REVIEW: Involvement of Thio-, Peroxi-, and Glutaredoxins in Cellular Redox-Dependent Processes
E. V. Kalinina1,2, N. N. Chernov3, and A. N. Saprin1*
1Center for Theoretical Problems of Physico-Chemical Pharmacology, Russian Academy of Sciences, ul. Kosygina 4, 119991 Moscow, Russia; fax: (495) 938-2533; E-mail: kevsan@orc.ru2Institute of Cytochemistry and Molecular Pharmacology, 6-ya Radial'naya ul. 24/14, 115404 Moscow, Russia
3Peoples' Friendship University of Russia, ul. Miklukho-Maklaya 8, 117198 Moscow, Russia
* To whom correspondence should be addressed.
Received November 7, 2007; Revision received July 24, 2008
Among the key antioxidant enzymes, thioredoxin and glutaredoxin systems play an important role in cell defense against oxidative stress and maintenance of redox homeostasis owing to the regulation of thiol-disulfide exchange. The thioredoxin isoforms Trx1 (cytoplasmic form) and Trx2 (mitochondrial form) can reduce inter- and intramolecular disulfide bonds in proteins, in particular, in oxidized peroxiredoxins, which disrupt organic hydroperoxides, H2O2, and peroxynitrite. NADPH-dependent thioredoxin reductase, which reduces a broad range of substrates including oxidized form of thioredoxin, can also directly reduce lipid hydroperoxides, H2O2, and dehydroascorbic and lipoic acids. Glutaredoxin, whose major isoforms in mammals are Grx1, Grx2, and Grx5, as well as thioredoxin, catalyzes S-glutathionylation and deglutathionylation of proteins to protect SH-groups from oxidation and restore functionally active thiols. However, in contrast to thioredoxin, glutaredoxin reduces GSH-mixed disulfides and catalyzes the reaction not only via a dithiol mechanism but also via monothiol reduction. In addition to the role in cellular antioxidant defense, all of the reviewed redox proteins (thioredoxin, thioredoxin reductase, peroxiredoxin, and glutaredoxin) have a number of significant functions required for cell viability: they regulate transcription factor activities, play the role of growth factors, serve as enzyme cofactors, take part in regulation of cell cycle, and are involved in antiapoptotic mechanisms.
KEY WORDS: oxidative stress, thioredoxin, thioredoxin reductase, peroxiredoxin, glutaredoxin, redox-dependent processesDOI: 10.1134/S0006297908130099
Abbreviations: AP-1, activator protein 1; ARE, antioxidant-responsive element; ASK-1, apoptosis signal-regulating kinase 1; Cdc2, cyclin-dependent kinase 2; ERK, extracellular signal regulated kinase; Grx, glutaredoxin; GSH and GSSG, glutathione reduced and oxidized, respectively; GST P1-1, glutathione transferase P1-1; IκB, inhibitor of κB; iNOS, inducible NO-synthase; JNK, c-Jun-N-terminal kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK/ERK kinase; Mn-SOD, Mn-dependent superoxide dismutase; NF-κB, nuclear factor κB; Nrf2, NF-E2-dependent factor 2; PDI, protein disulfide isomerase; PKC, protein kinase C; Prx, peroxiredoxin; ROS, reactive oxygen species; SAPK, stress-activated protein kinase; Sec, selenocysteine; SEK, SAPK/ERK kinase; Trx, thioredoxin; Trx1 and Trx2, cytoplasmic and mitochondrial Trx forms, respectively; TrxR, thioredoxin reductase.
Redox-dependent processes substantially influence the functional
activity of many proteins and participate in regulation of the most
important vital processes of the cell such as proliferation,
differentiation, and apoptosis. Considerable recent attention of
researchers has been focused on thiol-disulfide regulation, which is
realized by redox proteins, whose activities depend of a redox-active
site in the form of amino acid sequence containing one or two active
thiols. Among these proteins, two thiol-disulfide reductases stand out:
thioredoxin (Trx) and glutaredoxin (Grx), which are the members of
thioredoxin superfamily. These enzymes are multifunctional and comprise
thioredoxin- and glutaredoxin-dependent systems playing an important
role in maintenance of intracellular redox homeostasis. The first
system contains, apart from thioredoxin, a NADPH-dependent thioredoxin
reductase (TrxR), which reduces the oxidized form of thioredoxin. The
second system includes glutathione (GSH) as an agent reducing the
oxidized glutaredoxin and glutathione reductase reducing glutathione
from its oxidized form (GSSG).
Both of these systems contribute to the antioxidant defense of cells from destructive influence of oxidative stress, which causes formation of intra- and intermolecular disulfide bonds in proteins, oxidation of functional SH-groups with sulfonic acid formation, and subsequent proteosomal degradation of proteins [1, 2].
The antioxidant defense involves peroxiredoxins, which decompose H2O2, organic hydroperoxides, and peroxynitrite [3, 4]. Cofactors of these enzymes in most cases are distinct isoforms of thioredoxin. Worthy of mention is the antioxidative activity of thioredoxin reductase 1 (TrxR1), which can directly reduce many substrates, particularly lipid hydroperoxides, H2O2, and dehydroascorbic and lipoic acids [5-8].
In this review, the main attention is focused on functional properties of Trx and Grx, which maintain the redox-dependent thiol/disulfide state of proteins and influence their structure and functions. The regulatory mechanism for a series of vital cellular processes, such as regulation of cell cycle, inhibition of apoptosis, and regulation of transcription factor activities, is also discussed.
THIOREDOXINS
Thioredoxin (EC 1.8.4.8) is a multifunctional low-molecular-weight protein containing an active thiol/disulfide site and possessing oxidoreductase activity. Originally discovered in E. coli, Trx was later found in many prokaryotic and eukaryotic cells [9]. The major Trx isoforms are cytosolic Trx1 and mitochondrial Trx2. Moreover, there are the known Trx1-like protein (named p32TrxL) and recently found Trx2-like protein (Trl2) associated with cytoskeleton microtubules [10]. The Trx family now includes more than 10 proteins.
Human Trx1, a low-molecular-weight (12 kD) protein composed of 105 amino acid residues and substantially localized in cytoplasm is also found in the cell nucleus and blood plasma. Trx2 is primarily synthesized in the form of a precursor protein (18 kD) composed of 166 amino acid residues including a N-terminal 60-amino-acid sequence that is eliminated in the course of posttranslational proteolysis to form Trx2 (12.2 kD) and its transport into mitochondria. The p32TrxL protein is highly homologous to Trx1 and also localized in cytoplasm, but its function is not well understood.
Trx1 can undergo posttranslational modification; due to a limited proteolysis it turns into Trx80 composed of the 80 N-terminal amino acid residues. This enzyme is secreted from the cell and possesses a cytokine-like activity [11]. S-Nitrosylation at Cys69 is important for antiapoptotic effect of Trx1 [12], and its glutathionylation at Cys73 can prevent the dimerization of Trx1 caused by oxidative stress [1].
Both Trx1 and TrxR1 can be localized not only within the cells, but also in the intercellular space [13, 14]. Different cell types including tumor cells secrete Trx1 [15], with the secretion appearing not to be sensitive to oxidation [16]. However, the secretion process can change in presence of various xenobiotics including alkylating agents. A comparative study of secretion on normal liver cells and on HepG2 hepatocarcinoma cells showed that only normal cells demonstrate high secretion of Trx1 [17], whereas secretion of Trx1 by the HepG2 cells increases in presence of 80 µM 2-mercaptoethanol or 5 mM N-acetyl cysteine. However, these cells later undergo morphological changes accompanied by inhibition of their growth. Exogenous Trx1 (100 nM) inhibited the HepG2 cell proliferation, but did not induce secretion of endogenous Trx1.
It is still uncertain whether the secreted Trx1 can provide insurance of the cells (whole body) against xenobiotics and oxidative stress, although the extracellular Trx1 seems to play a certain role in development of inflammatory response. In particular, the Trx1 level in blood plasma increases in many diseases such as AIDS [18], rheumatoid arthritis [19], asthma [20], and hepatitis C [21]. The secreted Trx1 acts as a chemotactic factor for neutrophils, monocytes, and T-cells [22], but at the same time it expresses an inhibitory effect on the endotoxin-initiated chemotaxis of neutrophils [18].
