the very beginning of 2008, Aoi et al. [131] obtained clones of iPS cells from liver and gastric mucosa of adult mice. In late 2007 and the beginning of 2008, Takahashi et al. [132] and Nakagawa et al. [133] (from the same laboratory of Kyoto University) reported about successful dedifferentiation of human adult fibroblasts and preparation of human iPS cells using the same factors (Oct3/4, Sox2, c-Myc, and Klf4). Similar results (successful preparation of human iPS cells from fibroblasts using the same factors) were also reported by an American group [134]. It should be noted that resultant iPS cells exhibited similarity with human ES cells by the following signs: morphology, proliferating activity, expression of surface antigens, global gene expression, epigenetic status of genes responsible for pluripotency, telomerase activity, and (this is especially important) by normal diploid set of chromosomes. In addition, this cell line could differentiate into various cell types of all three germinal layers both in vitro, and in teratomas.
However, Nakagawa et al. [133] significantly improved the method for iPS cell preparation and excluded retroviral vector containing Myc gene from the experimental protocol. This approach significantly reduced the risk for tumor development in chimeric animals.
Yu et al. [135] obtained iPS cells from human fibroblasts by means of a rather different combination of factors. They used the following set of factors: Oct4, Sox2, NANOG, and LIN28. Absence of Oct4 or Sox2 completely prevented formation of iPS cell colonies, whereas lack of NANOG or LIN28 decreased their number by more than an order of magnitude. Finally, resultant clones of iPS cells also had normal diploid set of chromosomes and they insignificantly differed from human ES cells by morphology, proliferative and telomerase activity, expression of surface antigens, as well as by ability for differentiation into derivatives of the three germinal layers.
Figure 15 (see color insert) illustrates possibilities of preparation of iPS cells and their subsequent use for cell therapy of human diseases. It should be noted that not only skin fibroblasts but also other proliferating cells (available in reasonable quantities) might undergo dedifferentiation. The possibility of manipulations with cells from patients with inherited diseases is a very important aspect of this problem: dedifferentiation of these cells into iPS cells and correction of genetic defects in such cells by means of homologous recombination and subsequent transplantation into the same patients would be a powerful tool for treatment of these diseases.
However, this requires additional intensive studies for minimization of potential mutagenic effects during use of retroviral constructs in the processes of somatic cell dedifferentiation.Fig. 15. Preparation of inducible pluripotent stem cells from human somatic cells for subsequent transplantation to a patient.
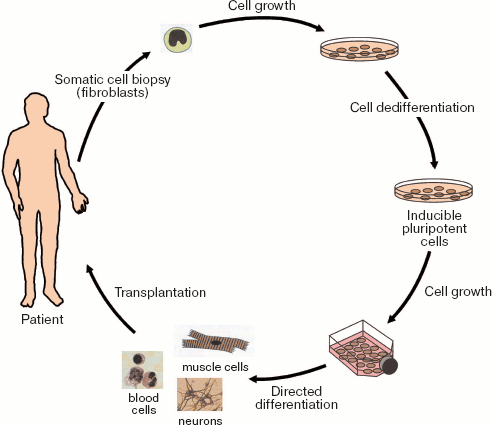
Efficiency of applicability of various approaches such as ES cells, iPS cells, and cells derived after nuclear transfer for cell therapy still remains unclear. Further studies are needed for elucidation of advantages of one of the above considered approaches.
The author is grateful to E. S. Manuilova, E. L. Arsen'eva, and E. V. Novosadova (Laboratory of Molecular Genetics of Somatic Cells, Institute of Molecular Genetics, Russian Academy of Sciences) for presentation of results and their help during preparation of this review.
This work was supported by a grant of the Program of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology” and by a grant from the Ministry of Education and Science of the Russian Federation (No. 02.512.12.2013).
REFERENCES
1.Evans, M. J., and Kaufman, M. H. (1981)
Nature, 292, 154-156.
2.Martin, G. R. (1981) Proc. Natl. Acad. Sci.
USA, 78, 7634-7638.
3.Bradley, A., Evans, M., Kaufman, M. H., and
Robertson, E. (1984) Nature, 309, 255-256.
4.Thomas, K. R., and Capecchi, M. R. (1987)
Cell, 51, 503-512.