Trx1 can move into the nucleus under the action of many factors. This was demonstrated by Western-blotting on cell cultures subjected to H2O2 [27], hypoxia [28], phorbol esters [29, 30], tumor necrosis factor (TNF) [30], UV [29, 31] and ionizing radiation [32], interleukin-1β? [33], and cisplatin [34], as well as in ischemic/reperfusion injury of the brain [35].
Trx2 has been cloned from heart cDNA libraries of rat [23] and human [24], osteosarcoma cells, and human embryonal stem cells [25]. The TRX2 gene is expressed in virtually all organs and tissues with the highest expression level in the brain [26]. It was also found that Trx2 is localized in mitochondria. The level of Trx2 mRNA therein corresponds to the Trx2 protein content [25].
It is notable that thioredoxins evolved similarly to chaperone-like proteins, whose function is maintenance of the dithiol/disulfide structure of proteins [36]. A highly conservative amino acid sequence of the active center (Trp-Cys-Gly-Pro-Cys-Lys) contains two active Cys residues (Cys32 and Cys35 in human Trx1 and Cys90 and Cys93 in human Trx2) that are oxidized into corresponding disulfides due to the transfer of two reducing equivalents from Trx to a disulfide-containing substrate (Fig. 1). The disulfides formed in the active centers of Trx1 and Trx2 are reduced by thioredoxin reductase (NADPH-dependent selenoflavoprotein), which has two major isoforms: cytosolic (TrxR1) and mitochondrial (TrxR2).
Unlike the mitochondrial Trx2, the cytosolic Trx1 has three Cys residues apart from two localized in the active center. Thus, another site of thiol-disulfide exchange exists, which contains residues Cys62 and Cys69 [37]. It is notable that Trx1 can bind with 15-deoxyprostaglandin at residues Cys35 and Cys69 [38]. Additional Cys residues in its molecule (particularly Cys72 localized in a loop adjacent to the active center) can be oxidized, resulting in dimer formation with the loss of catalytic activity [39]. It is obvious that higher resistance of Trx2 to oxidation is associated with the absence of additional cysteine residues [24]. The difference in resistance to oxidation can also be explained by higher level of intracellular Trx2 in tissues with high metabolic activity. Note that the active center of p32TrxL is analogous to that of Trx1 [40].Fig. 1. Scheme of reactions catalyzed by the thioredoxin-dependent system (Trx, thioredoxin; TrxR, thioredoxin reductase).
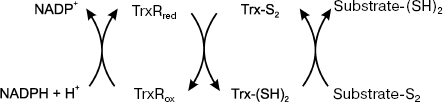
Holmgren and Fagerstedt [41] have offered to divide dithiol and disulfide forms of bacterial Trx via synthesis of iodoacetic acid derivatives followed by native gel electrophoresis. Using this method, the authors found that about 60% of E. coli Trx is in its reduced form during the exponential growth phase [41]. Fernando and coworkers [42] have improved this method with the use of antibodies against Trx1 and further developed so-called redox Western blotting for estimation of redox state of Trx1. Using this method, they have found that in endothelial cells virtually all Trx1 is in the reduced form, and not only under normal conditions, but being exposed to H2O2, 70-85% Trx1 remains in the completely reduced state. Das et al. [43], using the method of redox Western blotting for estimation of the redox-state of oxidized Trx1, when added to human A549 adenocarcinoma cells, found that 45% of Trx1 entered the cells was in completely or partially reduced state. This level increases to 80% and more when TrxR1 and NADPH are added to the culture medium.
The method of redox Western blotting is currently used for measurement of standard redox-potential (E0) of Trx1, which commonly is -230 mV [37], and for measurement of redox state of Trx1 in the cytoplasm [44, 45] and nucleus [46]. It was found that 95% of Trx1 is in the reduced state in both compartments (redox potential (Eh) is -280 mV).
Trx1 is multifunctional: it plays a role of growth factor, enzyme cofactor (see below), regulates activity of a series of transcription factors, and contributes to maintenance of protein folding. Besides, it is involved in development of cancer cell resistance against antitumor drugs and secures cells against oxidative stress. The latter was particularly observed in the case of lung injury caused by bleomycin and doxorubicin-induced cardiotoxicity [47-51]. Mechanisms of the protective effect of Trx1 seem to be associated with its role in redox regulation of cell signaling to a greater extent than with its direct involvement in redox-dependent reactions, because its content in the cell is lower compared with other endogenous antioxidants, particularly GSH.
Both thioredoxin isoforms significantly contribute to antioxidant cell defense due not only to their capability to repair the catalytic activity of peroxiredoxins and glutathione peroxidases decomposing hydroperoxides and H2O2 [52], but also by the ability of Trx1 to directly reduce both H2O2 and GSSG [53]. Moreover, both isoforms can play a role of “trap” for •OH radicals [54].
It is known that Trx1 serves as a cofactor of a series of enzymes [55], such as peroxiredoxins [52], ribonucleotide reductases, and methionine sulfoxide reductases, and is involved in DNA repair [9].
Most human tumor cells are characterized by increased level of Trx1. Trx1 seems to be an atypical growth factor because it has no specific receptor. It is likely to be considered as an important growth cofactor rather than a true growth factor. Mechanisms underlying the pro-growth activity of Trx1 can be realized due to its ability to prevent inactivation or elevate activity of other endogenous growth factors.
Multifunctionality of Trx entitles us to believe that it is very important for maintenance of cell viability. This conclusion is supported by the data on early embryonal lethality of both TXN1 [56] and TXN2 [57] knockout mice. Fibroblasts of TXN2 knockout embryos undergo extensive apoptosis. Interestingly, in this case lethality develops in about the middle of development. In contrast, the death of TXN1 knockout mice occurs shortly after implantation [56]. It was concluded that the main cause of death of TXN2 knockout mice is elevation in the level of reactive oxygen species (ROS) and, hence, Trx2 is a critical component of the antioxidant-based defense system. In general, although the main redox-dependent reactions of Trx1 and Trx2 are very much alike, and both proteins are essential for embryogenesis, the difference in stages of embryonal death suggests some difference in their functions.
Several functions of Trx2 have been identified. The Trx2 isolated from porcine brain mitochondria catalyses a reduced-to-oxidized form transition of native E. coli 4-aminobutyrate aminotransferase [58] and ribonucleotide reductase [59]. In yeasts, Trx2 acts as a reducer of the oxidized form of mitochondrial peroxiredoxin 3 (Prx3), which, in turn, reduces peroxides. Mutant yeast cells deficient in Trx2 are more sensitive to H2O2 [60]. Reddy and coworkers found that in the eye lens of the Emory mouse used as a model for studies on human senile cataracts, an increase of Trx1 mRNA and the protein was observed three weeks after photochemical impact, whereas six weeks after a decrease in the level of Trx2 mRNA was observed. It is hypothesized that inability to maintain or elevate the Trx2 level is critical for development of this kind pathology [61]. Moreover, Trx2, like Trx1, can reduce insulin in the presence of NADPH and TrxR2. This reaction is used for estimation of the total activity of thioredoxin isoforms in vitro [62].
The indices of Trx1 level in blood plasma or serum can be used for diagnosis of several diseases [63]. In particular, the level of Trx1 in blood serum significantly increases in cardiovascular pathology: not only in ischemic heart disease [64, 65], but also in chronic heart insufficiency [66].
It proved that C57BL/6 transgenic mice with overexpression of human Trx1 are more resistant to various oxidative stress types and are characterized by extended lifetime compared with control mice [67]. They demonstrate resistance to cerebral infarction [68], doxorubicin- and bleomycin-evoked cytotoxicity [50], pneumonia [70], and renal ischemia-reperfusion [71]. Embryos of Trx1 transgenic mice can develop normally even under conditions of oxidative stress [72].