5.Doetschman, T. C., Eistetter, H., Katz, M.,
Schmidt, W., and Kemler, R. J. (1985) Embryol. Exp. Morphol.,
87, 27-45.
6.Keller, G. (2005) Genes Dev., 19,
1129-1155.
7.Geijsen, N., Horoschak, M., Kim, K., Gribnau, J.,
Eggan, K., and Daley, G. Q. (2004) Nature, 427,
106-107.
8.Hubner, K., Fuhrmann, G., Christenson, L. K.,
Kehler, J., Reinbold, R., de la Fuente, R., Wood, J., Strauss, J. F.,
Boiani, M., and Scholer, H. R. (2003) Science, 300,
1251-1256.
9.Nayernia, K., Notle, J., Michelmann, H. W., Lee, J.
H., Rathsack, K., Drusenheimer, N., Dev, A., Wulf, G., Ehrmann, I. F.,
Elliot, D. J., Okpanyi, V., Zechner, U., Haaf, T., Meinhardt, A., and
Engel, W. (2006) Dev. Cell, 11, 125-132.
10.Martin, G. R., and Evans, M. J. (1975) Proc.
Natl. Acad. Sci. USA, 72, 1441-1445.
11.Resnick, J. L., Bixler, L. S., Cheng, L., and
Donovan, P. J. (1992) Nature, 359, 550-551.
12.Stewart, C. L., Gadi, I., and Bhatt, H. (1994)
Dev. Biol., 161, 626-628.
13.Joyner, A. L. (1993) IRL PRESS at Oxford
University Press, p. 109.
14.Robertson, E., Bradley, A., Kuehn, M., and Evans,
M. (1986) Nature, 323, 445-448.
15.Mitalipov, Sh. M., Mitalipova, M. M., and Ivanov,
V. I. (1994) Ontogenez, 25, 19-27.
16.Savatier, P., Lapillone, H., van Grunsven, L. A.,
Rudkin, B. B., and Samarut, J. (1996) Oncogene, 12,
309-322.
17.Rathjen, P., Nichols, J., Toth, S., Edwards, D.
R., Heath, J. K., and Smith, A. G. (1990) Genes Dev., 4,
2308-2318.
18.Zhang, J. G., Owerzarek, C. M., Ward, L. D.,
Howlett, G. J., Fanri, L. J., Roberts, B. A., and Nicola, N. A. (1997)
Biochem. J., 325, 693-700.
19.Burdon, T., Chambers, I., Stracey, C., Niwa, H.,
and Smith, A. (1999) Cells Tissues Organs, 165,
131-143.
20.Niwa, H., Burdon, T., Chambers, I., and Smith, A.
(1998) Genes Dev., 12, 2048-2060.
21.Ying, Q. L., Nichols, J., Chambers, I., and
Smith, A. (2003) Cell, 115, 281-292.
22.Burdon, T., Smith, A., and Savatier, P. (2002)
Trends Cell Biol., 12, 432-438.
23.Sato, N., Meijer, L., Skaltsounis, L., Greengard,
P., and Brivanlou, A. H. (2004) Nat. Med., 10, 55-63.
24.Solter, D., and Knowles, B. B. (1978) Proc.
Natl. Acad. Sci. USA, 75, 5565-5569.
25.Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S.
S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., and Jones, J. M.
(1998) Science, 282, 1145-1147.
26.Armstrong, L., Lako, M., Lincoln, J., Cairns, P.
M., and Hole, N. (2000) Mech. Dev., 97, 109-116.
27.Astashkin, E. I., Manuilova, E. S., Arsen'eva, E.
L., Grivennikov, I. A., Tarantul, V. Z., and Grachev, S. V. (2005)
Dokl. Ros. Akad. Nauk, 401, 380-382.
28.Niwa, H., Miyazaki, J., and Smith, A. G.
(2000) Nat. Genet., 24, 372-376.
29.Gordeeva, O. F., Manuilova, E. S., Grivennikov,
I. A., Gulyaev, D. V., Smirnova, Yu. A., Zinov'eva, R. D., and
Khruschov, N. G. (2002) Dokl. Ros. Akad. Nauk, 386,
555-558.
30.Gordeeva, O. F., Manuilova, E. S., Grivennikov,
I. A., Smirnova, Yu. A., Krasnikova, N. Yu., Zinov'eva, R. D., and
Khruschov, N. G. (2003) Ontogenez, 34, 174-182.
31.Mitsui, K., Tokuzawa, Y., Itoh, H., Segawa, K.,
Murakami, M., Takahashi, K., Maruyama, M., Maeda, M., and Yamanaka, S.
(2003) Cell, 113, 631-642.
32.Nichols, J., Zevnik, B., Anastassiadis, K., Niwa,
H., Klewe-Nebenius, D., Chambers, I., Scholer, H., and Smith, A. (1998)
Cell, 95, 379-391.
33.Chambers, I., Colby, D., Robertson, M., Nichols,
J., Lee, S., Tweedie, S., and Smith, A. (2003) Cell, 113,
643-655.
34.Pyle, A. D., Lock, L. F., and Donovan, P. J.
(2006) Nat. Biotechnol., 24, 344-350.
35.Wei, H., Juharsz, O., Li, J., Tarasova, Y. S.,
and Boheler, K. R. (2005) J. Cell. Mol. Med., 9,
804-817.
36.Reubinoff, B. E., Pera, M. F., Fong, C. Y.,
Trounson, A., and Bongso, A. (2000) Nat. Biotechnol., 18,
399-404.
37.Krylova, T. A., Zenin, V. V., Musorina, N. S.,
Baranov, V. S., Bichevaya, N. K., Korsak, B. S., Nikolskii, N. N.,
Pinaev, G. P., and Polyanskaya, G. G. (2003) Tsitologiya,
45, 1172-1178.
38.Pryzhakova, M. V., Lagar'kova, M. V., Lyakisheva,
A. V., Revazova, E. S., Gnuchev, N. V., and Kiselev, S. L. (2004)
Inform. Byul. Klet. Kultury, 19, 22-25.
39.Mitalipova, M., Calhoun, J., Shin, S., Wininger,
D., Schulz, T., Nogglee, S., Venabler, A., Lyons, I., Robins, A., and
Stice, S. (2003) Stem Cells, 21, 521-526.
40.Amit, M., Carpenter, M. K., Inokuma, M. S., Chiu,
C. P., Harris, C. P., Waknitz, M. A., Itskovitz-Eldor, J., and Thomson,
J. A. (2000) Dev. Biol., 227, 271-278.
41.Odorico, J. S., Kaufman, D. S., and Thomson, J.
A. (2001) Stem Cells, 19, 193-204.
42.Cowan, C. A., Klimanskaya, I., McMahon, J.,
Atienza, J., Witmyer, J., Zucker, J. P., Wang, S., Morton, C. C.,
McMahon, A. P., Powers, D., and Melton, D. A. (2004) N. Engl. J.
Med., 350, 1353-1356.
43.Daheron, L., Opitz, S. L., Zaehres, H., Lensch,
W. M., Andrews, P. W., Itskovitz-Eldor, J., and Daley, G. Q. (2004)
Stem Cells, 22, 770-778.
44.Xu, R. H., Chen, X., Li, D. S., Addicks, G. C.,
Glennon, C., Zwaka, T. P., and Thomson, J. A. (2002) Nat.
Biotechnol., 20, 1261-1264.
45.Hay, D. C., Surtherland, L., Clark, J., and
Burdon, T. (2004) Stem Cells, 22, 225-235.
46.Zaehres, H., Lensch, M. W., Daneron, L., Stewart,
S., Itskovitz-Eldor, J., and Daley, G. Q. (2005) Stem Cells,
23, 299-305.
47.Amit, M., Shariki, C., Margulets, V., and
Itskovitz-Eldor, J. (2004) Biol. Reprod., 70,
837-845.
48.Chang, I. K., Jeong, D. K., Hong, Y. H., Park, T.
S., Moon, Y. K., Ohno, T., and Han, J. Y. (1997) Cell Biol.
Int., 21, 495-499.
49.Doetschman, T., Williams, P., and Maeda, N.
(1998) Dev. Biol., 127, 224-227.
50.Schoonjans, L., Albright, G. M., Li, J. L.,
Collen, D., and Moreadith, R. W. (1996) Mol. Reprod.
Dev., 45, 439-443.