Note that the exogenous Trx1 can penetrate into cells, decreasing ROS production and hindering emergence of apoptosis [73], which is currently used for creation of Trx1-based pharmaceuticals, in particular, for therapy of acute inflammatory processes in lungs [69]. Moreover, exogenous Trx1 suppresses development of autoimmune myocarditis [74]. A prolonged intravenous infusion of recombinant Trx1 involutes cerebral stroke locus [75]. The level of Trx1 was found to decrease within aorta of rats with spontaneous hypertension [76]. It was also found that the use of Trx1-inducing substances might be promising for prevention of the progression of hypertension.
As mentioned above, overexpression of the TXN1 gene enables defense against oxidative stress and reduces toxicity of many xenobiotics [48-51]. At the same time, overexpression of the TXN2 gene can facilitate development of resistance to ROS-induced apoptosis [25], as well as resistance of human embryo kidney cells to etoposide [24]. The latter fact evokes great interest because etoposide somehow influences in dose-dependent manner the signaling pathways of mitochondrial apoptosis [77], which are sensitive to the effect of Trx2. Besides, Trx2 can interact with mitochondrial respiratory chain components [78]. In particular, it has been ascertained that Trx2 can form a complex with cytochrome c [79], whereas overexpression of its gene (TXR2) leads to increase of mitochondrial Δψm [24]. The data on ability of Trx2 to bind with cytochrome c [79], as well as the data on important role of cytochrome c in development of apoptosis [80], suggest that Trx2 can be released in response to stimuli activating apoptosis and play a protective role in regulation of programmed cell death, primarily through its effect on formation of apoptosomes and activation of caspases, whose active state requires reduced Cys residues [81].
Like Trx2, Trx1 can exert a control over the apoptosis mechanism [9]. Introduction of Trx1 into a culture medium prevents apoptosis induced by a series of oxidants in neuroblastoma cells [48], whereas overexpression of the TXN1 gene leads to abatement of tumor cell apoptosis in gastric carcinoma [82]. Contrariwise, transfected lymphatic cells expressing the redox-inactive Trx1 are more sensitive to apoptosis induced by various xenobiotics [83].
Mechanisms of the Trx1 effect on apoptosis are still poorly understood. Possibly, it interacts directly with ROS, although its effect on signaling pathways might be more important for this function of Trx1. Thus, Trx1 can bind with apoptotic signal-regulating kinase 1 (ASK-1) to form inactive complex [84]. Some factors inducing apoptosis, particularly oxidative stress, can disrupt this complex, thus activating kinase 1, which leads to activation of JNK/p38 MAP kinases and apoptosis [85].
JNK and p38-dependent pathways of cell signaling have been studied in detail and include MAP-kinase-dependent signaling cascade, which usually includes three levels of protein kinases: MAPKKK, MAPKK, and MAPK [86-88]. The latter enzyme is activated via sequential phosphorylation and then, in turn, regulates activities of downstream transcription factors and/or other kinases, thus exercising a control over expression of distinct genes.
ASK-1 identified as MAPKKK is activated by SEK1-JNK and MKK3/MKK6-p38 signaling cascades [89]. ASK-1 is supposed to be a key element in the mechanism of cytokine-dependent and stress-induced apoptosis. It has been found that overexpression of ASK-1 induces apoptosis, whereas its inactive mutant (ASK-1-K709R) causes suppression of apoptosis induced by TNF-α (tumor necrosis factor) [89, 90]. The data obtained on ASK-1-deficient mice also show that prolonged activation of JNK/p38 and apoptosis induced by TNF-α and ROS require activation of ASK-1 [85].
Trx1 is a key regulator of ASK-1 functions. Its reduced form directly binds with the ASK-1 N-terminal fragment to inhibit both the ASK-1 activity and the ASK-1-dependent apoptosis. Decrease in the level of Trx1 activates endogenous ASK-1 [84]. It has been established that the HIV Nef protein inhibits the ASK-1 activity, thus preventing release of Trx1 from the Trx1-ASK-1 complex [91]. It has been also found that Trx1 is sensitive to S-nitrosylation, which can lead to dissociation of the Trx1-ASK-1 complex and activation of ASK-1 [92].
Some data suggest that Trx1 can facilitate ubiquitinylation of ASK-1 and its degradation in endothelial cells [93]. As mentioned above, the inhibitory effect of Trx1 on ASK-1 depends on the reduced state of Trx1 [84]. Trx1 seems to form a stable complex with ASK-1 via any of its Cys residues [93]. A possibility of this kind of complex formation between Cys-residues of Trx1 and TrxR1 (via Cys32) or Trx1 and NF-κB (via Cys35) is also established [94, 95].
Regulation of transcription factor activities by Trx1 is associated with its capability for reduction of thiols, which are critical for binding with DNA [96]. At least 64 redox-regulated transcription factors have been identified [97, 98]. Many of them have critical Cys residues and can be regulated by a Trx-dependent system [99, 100]. In particular, this has been established for the transcription factors p53, NF-κB, AP-1, and Nrf2, each of which is thiol-dependent and associated with cell proliferation and mechanism of apoptosis [101-106].
The most exhaustively studied is the process of NF-κB activation, which includes three steps: 1) phosphorylation of the inhibitory IκB subunit; 2) its dissociation from inactive complex; and 3) degradation of the IκB subunit. In distinct cell types, ROS can activate NF-κB, whereas antioxidants counteract this activation. Besides, NF-κB inducers, such as TNF-α, ?promote increase of the ROS production level. Reduction of critical Cys residues is necessary for translocation of NF-κB across the nuclear membrane and its binding with DNA. So, the cellular redox status is crucial for the control over the NF-κB activation state [107]. It has been established that both GSH and Trx1 are necessary for NF-κB reduction [30, 108]. For instance, the Trx1 overexpression and translocation into the nucleus lead to strengthening of the NF-κB-mediated gene expression [30]. It has been also established that Trx1 can activate NF-κB to elevate IκB degradation indirectly, via the JNK-signaling pathway [109, 110].
DNA binding with the AP-1 transcription factor, which is either a homodimeric (Jun-Jun) or heterodimeric (Fos-Jun) complex, is mediated by factor Ref-1. Together with the reduced form of Trx1, Ref-1 potentiates the binding of DNA with AP-1 [29].
The Trx-system can influence p53, a multifunctional redox-sensitive tumor suppressor [111]. Protein p53 has several crucial Cys residues in the DNA-binding domain, some of which are necessary for coordination of zinc binding to form the zinc finger domain, whereas others are used for DNA binding [112]. Since both binding types require the reduced state of SH-groups, Trx1 can elevate activity of p53 in the nucleus either directly or via Ref-1 factor [81].
Because all transcription factors possessing critical Cys residues in DNA-binding domains are sensitive to oxidation, Trx1 can maintain them in reduced, functionally active state. A majority of data suggests that the main redox-dependent regulation of Trx1 most likely occurs in cytoplasm, in which key processes of signaling mechanisms are realized. At the same time, a series of studies has shown that Trx1 can implement a regulation of transcription of certain genes in the nucleus [55]. Additional studies are required for decisive elucidation of the functions of nuclear and plasmatic Trx1 in regulation of the expression of the gene.
A series of genes, whose products play an important role in defense of organisms against oxidative stress or xenobiotics, contain in a promoter region an antioxidant responsive element (ARE), whose regulation occurs via the binding with Nrf2 forming heterodimer with low-molecular-weight Maf proteins [113]. Normally, Nrf2 is in the cytoplasm in a complex with Keap1 protein. When oxidative stress occurs, Cys residues in Keap1 oxidize, and Nrf2 is released, translocated into the nucleus, and binds with ARE-containing gene promoters [114]. Although oxidative stress in the cytoplasm contributes to Nrf2 activation, it should be noted that oxidative conditions in the nucleus inhibit the binding of Nrf2 with ARE [104]. At the same time, it is established that Trx1 in reduced state can enhance the binding of Nrf2 with ARE, thus activating it [68]. It is worth noting that transcription of the TRX1 gene, in turn, is regulated via ARE. It is hypothesized that such mechanism of self-heightening effect of Trx1 allows acceleration of cell response to oxidative effects and effective maintenance of redox balance [115]. In mammalian cells, among TrxR isoforms a plasmatic selenium-containing TrxR is inducible, whose synthesis is also regulated by ARE on a transcription level. Elevation of TrxR activity in murine and human hepatoma cells (Hepa1c1c7 and Hep2) was observed under the action of “classic” ARE activators (sulforan, tert-butylhydroquinone, and β-naphthoflavone) and accompanied by enhancement of TrxR1 mRNA synthesis [79]. Cadmium ions, diethylmaleate, and arsenite induced synthesis of Trx1 and TrxR1 in endothelial cells of bovine arteries via stimulation of the Nrf2-ARE regulatory element [116].