51.Brenin, D., Look, J., Bader, M., Hubner, N.,
Levan, G., and Iannaccone, P. (1997) Transplant. Proc.,
29, 1761-1765.
52.Iannaccone, P. M., Taborn, G. U., Garton, R. L.,
Caplice, M. D., and Brenin, D. R. (1994) Dev. Biol., 163,
288-292.
53.Sukoyan, M. A., Golubitsa, A. N., Zhelezova, A.
I., Shilov, A. G., Vatolin, S. Y., Maximovky, L. P., Andreeva, L. E.,
McWhire, J., Pack, S. D., Bayborodin, S. I., Kerkis, A. Y., Kizilova,
H. I., and Serov, O. L. (1992) Mol. Reprod. Dev., 33,
418-431.
54.Wheeler, M. B. (1994) Reprod. Fertil.
Dev., 6, 563-568.
55.Notarianni, E., Galli, C., Laurie, S., Moor,
R. M., and Evans, M. J. (1991) J. Reprod. Fertil. Suppl.,
43, 255-260.
56.First, N. L., Sims, M. M., Park, S. P., and
Kent-First, M. J. (1994) Reprod. Fertil. Dev., 6,
553-562.
57.Stice, S. L., Strelchenko, N. S., Keefer, C. L.,
and Matthews, L. (1996) Biol. Reprod., 54, 100-110.
58.Mitalipova, M., Beyhan, Z., and First, N. L.
(2001) Cloning, 3, 59-67.
59.Pau, K. Y., and Wolf, D. P. (2004) Reprod.
Biol. Endocrinol., 2, 41.
60.Thomson, J. A., Kalishman, J., Golos, T. G.,
Durning, M., Harris, C. P., Becke, R. A., and Hearn, J. P. (1995)
Proc. Natl. Acad. Sci. USA, 92, 7844-7848.
61.Mitalipov, S., Kuo, H. C., Byrne, J., Clepper,
L., Meisner, L., Johnson, J., Zeier, R., and Wolf, D. (2006) Stem
Cells, 24, 2177-2186.
62.Thomson, J. A., Kalishman, J., Golos, T. G.,
Durning, M., Harris, C. P., and Hearn, J. P. (1996) Biol.
Reprod., 55, 254-259.
63.Suemori, H., Tada, T., Torii, R., Hosoi, Y.,
Kobayashi, K., Imahie, H., Kondo, Y., Iritani, A., and Nakatsuji, N.
(2001) Dev. Dyn., 222, 273-279.
64.Vrana, K. E., Hipp, J. D., Goss, A. M., McCool,
B. A., Riddle, D. R., Walker, S. J., Wettstein, P. J., Studer, L. P.,
Tabar, V., Cunniff, K., Chapman, K., Vilner, L., West, M. D., Grant, K.
A., and Cibelli, J. B. (2003) Proc. Natl. Acad. Sci. USA,
100, 11911-11916.
65.Hernandez, L., Kozlov, S., Piras, G., and
Stewart, C. L. (2003) Proc. Natl. Acad. Sci. USA, 100,
13344-13349.
66.Pavlova, G. V., Manuilova, E. S., Arsen'eva, E.
L., Grivennikov, I. A., Murkin, E. V., Kanaikina, N. V., Revischin, A.
V., Tarantul, V. Z., and Korochkin, L. I. (2005) Klet. Tekhnol.
Biol. Med., 2, 99-102.
67.Schmitt, T. M., de Pooter, R. F., Gronski, M. A.,
Cho, S. K., Ohashi, P. S., and Zuniga-Pflucker, J. C. (2004) Nat.
Immunol., 5, 410-417.
68.Rohwedel, J., Maltsev, V., Bober, E., Arnold, H.
H., Hescheler, J., and Wobus, A. M. (1994) Dev. Biol.,
164, 87-101.
69.Bagutti, C., Wobus, A. M., Fassler, R., and Watt,
F. M. (1996) Dev. Biol., 179, 184-196.
70.Maltsev, V. A., Wobus, A. M., Rohwedel, J.,
Bader, M., and Hescheler, J. (1994) Circ. Res., 75,
233-244.
71.Wobus, A. M., Kaomei, G., Shan, J., Wellner, M.