Electrophilic substances constantly produced during metabolism of xenobiotics can possess significant toxic effect due to their high reactive capability. One of the most electrophilic aldehydes is acrolein [117] resulting from activation of lipid peroxidation [118] and oxidation of OH-containing amino acids by myeloperoxidase [119]. Acrolein is also a metabolic product of cyclophosphamide, spermine, spermidine, allyl alcohol, and allyl amine [120]. At sublethal doses, it decreases proliferation [121, 122], and at high doses causes significant damage to cells and tissues.
Acrolein induces most genes regulated by ARE [123] and easily reacts with nucleophiles, particularly with cellular thiols, such as GSH [121, 124]. Moreover, acrolein interacts with SH-groups of Trx1 and TrxR1 [125]. Acrolein causes a fast synthesis of Trx1, which is in agreement with the data on induction of the TXN1 gene expression by electrophiles [104].
A decrease in the levels of Trx1 and GSH by electrophiles influences activation of transcription factors. For instance, inhibition of NF-κB activity results from both the loss of activities of regulatory thiols, Trx1 and GSH, after the action of acrolein [124, 126] and direct binding of acrolein with critical Cys residues in subunits of the transcription factor itself [127]. The effect of acrolein on p53 and AP-1 is also realized via its direct binding with critical Cys residues of the transcription factor [124, 128, 129].
Thus, despite incomplete understanding of the mechanism of Trx involvement in processes of toxicity modulation and signaling activity of xenobiotics, the growing number of studies in this branch manifests a particular role of Trx in these processes.
THIOREDOXIN REDUCTASES
TrxR (EC 1.8.1.9, systematic name thioredoxin:NADPH oxidoreductase) is an NADPH-dependent homodimer of oxidoreductase (with one FAD per subunit), which reduces the active center of disulfide in oxidized Trx [55, 62, 130, 131]. The molecular mass of the TrxR subunit is 55-65 kD in mammals and 35 kD in prokaryotes, plants, and yeasts [55, 62, 132].
Three mammalian TrxR isoforms have been found and characterized: cytoplasmic (TrxR1), mitochondrial (TrxR2), and TrxR catalyzing reduction of both Trx and GSSG and named thioredoxin glutathione reductase (TGR), high level of which is found in testes [133-135]. All three isoforms possess the same domain structure and are characterized by presence of selenocysteine (Sec) in the C-terminal active center (Gly-Cys-Sec-Gly-COOH). At the same time, three 5′-non-translated regions in cytosolic TrxR1 mRNA and alternative ATG codon in the gene sequence are found [136, 137], but their role remains unknown. An additional N-terminal monothiol domain features the TGR isoform [135].
Existence of TGR illustrates a functional overlap of Trx- and GSH-systems. For example, glutathione reductase is absent in Drosophila melanogaster, and reduction of GSSG is only fulfilled by the Trx-dependent system [138].
The mammalian TrxR1 was first purified and characterized in 1977 [139]. It should be emphasized that Se is important for the TrxR enzymatic activity [140-143]. The presence of Se is supposed to decrease Cys redox potential, elevate efficacy of enzymes at low pH values, and broaden the spectrum of substances being reduced [144]. It is also observed that the TrxR activity drops with a deficit of Se in mammalian and human cell lines [145, 146]. At the same time, addition of Se to tumor cell cultures several times increases activity of TrxR, unlike other Se-containing enzymes including glutathione peroxidase [147].
Mammalian TrxR closely resembles other disulfide oxidoreductases, such as glutathione reductase and lipoamide dehydrogenase, both in primary structure [141, 148] and characteristics of catalytic mechanism [149]. However, TrxR has a C-terminal Sec-containing domain, which is absent both in lipoamide dehydrogenase and glutathione reductase [141], which explains many unique properties of this enzyme.
The catalytic mechanism of mammalian TrxR includes TrxR reduction with involvement of NADPH, which is similar to that of glutathione reductase and lipoamide dehydrogenase [143, 149, 150]. Initially, the electron moves from NADPH via FAD to the active center disulfide formed by Cys residues at positions 59 and 64 in the N-terminal domain, which is identical to the analogous domain in glutathione reductase. Then, electrons are transferred from the formed dithiol of one subunit of the dimeric enzyme to selenyl sulfide of the C-terminal sequence of another subunit. Such electron transfer to another subunit can occur, when GSSG is reduced by glutathione reductase [143]. The reduced C-terminal Sec participates in reduction of multiple substrates of mammalian TrxR1.
The active center of TrxR seems to be largely protected due to oxidation with formation of selenyl sulfide. In this oxidized state, the enzyme is resistant to carboxypeptidase and trypsin [141, 151] as well as to formation of derivatives with electrophilic substances [152]. These facts point to the possibility of certain conformational changes in the active center region during reduction/oxidation, which apparently enables TrxR to reduce a great number of various substrates due to easy accessibility of the enzyme active center in the reduced state.
Actually, mammalian TrxR isoforms are characterized by broad substrate specificity and capability of direct reduction of various protein disulfides, many low-molecular-weight disulfides, and substances that are not disulfides. A great number of disulfides are reduced indirectly, via thioredoxin reduced by TrxR. In particular, although GSSG and insulin are not substrates for TrxR1, both they can be effectively reduced by Trx1 [139, 153].
Peroxides including lipid hydroperoxides and H2O2 can be directly reduced by TrxR1 [5, 6]. By this mechanism, TrxR1 acts as an alternative pathway of enzymatic detoxification of lipid hydroperoxides. However, the high Km of TrxR1 for H2O2 (2.5 mM) makes the role of this enzyme significant only with elevation of H2O2 level [5].
Protein disulfide isomerases (PDI) comprise a family of proteins localized in endoplasmic reticulum and playing an important role in posttranslational protein folding and processing. These proteins contain one or more thioredoxin domains, some of which have redox-active dithiol-disulfide regions [154] structurally homologous to the active center of Trx and reducible by both Trx1 and TrxR1 [155, 156].
TrxR1 also reduces various low-molecular-weight substances including antibacterial polypeptides [157], cystine, alloxan [158], and vitamin K [159].
Ascorbate filling due to reduction of dehydroascorbic acid plays an important role in the antioxidant defense system because ascorbate is one of the main water-soluble low-molecular-weight antioxidants. Ascorbate reduces α-tocopherol, peroxides, and O2•- [160] and prevents lipid hydroperoxide formation, which is of great importance because it decreases the risk of atherosclerotic plaque formation [161]. When intracellular GSH pool drastically decreases, the function of the Trx-dependent system becomes the alternative mechanism of dehydroascorbate reduction [7]; however, a question on its contribution into reduction of dehydroascorbate in different cells is still not resolved. In some cells dehydroascorbic acid is reduced independently of GSH and NADPH, thus implying existence of an additional mechanism of ascorbate metabolism [162].
It is worth noting that, since ascorbate is involved in α-tocopherol reduction, and TrxR1 reduces dehydroascorbate, TrxR1 can play an important role in regulation of antioxidant function of vitamins C and E as a link connecting these systems [163].