C., Rohwedel, J., Ji, G., Fleischmann, B., Katus, H. A., Hescheler, J.,
and Franz, W. M. (1997) J. Mol. Cell. Cardiol., 29,
1525-1539.
72.Ventura, C., and Maioli, M. (2000) Circ.
Res., 87, 189-194.
73.Xu, C., Police, S., Rao, N., and Carpenter, M. K.
(2002) Circ. Res., 91, 501-508.
74.Takahashi, T., Lord, B., Schulze, P. C., Fryer,
R. M., Sarang, S. S., Gullans, S. R., and Lee, R. T. (2003)
Circulation, 107, 1912-1916.
75.Kanno, S., Kim, P. K., Sallam, K., Lei, J.,
Billiar, T. R., and Shears, L. L. (2004) Proc. Natl. Acad. Sci.
USA, 101, 12277-12281.
76.E, L. L., Zhao, Y. S., Guo, X. M., Wang, C. Y.,
Jiang, H., Li, J., Duan, C. M., and Song, Y. (2006) J. Heart Lung
Transplant., 25, 664-674.
77.Coraux, C., Hilmi, C., Rouleau, M., Spadafora,
A., Hinnrasky, J., Ortonne, J. P., Dani, C., and Aberdam, D. (2003)
Curr. Biol., 13, 849-853.
78.Kawasaki, H., Mizuseki, K., Nishikawa, S.,
Kaneko, S., Kuwana, Y., Nakanishi, S., Nishikawa, S. I., and Sasai, Y.
(2000) Neuron, 28, 31-40.
79.Gottlieb, D. I., and Huettner, J. E. (1999)
Cells Tissues Organs, 165, 165-172.
80.Ying, Q. L., and Smith, A. G. (2003) Meth.
Enzymol., 365, 327-341.
81.Levenberg, S., Burdick, J. A., Kraehenbuehl, T.,
and Langer, R. (2005) Tissue Eng., 11, 506-512.
82.Gerrard, L., Rodgers, L., and Cui, W. (2005)
Stem Cells, 23, 1234-1241.
83.Kitagawa, A., Nakayama, T., Takenaga, M.,
Matsumoto, K., Tokura, Y., Ohta, Y., Ichinohe, M., Yamaguchi, Y.,
Suzuki, N., Okano, H., and Igarashi, R. (2005) Biochem. Biophys.
Res. Commun., 328, 1051-1057.
84.Li, Z., Theus, M. H., and Wei, L. (2006) Dev.
Growth Differ., 48, 513-523.
85.Amura, C. R., Marek, L., Winn, R. A., and
Heasley, L. E. (2005) Mol. Cell. Biol., 25,
10791-10802.
86.Kitazawa, A., and Shimizu, N. (2005) J.
Biosci. Bioeng., 100, 94-99.
87.Cazillis, M., Gonzalez, B. J., Billardon, C.,
Lombet, A., Fraichard, A., Samarut, J., Gressens, P., Vaudry, H., and
Rostene, W. (2004) Eur. J. Neurosci., 19, 798-808.
88.Kim, E., Clark, A. L., Kiss, A., Hahn, J. W.,
Wesselschmidt, R., Coscia, C. J., and Belcheva, M. M. (2006) J.
Biol. Chem., 281, 33749-33760.
89.Keller, G. (1995) Curr. Opin. Cell Biol.,
7, 862-869.
90.Kabrun, N., Buhring, H. J., Choi, K., Ullrich,
A., Risau, W., and Keller, G. (1997) Development, 124,
2039-2048.
91.Moore, M. A., Shieh, J. H., and Lee, G. (2006)
Meth. Enzymol., 418, 208-242.
92.Palis, J., Roberston, S., Kennedy, M., Wall, C.,
and Keller, G. (1999) Development, 126, 5073-5084.
93.Robertson, S. M., Kennedy, M., Shannon, J. M.,
and Keller, G. (2000) Development, 127, 2447-2459.
94.Nakano, T., Kodama, H., and Honjo, T. (1994)
Science, 265, 1098-1101.
95.Cho, S. K., Webber, T. D., Carlyle, J. R.,
Nakano, T., Lewis, S. M., and Zuniga-Pflucker, J. C. (1999) Proc.
Natl. Acad. Sci. USA, 96, 9797-9802.
96.Burt, R. K., Verda, L., Kim, D. A., Oyama, Y.,
Luo, K., and Link, C. (2004) J. Exp. Med., 199,
895-904.
97.Kyba, M., Perlingeiro, R. C., and Daley, G. Q.
(2002) Cell, 109, 29-37.
98.Assady, S., Maor, G., Amit, M., Itskovitz-Eldor,
J., Skorecki, K. L., and Tzukerman, M. (2001) Diabetes,
50, 1691-1697.
99.Lumelsky, N., Blondel, O., and Laeng, P. (2001)
Science, 242, 1389-1394.
100.Shi, Y., Hou, L., Tang, F., Jiang, W., Wang,
P., Ding, M., and Deng, H. (2005) Stem Cells, 23,
656-662.
101.Kuai, X. L., Cong, X. Q., Li, X. L., and Xiao,
S. D. (2003) Liver Transpl., 9, 1094-1099.
102.Hirata, S., Senju, S., Matsuyoshi, H., Fukuma,
D., Uemura, Y., and Nishimura, Y. (2005) J. Immunol.,
174, 1888-1897.
103.Kim, D. W., Chung, S., Hwang, M., Ferree, A.,
Tsai, H. C., Park, J. J., Chung, S., Nam, T. S., Kang, U. J., Isacson,
O., and Kim, K. S. (2006) Stem Cells, 24, 557-567.
104.Shim, J. W., Koh, H. C., Chang, M. Y., Roh, E.,
Choi, C. Y., Oh, Y. J., Son, H., Lee, Y. S., Studer, L., and Lee, S. H.
(2004) J. Neurosci., 24, 843-852.
105.Wei, L., Cui, L., Snider, B. J., Rivkin, M.,
Yu, S. S., Lee, C. S., Adams, L. D., Gottlieb, D. I., Johnson, E. M.,
Jr., Yu, S. P., and Choi, D. W. (2005) Neurobiol. Dis.,
19, 183-193.
106.Kamochi, H., Kurokawa, M. S., Yoshikawa, H.,
Ueda, Y., Masuda, C., Takada, E., Watanabe, K., Sakakibara, M., Natuki,
Y., Kimura, K., Beppu, M., Aoki, H., and Suzuki, N. (2006)
Transplantation, 82, 516-526.
107.Shiroi, A., Ueda, S., Ouji, Y., Saito, K.,
Moriya, K., Sugie, Y., Fukui, H., Ishizaka, S., and Yoshikawa, M.
(2005) World J. Gastroenterol., 11, 4161-4166.
108.Tabata, Y., Ouchi, Y., Kamiya, H., Manabe, T.,
Arai, K., and Watanabe, S. (2004) Mol. Cell. Biol., 24,
4513-4521.
109.Guo, Y., Hangoc, G., Bian, H., Pelus, L. M.,
and Broxmeyer, H. E. (2005) Stem Cells, 23,
1324-1332.
110.Klein, D., Schmandt, T., Muth-Kohne, E.,
Perez-Bouza, A., Segschneider, M., Gieselmann, V., and Brüstle, O.
(2006) Gene Ther., 13, 1686-1695.
111.Wu, L. C., Sun, C. W., Ryan, T. M., Pawlik, K.
M., Ren, J., and Townes, T. M. (2006) Blood, 108,
1183-1188.
112.Hanna, J., Wernig, M., Markoulaki, S., Sun, C.
W., Meissner, A., Cassady, J. P., Beard, C., Brambrink, T., Wu, L. C.,
Townes, T. M., and Jaenisch, R. (2007) Science, 318,
1920-1923.
113.Manuilova, E. S., Arsen'eva, E. L., Khaidarova,
N. V., Shugurova, I. M., Gornostaeva, S. N., Inozemtseva, L. S.,
Katrukha, A. M., Grivennikov, I. A., and Tarantul, V. Z. (2003)
Ontogenez, 34, 224-230.
114.Shugurova, I., Bobrisheva, I., Surkova, I.,
Grivennikov, I., and Tarantul, V. (2002) Cell. Prolif.,
35, 237-245.
115.Manuilova, E. S., Arsenyeva, E. L., Khaidarova,
N. V., Shugurova, I. M., Inozemtseva, L. S., Tarantul, V. Z., and
Grivennikov, I. A. (2008) Int. J. Biomed. Sci., 4.