Lipoic acid, which reduces GSSG [164], is a metal chelator and can play a role as a trap for free radicals [165]; it is also involved in reduction of vitamins C and E. Lipoic acid acquires high reducing capability after its reduction to dihydrolipoic acid [166] by lipoamide dehydrogenase, glutathione reductase, TrxR1, and TrxR2 [8, 167, 168]. A comparison of kinetic parameters shows that the ratio kcat/Km for TrxR1 is higher than that for glutathione reductase and lower than that for lipoamide dehydrogenase, whereas Km for lipoamide dehydrogenase is significantly higher than that for TrxR1, which is indicative of an important role of TrxR1 in lipoic acid reduction. Compared to cytosolic TrxR1, mitochondrial TrxR2 is less effective [8].
TrxR1 reduces many Se-containing substances including selenite [169] and Se-containing active center of glutathione peroxidase in plasma [170]. A transition of inorganic Se, for instance in the form of selenite (SeO32-) to selenide (HSe-), is necessary for active incorporation of Se into Se-containing proteins. This reduction can be catalyzed either by TrxR1 regardless of involvement of Trx1 [169] or include formation of selenoglutathione (GS-Se-SG) as an intermediate. Formation of GS-Se-SG requires the presence of GSH, whose level is maintained by glutathione reductase, which also can directly reduce GS-Se-SG [171]. However, TrxR1 is also highly effective in GS-Se-SG reduction [172]. Thus, TrxR1 plays an essential role in metabolism of Se-containing substances [173] and is an important link between metabolism of Se and antioxidative processes.
PEROXIREDOXINS
Peroxiredoxins (Prx, EC 1.11.1.15), discovered about 10 years ago, are members of a superfamily of Se-independent peroxidases. Peroxiredoxins execute enzymatic degradation of H2O2, organic hydroperoxides, and peroxynitrite [3, 4]. They are found in bacteria, plants, and mammals. Unlike Trx possessing the active double-cysteine region and forming the intramolecular disulfide bond when oxidized, Prx have no such regions; however, the easily oxidized Cys residues present in their structure can form intermolecular disulfide bonds [144]. By the number of active Cys residues, mammalian peroxiredoxins fall into typical double-cysteine (Prx1-Prx4), atypical double-cysteine (Prx5), and single-cysteine (Prx6) classes. All of them contain fixed Cys residues on the N-terminal regions of the molecules, and the isoforms Prx1-Prx4 have additional analogous Cys residues on the C-ends of the molecules [4].
Prx1-Prx5 use Trx as a donor of electrons, whereas Prx6 uses GSH [174]. Although the catalytic activity of Prx towards H2O2 (105-106 M-1⋅sec-1) is lower than that of glutathione peroxidase (108 M-1⋅sec-1) and catalase (106 M-1⋅sec-1), they play an important role in detoxification of H2O2 [144]. Reduction of H2O2 by all Prx isoforms passes through formation of sulfenic acid (Cys-SOH) due to oxidation of SH-group of the Cys residue; however, the mechanism of the peroxidase reaction slightly differs in different Prx isoforms.
The typical double-cysteine Prx1-Prx4 are homodimers, and their interaction with H2O2 leads to formation of sulfenic acid, which can participate in formation of inter-peptide disulfide bond reduced by Trx. A similar mechanism was ascertained for Prx5, but the latter is a monomer, and the intramolecular disulfide bond is formed between Cys47 and Cys151 [144, 175]. Prx6 uses low-molecular-weight thiols including GSH as sources of electrons rather than Trx, as do other peroxiredoxins [174]. Moreover, Prx6 reduces phospholipid hydroperoxides and exhibits activity of phospholipase A2 [174]. The mechanism of H2O2 reduction by Prx6 includes oxidation of the active Cys47 into sulfenic acid followed by its reduction to disulfide by means of S-glutathionylation if heterodimerization of Prx6 with glutathione transferase P1-1 takes place [176]. The disulfide formed is further non-enzymatically reduced by GSH to restore the functional activity of Prx6.
Since H2O2 can rapidly transform into highly toxic ROS, such as O2•- radicals, elevation of its level can lead to development of oxidative stress [177], which causes DNA breaks, linkages in protein molecules, and activation of lipid peroxidation [178]. A physiological role of Prx associated with enzymatic degradation of H2O2 is particularly significant in erythrocytes, in which these enzymes are in second or third place in protein content [179].
An important role of Prx in defense against oxidative stress was demonstrated in a series of studies with knockout of genes corresponding to Prx. Hemolytic anemia, characterized by hemoglobin instability developed, in PRDX1 gene knockout mice [180]. In PRDX2 gene knockout mice, a significant decrease of lifespan was also accompanied by development of anemia [181]. In both cases, the knockout of the corresponding gene caused a significant elevation of ROS in erythrocytes. The PRDX6 gene knockout mice were characterized by low survival, high level of protein oxidation, and significant injury of kidneys, liver, and lungs. It should be noted that in this case the expression of antioxidant enzymes, such as catalase, glutathione peroxidase, and Mn-SOD did not differ from that in wild-type mice. The data of these studies suggest that function of Prx6 cannot be compensated by expression of other genes [182].
Nonetheless, H2O2 not only contributes to the development of oxidative stress, but at low concentrations it can play a role of secondary messenger involved in intracellular transmission of signals from various surface receptors [183, 184]. H2O2 produced with the action of extracellular signals is rapidly eliminated after accomplishment of its function. According to this conception, Prx can regulate pathways of cellular signal transduction by control over the level of H2O2 [183]. In fact, it was found that overexpression of the PRDX1 and PRDX2 genes in transfected cells led to decrease in the level of intracellular H2O2 caused by epidermal growth factor and inhibited H2O2- and TNFα-dependent activation of the NF-κB transcription factor [185]. It has been shown on the embryonic fibroblast cell culture that overexpression of the PRDX2 gene causes a clear modification of H2O2-dependent activation of JNK and p38 kinases in response to TNFα [186]. The authors concluded that Prx can complement effects of other antioxidant enzymes as a modulator of intracellular redox-dependent signaling cascades [183, 186]. Similar results were obtained for the TNFα-dependent activation of the AP-1 transcription factor, which decreased with overexpression of the PRDX2 gene in transfected endothelial cell culture [187]. In thyroid cell culture, overexpression of the PRDX1 and PRDX2 genes eliminated H2O2 (whose level was significantly increased under the action of thyrotropin) and protected the cells from H2O2-induced apoptosis [188].
Studies on crystalline structure of Prx have shown that two functionally active Cys residues act as potential cellular sensor systems determining the role of H2O2 either as toxic oxidant or signaling molecule [189]. A model has been proposed in which sensitivity of peroxiredoxins to H2O2 correlates with structural changes of these proteins. This model supposes that high intracellular level of Prx with two functionally active Cys residues can retain low level of H2O2 in quiescent cells. Alternatively, when the level of H2O2 increases (for example, in cells treated with TNFα) oxidation of redox-sensitive Cys residues reduces their peroxidase activity, and high level of H2O2 activates distinct cellular redox-dependent signaling pathways. Here the authors consider the mammalian Prx containing two active Cys residues as “gates” for peroxides, which are open under certain conditions.
The expression of genes encoding different Prx isoforms has cellular, tissue, and organ specificity. Prx1 is the most widely represented and highly expressed member of the peroxiredoxin family in virtually all organs and tissues of mice and humans, both in normal tissues and malignant tumors [190-192]. In particular, it should be noted that the PRDX1 gene is widely expressed in various areas of the central and peripheral nervous system with expression specificity depending on the cell type [193]. High expression of the PRDX4 gene is characteristic of liver, testes, ovaries, and muscles, whereas low expression is observed in small intestine, placenta, lung, kidney, spleen, and thymus [190]. The genes encoding Prx1, Prx2, and Prx3 isoforms are universally expressed in rat skin [194]. In epidermis, the genes encoding Prx1 and Prx2 are expressed in all layers with elevation from basal to granular layer. The Prx3 protein is also found in all layers, but in reverse order (maximum concentration in basal layer). UV irradiation leads to significant increase in the level of Prx2 only [194].