116.Novosadova, E. V., Manuilova, E. S., Arsen'eva
E. L., Khaidarova, N. V., Dolotov, O. V., Inozemtseva, L. S.,
Sitnikova, O. V., Tarantul, V. Z., and Grivennikov, I. A. (2005)
Vestnik Biotekhnol. Fiz.-Khim. Biol., 2, 153-158.
117.Novosadova, E. V., Manuilova, E. S., Arsen'eva,
E. L., Khaidarova, N. V., Dolotov, O. V., Inozemtseva, L. S.,
Kozachenkov, K., Grivennikov, I. A., and Tarantul, V. Z. (2005)
Klet. Tekhnol. Biol. Med., 3, 174-179.
118.Liu, Y. P., Dambaeva, S. V., Dovzhenko, O. V.,
Garthwaite, M. A., and Golos, T. G. (2005) Stem. Cells Dev.,
14, 487-492.
119.Zou, G. M., and Yoder, M. C. (2005) Biol.
Cell., 97, 211-219.
120.Velkey, J. M., Slawny, N. A., Gratsch, T. E.,
and O'Shea, K. S. (2006) Meth. Mol. Biol., 329,
233-261.
121.Chen, S., Choo, A. B., Nai-Dy, W., Heng-Phon,
T., and Oh, S. K. (2007) Stem Cells Dev., 16,
413-420.
122.Greber, B., Lehrach, H., and Adjaye, J. (2007)
BMC Dev. Biol., 7, 46.
123.Zeineddine, D., Papadimou, E., Chebli, K.,
Gineste, M., Liu, J., Grey, C., Thurig, S., Behfar, A., Wallace, V. A.,
Skerjanc, I. S., and Puceat, M. (2006) Dev. Cell, 11,
535-546.
124.Manabe, T., Tatsumi, K., Inoue, M., Makinodan,
M., Yamauchi, T., Makinodan, E., Yokoyama, S., Sakumura, R., and
Wanaka, A. (2007) Cell Death Differ., 14, 1080-1085.
125.Wang, Y. Y., Deng, X., Xu, L., Gao, F., Flagg,
T., and May, W. S. (2007) Exp. Hematol., available online 26
November 2007.
126.Jeong, C. H., Lee, H. J., Cha, J. H., Kim, J.
H., Kim, K. R., Kim, J. H., Yoon, D. K., and Kim, K. W. (2007) J.
Biol. Chem., 282, 13672-13679.
127.Wang, B. B., Lu, R., Wang, W. C., and Jin, Y.
(2006) Biochem. Biophys. Res. Commun., 347,
1129-1137.
128.Takahashi, K., and Yamanaka, S. (2006)
Cell, 126, 663-676.
129.Okita, K., Ichisaka, T., and Yamanaka, S.
(2007) Nature, 448, 313-317.
130.Yamanaka, S. (2008) Cell Prolif.,
41, Suppl. 1, 51-56.
131.Aoi, T., Yae, K., Nakagawa, M., Ichisaka, T.,
Okita, K., Takahashi, K., Chiba, T., and Yamanaka, S. (2008)
Science, Feb 14.
132.Takahashi, K., Tanabe, K., Ohnuki, M., Narita,
M., Ichisaka, T., Tomoda, K., and Yamanaka, S. (2007) Cell,
131, 861-872.
133.Nakagawa, M., Koyanagi, M., Tanabe, K.,
Takahashi, K., Ichisaka, T., Aoi, T., Okita, K., Mochiduki, Y.,
Takizawa, N., and Yamanaka, S. (2008) Nat. Biotechnol.,
26, 101-106.
134.Park, I. H., Zhao, R., West, J. A., Yabuuchi,
A., Huo, H., Ince, T. A., Lerou, P. H., Lensch, M. W., and Daley, G. Q.
(2008) Nature, 451, 141-146.
135.Yu, J., Vodyanik, M. A., Smuga-Otto, K.,
Antosiewicz-Bourget, J., Frane, J. L., Tian, S., Nie, J., Jonsdottir,
G. A., Ruotti, V., Stewart, R., Slukvin, I. I., and Thomson, J. A.
(2007) Science, 318, 1917-1920.