Bast and coworkers found Prx1 and Prx2 in pancreatic β-cells of the islets of Langerhans, whereas expression of their genes was absent in the α-cells [195]. Differing expression patterns of genes encoding Prx isoforms have been found in lungs and bronchi [196]. Moderate or high levels of Prx1, Prx3, Prx5, and Prx6 are found in bronchial epithelial cells, mainly Prx5 and Prx6 in alveolar epithelial cells, and Prx1 and Prx6 in alveolar macrophages. It should be noted that the contribution of Prx6 to the antioxidant defense system of the mammalian upper respiratory tract is up to 75% [197, 198], so in acute inflammatory processes application of Prx6 significantly diminishes the tissue regeneration time. In this connection, attempts to create Prx6-based pharmaceuticals are currently underway.
Prx is present in all subcellular compartments, with some specificity of various isoform gene expression being observed [4]. In intracellular organelles, Prx1 is most widely represented [199]. Besides Prx1, Prx5 is found in cytoplasm, peroxisomes, mitochondria, and nuclei [200, 201]. Unlike Prx1 and Prx5, other isoforms have more restricted subcellular localization. In particular, Prx2 is present both in the nucleus and cytoplasm, secreted Prx4 in cytoplasm and lysosomes, Prx3 in mitochondria, and Prx6 in cytoplasm [4].
Regulation of expression of Prx-encoding genes can occur both on the level of transcription and due to posttranslational modification [202]. A variety of factors stimulating oxidative stress in murine macrophages influences expression of the PRDX1 gene [203]. It is induced by heavy metals in hepatocyte culture and by lipopolysaccharides in rat macrophages [204]. It was found in all cases that induction of expression of this gene was observed together with expression of stress-inducible gene HO-1, whose product is heme oxygenase-1, the rate-limiting enzyme of heme degradation [205, 206]. A parallel induction of PRDX1 and HO-1 gene expression was found in smooth muscle vessel cell culture under the action of oxidized low-density lipoproteins [207] and in experiments in vivo in ischemic loci of rat brain [208]. A concerted induction of the PRDX1 and HO-1 genes seems to be a common adaptive response of cells as a defense against oxidative stress. Moreover, the stress-induced induction of gene expression was also marked for other Prx isoforms, such as Prx2 and Prx6 [195, 209].
It is worth noting that the Nrf2 transcription factor plays the leading role in regulation of the PRDX1 gene expression by electrophilic and ROS-producing agents [210]. This is supported by data on the absence of expression of this gene under the effect of stress-inducing factors in NRF2 knockout mice [211]. Although Nrf2 is a key regulator of PRDX1 gene expression, various data point to involvement of other transcription factors in regulation of this gene. In particular, expression on the PRDX1 gene in culture of rat macrophages occurs via an AP-1-dependent mechanism when 12-O-tetradecanoylphorbol-13-acetate (TPA) is added [212]. Protein kinase C and Ras protein activating the p38 MAPK-signaling cascade are involved in this process [213, 214]. It has been found that PKCδ participates in posttranslational induction of Prx1, when sodium arsenate is added to an osteoblast culture [191]. Besides, in rat macrophage culture lipopolysaccharides can induce expression of the PRDX1 gene via the NO-dependent signaling cascade, possibly by means of induction of iNOS [215]. The regulatory role of the NO-dependent signaling pathway was also discovered from the study of the mechanism of induction of PRDX1 and PRDX2 gene expression in pancreatic cell culture [195].
The activity of Prx can be modified by posttranslational mechanisms, such as phosphorylation, redox-dependent oligomerization, proteolysis, and ligand binding. Phosphorylation of Prx1, Prx2, Prx3, and Prx4 at Thr residues by Cdc2 cyclin-dependent kinase [216] inhibits their peroxidase activities. The mechanism of this inhibition can be explained as a negative modulating effect of negatively charged phosphate group on the Prx active center through electrostatic interaction [216]. Prx can also form dimers and decamers upon change in ionic strength and at low pH values. Activation of Prx oligomerization is evoked by a change in the state of the redox-active disulfide center. A direct functional connection between the redox state and oligomerization has been established for Prx in bacteria [217]. Moreover, a restricted proteolysis of typical double-cysteine Prx from the C-end elevates their resistance to oxidation and subsequently to inhibition of peroxidase activity [218]. The Prx activity can also change due to the noncovalent binding with ligands, such as heme and cyclophilin A. Prx1 was first identified as a protein with high affinity to heme [219], and the binding with heme appreciably decreased its activity [220]. In contrast, the peroxidase activity of Prx6 increased after interaction with cyclophilin A [221]. In general, the posttranslational modifications of Prx result in structural and associated functional changes, which seem to have functional significance for these enzymes as regulators of cellular redox homeostasis.
It is well known that ROS production and cellular redox state play an important role in regulation of the cell cycle and cell proliferation [222, 223]. It has been found that antioxidant enzymes, such as glutathione peroxidase and Mn-SOD, are involved in cell cycle regulation [224]. At the same time, Irani and coworkers found ROS-dependent regulation of the cell cycle [225] and established that increase in ROS production accelerates the cell cycle in fibroblast culture, whereas the action of the antioxidant N-acetyl-L-cysteine decelerates it [225]. Also, in embryonic murine fibroblasts the cellular level of ROS correlates with the cell cycle time, whereas overexpression of the SOD2 gene inhibits cell proliferation [226].
Data on association of Prx1 with cell proliferation date from early studies [227]. In particular, it was shown that expression of the PRDX1 gene was appreciably higher in Ras-transfected epithelial cells compared with the wild-type cells [227]. Correlation of PRDX1 gene expression level with cell proliferation was confirmed in experiments on HL 60 promyelocytes treated with dimethyl sulfoxide [227]. Moreover, it was found that Prx1 interacts with c-Abl and c-Myc protein kinases playing an important role in regulation of cell proliferation [228]. In yeasts, Prx1 can regulate the tyrosine kinase activity of c-Abl via binding with its third structural domain [228], which leads to restriction of the transforming ability of c-Abl. In this connection, it has been supposed that the reversible binding of Prx1 with c-Abl can serve as a key cell cycle regulator. It has been also found that Prx1 can bind with c-Myc via the c-Myc-transactivating domain [229]. Moreover, a decrease in expression of a series of genes specific for activity of c-Myc is observed in the case of overexpression of the PRDX1 gene [229].
As noted above, Prx can be specifically phosphorylated at the Thr90 residue via the Cdc2 cyclin-dependent kinase, which leads to decrease of the enzyme activity [216]. Prx1 phosphorylation occurs during mitosis rather than in interphase. In this connection, Prx phosphorylation is supposed to play an important role of “switch” in direction to elevation of H2O2 level, which, in turn, leads to acceleration of the cell cycle [216]. Another peculiarity of the Prx functional activity merits attention: like other antioxidant enzymes, such as Mn-SOD, these proteins, due to their peroxidase activity, can inhibit proliferation of various tumor cells [224]. Thus, progression of malignant tumors such as lymphomas, sarcomas, and carcinomas is observed in PRDX1 knockout mice [180]. Prx are thus supposed to play a role as tumor suppressors.
Cell cycle development and apoptosis are related processes, and disturbance of regulation of Cdc2-kinase activity in mammalian cells can result in initiation of apoptosis [230]. It is known that one of the cytokines inducing ROS production during intracellular signal transmission is TNFα, which induces apoptosis by binding with the death-domain of the TNFα receptor [231]. In this process, TNFα activates the NF-κB transcription factor involved in redox-dependent gene regulation [184, 232]. It was found that overexpression of the PRDX2 gene inhibits NF-κB activation after stimulation of cells with H2O2 [185], and overexpression of this gene in Molt-4 leukemia cells has a protective effect against apoptosis induced by ceramide or etoposide [233]. In so doing, Prx2 prevents the leak of cytochrome c out of mitochondria and inhibits lipid peroxidation. Interestingly, the overexpression of the PRDX1 gene also has a protective effect on cells exposed to peroxides [200, 234]. However, it has been found that PRDX1 gene overexpression can suppress the development of apoptosis and enhance cell resistance to radiation via inhibition of the JNK kinase activity [235]. It has thus been found that Prx1 can directly bind with the GSTP/JNK complex to elevate its stability.
Based on the data described above, one can conclude that the elevation of the peroxiredoxin level counteracts apoptosis, enhances antioxidant effect, and regulates cell proliferation.
GLUTAREDOXINS
Glutaredoxins (Grx, EC 1.20.4.1), GSH-dependent oxidoreductases with low molecular masses (9-14 kD), are members of the glutaredoxin superfamily in their structure, which play an important role in cellular redox-dependent processes. Grx are found in virtually all taxonomic groups including prokaryotes and eukaryotes (from yeasts to plants and humans) [2]. Unlike Trx, Grx of various species exhibit high degree of homology of amino acid sequence, particularly, in the active center region [2].
The electron transfer in the Grx-dependent system occurs from NADPH-dependent glutathione reductase to the oxidized glutathione to form GSH, which, in turn, reduces the oxidized glutaredoxin (Fig. 2). The Grx substrates are disulfides and mixed disulfides. The disulfide reduction catalyzed by Grx can proceed in two pathways: monothoiol and dithiol ones involving one or two Cys residues in the active center, respectively.
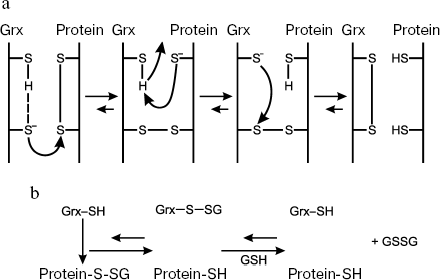
The structure of dithiol Grx has three features: (i) a CXXC fragment of amino acid sequence (commonly Cys-Pro-Tyr-Cys) with two functionally active thiols in the active center; (ii) a certain surface hydrophobic area; and (iii) the binding site for GSH [236, 237].Fig. 2. Scheme of reactions catalyzed by the glutaredoxin-dependent system.

In the dithiol mechanism (Fig. 3a), the N-terminal Cys residue of the active center executes nucleophilic substitution, thus initiating formation of mixed disulfide between glutaredoxin and substrate, a protein carrying a disulfide bond [2]. Another, free, C-terminal Cys residue undergoes deprotonation and then interacts with the N-terminal S atom yielding the oxidized enzyme Grx-S2 and reduced substrate. The oxidized form of the enzyme is reduced via interaction with GSH to form a mixed disulfide (between GSH and the N-terminal Cys residue of the active center), which, in turn, is reduced by the second GSH molecule. By the dithiol mechanism, Grx reduces both low-molecular-weight and protein disulfides.
Reduction of mixed disulfides and glutathionylated proteins occurs by the monothiol mechanism (Fig. 3b) called deglutathionylation; in this case, Grx only uses the N-terminal Cys residue [2]. In this reaction, due to high affinity to GSH, the enzyme specifically interacts with the GSH residue of mixed disulfide protein-S-SG [236, 237] to form an intermediate Grx-S-SG, which is reduced using GSH. The GSSG produced is reduced to GSH by glutathione reductase. Since the recognition of only GS-residues rather than a total substrate is necessary for reduction of glutathionylated proteins, the monothiol mechanism leading to deglutathionylation seems to be the most common function of Grx. Unlike Grx, Trx demonstrate extremely low, if any, activity towards mixed disulfides.Fig. 3. Dithiol (a) and monothiol (b) catalytic mechanisms of glutaredoxin.
Glutaredoxins are functionally coupled with glutathione reductase and with the GSH/GSSG ratio. The GSH/GSSG ratio is an indicator of the cell redox state and important determinant of redox-potential, which correlates with biological state of the cell and reaches 240 mV in proliferation, 200 mV in differentiation, and 170 mV in apoptosis [238].
S-Glutathionylation of proteins is an important regulatory mechanism of biological processes due to the reversible modification of protein thiols [239, 240] that can be catalyzed by Grx and glutathione S-transferase [241]. A series of proteins, such as chaperones, cell cytoskeleton proteins, and cell cycle regulators [242, 243], undergoes reversible S-glutathionylation with change in intracellular redox state. It has been demonstrated that S-glutathionylation of Cys62 in the NF-κB p50 subunit and Cys269 of c-Jun kinase leads to the loss of their DNA-binding activity [89, 244]. At the same time, other enzymes, such as tyrosine hydroxylase, protein phosphatase 2A, tyrosine phosphatase 1B, and α-ketoglutarate dehydrogenase are reversibly inhibited due to S-glutathionylation [245-248]. It has been also found that G-actin can be glutathionylated and deglutathionylated at Cys374. Human GLRX1 gene knockout significantly influences the capability of G-actin for polymerization and translocation, as well as cell cytoskeleton reorganization [249, 250].
S-Glutathionylation also serves for prevention of irreversible oxidation of protein SH-groups. Depending on the SH-group oxidation degree, either sulfenic (Cys-SOH), sulfinic (Cys-SO2H), or sulfonic (Cys-SO3H) acid is formed. Sulfonic acid is not reduced, and its appearance leads to the proteosomal degradation of a protein, whereas sulfenic and sulfinic acids can be reduced by Grx and Trx [241]; Trx only reduces the former acid, whereas Grx reduces both acids to form the functionally active SH-groups [1, 2].
In general, it is now taken for granted that formation of mixed disulfides from protein thiols and GSH is a key event in regulation of cell response to oxidative stress. Grx is the main enzyme catalyzing both production and reduction of mixed disulfides [251, 252]. As a general rule, the process of protein mixed disulfide reduction is preferable, but the production of mixed disulfides predominates over their reduction, when GSH concentration is dropped or GSSG level is elevated (for instance, under oxidative stress conditions) [253].
Three Grx isoforms have been found in mammals including humans: cytosolic Grx1 with the active center containing the Cys-Pro-Tyr-Cys motif and two mitochondrial, Grx2 and Grx5, whose active centers contain the sequences Cys-Ser-Tyr-Cys and Cys-Gly-Phe-Ser, respectively [254, 255]. Grx1 and Grx2 are dithiol isoforms and contain two Cys residues in their active centers, whereas Grx5 has only one Cys residue. The Grx3 and Grx4 isoforms are only found in lower eukaryotes.
Grx1, a protein with molecular mass of 12 kD localized mainly in cytosol (although also found in nucleus) [256], was discovered as a GSH-dependent donor for ribonucleoside reductase [257]. Compared with Trx, whose level (10 µM) commonly exceeds Grx1 level in the cell (1 µM), Grx1 has tenfold lower Km value for ribonucleoside reductase [258, 259]. The studies have shown that the main contribution to deoxyribonucleotide production in E. coli belongs to Grx1 and, to lesser extent, to Trx1 [260].
The Grx1 isoform reduces disulfides by the dithiol mechanism and plays an important role in thiol-disulfide exchange [130], by which it is involved, in particular, in cell differentiation [261], regulation of transcription factor activities [262-264], and apoptosis [265, 266]. Grx1 protects neurons from dopamine-induced apoptosis via activation of NF-κB with participation of regulatory factor 1 [239, 266]. It has been found in a series of studies that Grx1 through catalysis of deglutathionylation restores the functional activities of proteins such as glyceraldehyde-3-phosphate dehydrogenase, protein tyrosine phosphatase 1B, creatine kinase, c-Jun, NF-κB p50 subunit, and caspase-3 [245, 249, 267, 268]. It has been also found that expression of the GLRX1 gene can be significantly increased in tumor cells [269].
The Grx2 isoform (14 kD) is found in two splicing forms, Grx2α and Grx2β, localized in mitochondria and nucleus, respectively [254, 270]. The Grx2 efficacy (kcat/Km) in catalysis of deglutathionylation of protein mixed disulfides is 1.5-3.0-fold higher than that of Grx1 [271]. Grx2 regulates the mitochondrial ROS level via catalysis of reversible S-glutathionylation of mitochondrial complex I, whose functional activity is accompanied by formation of O2•- [272, 273]. Besides, Grx2 catalyzes the reversible glutathionylation of inner membrane proteins, depending on changes in the GSH/GSSG ratio [272].
The oxidized form of Grx2 is reduced both by GSH and TrxR2; the latter reduces not only the disulfide formed by Cys residues in the Grx2 active center, but also the mixed disulfide of Grx2 and GSH (an intermediate in the reaction of deglutathionylation catalyzed by Grx2), as well as a series of low-molecular-weight disulfides [271, 274], such as GSSG, CoA-S-S-CoA, CoA-S-SG, Cys-S-SG, and dehydroascorbate [271]. Reduction of oxidized Grx2 at the expense of TrxR2 can occur under oxidative stress conditions, when the GSSG level significantly increases, and reduction of functionally active SH-groups in the Grx2 active center by reduced glutathione is hampered.
It is notable that Grx2 is the first member of the thioredoxin family [275] that contains [2Fe-2S] cluster binding two Grx2 molecules [276]. The dimeric holoenzyme is not active, but degradation of the cluster to form monomers leads to activation of Grx2, which occurs with increase in GSSG level and in the presence of oxidants [277]. It is thus supposed that the Grx2 isoform serves as a redox-sensor, and functionally active Grx2 is formed when necessary for cell defense, in particular, against activation of ROS-dependent apoptosis.
Grx2 can suppress apoptosis by prevention of both the leak of cytochrome c from mitochondria and cardiolipin oxidation [278, 279]. The use of siRNA leads to decrease in the intracellular level of Grx2 and significant elevation of HeLa cell apoptosis under the influence of oxidative stress caused by doxorubicin and phenylarsine oxide, which is accompanied by 60-fold enhancement of toxic effect of doxorubicin and 40-fold enhancement of that of phenylarsine [278]. Alternatively, overexpression of the GLRX2 gene in these cells appreciably decreases their sensitivity to apoptosis induced by 2-deoxy-D-glucose or doxorubicin, prevents oxidation of cardiolipin, and inhibits both the cytochrome c leak from mitochondria and activation of caspases [279]. Overexpression of mitochondrial Grx2 has more expressed protective effect because of the significant role of Grx2 in maintenance of mitochondrial redox homeostasis than has overexpression of cytosolic mutant devoid of signal sequence region of mitochondrial translocation [280].
The human Grx5 isoform is highly homologous to the yeast Grx5, so these proteins are homonymous [255]. The human Grx5 sequence contains the N-terminal region providing translocation of Grx5 into mitochondria. Like in humans, the yeast and E. coli Grx5 are involved in maintenance of homeostasis of iron [255, 281, 282]. GRDX5 gene knockout in yeasts leads to oxidative stress, accumulation of iron in the cell, and inactivation of enzymes containing the [Fe-S] clusters [281]. In yeasts, Grx5 seems to be required at the stage following [Fe-S] cluster formation, when the cluster is binding with apoenzyme [283]. It has been recently shown that the human Grx5 has the same function [255, 284]. Moreover, human Grx5 is active towards mixed protein disulfides and protein disulfides [285].
Existence of a specific category of Grx isoforms the yeast Grx2 belongs to evokes great interest. Their tertiary structure closely resembles that of glutathione S-transferase and, at the same time, has a Grx-like domain with distinctive amino acid sequence Cys-Pro-Tyr-Cys with two functionally active thiols determining the Grx-like activity of such proteins [286].
Grx together with Trx significantly contribute to the system of cell defense upon development of cardiovascular diseases [287]. Like Trx1, Grx1 is expressed in vessel endothelial and smooth-muscle cells and fibroblasts [288]. Grx1 is also found in plasma [289], whereas Grx2 is not secreted from the cells [290]. Grx1 is hypothesized to implement a protective function in atherosclerosis: infiltrating macrophages exhibit high level of the GLRX1 gene expression in zones of atherosclerosis development [288]. It is also found that overexpression of the GLRX1 gene leads to deglutathionylation of Ras protein, thus inhibiting its activation [291]. At the same time, the ROS-dependent activation of Ras is found to be one of the causes of development of atherosclerosis and restenosis [292]. Grx2 and Grx5 localized in mitochondria of cardiomyocytes can also contribute to development of cardiovascular diseases because they are involved in maintenance of iron homeostasis [255, 278, 284].
It should be noted that glutaredoxin, like thioredoxin, plays an important role in development of drug resistance of tumor cells to pharmaceuticals possessing prooxidant activity, by enhancement of the antioxidant defense system [25, 293-295].
The two enzymes are functionally interrelated and complement each other, being involved in control over cell redox balance. There is evidence for cross-talk regulation between these two systems in mammalian cells [1]. Herein, it is shown that glutaredoxin can inactivate human thioredoxin by means of glytathionylation at the Cys73 residue.
The data presented in this review suggest an important role of thiol-containing proteins such as thioredoxin, peroxiredoxin, and glutaredoxin in maintenance of cellular redox homeostasis and in redox-dependent regulation of a series of intracellular processes including proliferation, differentiation, and apoptosis. The combination of antioxidant properties and ability of both gene transcription activation, including the genes that encode some antioxidant enzymes, and inhibition of redox-dependent pathways of apoptosis induction suggest an important contribution of these enzymes to the antioxidant defense system. They elevate cell resistance to oxidative stress, thus preventing development of many diseases including cardiovascular diseases, acute lung inflammation, and cerebral stroke.
REFERENCES
1.Casagrande, S., Bonetto, V., Fratelli, M.,
Gianazza, E., Eberini, I., Massignan, T., Salmona, M., Chang, G.,
Holmgren, A., and Ghezzi, P. (2002) Proc. Natl. Acad. Sci. USA,
99, 9745-9749.
2.Fernandes, A. P., and Holmgren, A. (2004)
Antioxid. Redox Signal., 6, 63-74.
3.Hofmann, B., Hecht, H.-J., and Flohe, L. (2002)
J. Biol. Chem., 383, 347-364.
4.Wood, Z. A., Schroder, E., Harris, J. R., and
Poole, L. B. (2003) Trends Biochem. Sci., 28, 32-40.
5.Zhong, L., and Holmgren, A. (2000) J. Biol.
Chem., 275, 18121-18128.
6.Bjornstedt, M., Hamberg, M., Kumar, S., Xue, J.,
and Holmgren, A. (1995) J. Biol. Chem., 270,
11761-11764.
7.May, J. M., Mendiratta, S., Hill, K. E., and Burk,
R. F. (1997) J. Biol. Chem., 272, 22607-22610.
8.Watabe, S., Makino, Y., Ogawa, K., Hiroi, T.,
Yamamoto, Y., and Takahashi, S. Y. (1999) Eur. J. Biochem.,
264, 74-84.
9.Powis, G., and Montfort, W. R. (2001) Annu. Rev.
Pharmacol. Toxicol., 41, 261-295.
10.Sadek, C. M., Jimenez, A., Damdimopoulos, A. E.,
Kieselbach, T., Nord, M., Gustalsson, J. A., Spyron, G., Davis, E. C.,
Oko, R., van der Hoor, F. A., and Miranda-Vizuete, A. (2003) J.
Biol. Chem., 278, 13133-13142.
11.Bodnar, J. S., Chatterjee, A., Castellani, L. W.,
Ross, D. A., Ohmen, J., Cavalcoli, J., Wu, C., Dains, K. M., Catanese,
J., Chu, M., Sheth, S. S., Charugundla, K., Demant, P., West, D. B.,
DeJong, P., and Lusis, A. J. (2002) Nat. Genet., 30,
110-116.
12.Haendeler, J., Hoffmann, J., Tischler, V., Berk,
B. C., Zeiher, A. M., and Dimmeler, S. (2002) Nat. Cell Biol.,
4, 743-749.
13.Rubartelli, A., Bajetto, A., Allavena, G.,
Wollman, E., and Sitia, R. (1992) J. Biol. Chem., 267,
24161-24164.
14.Soderberg, A., Sahaf, B., and Rosen, A. (2000)
Cancer Res., 60, 2281-2289.
15.Powis, G., Mustacich, D., and Coon, A. (2000)
Free Rad. Biol. Med., 29, 312-322.
16.Tanudji, M., Hevi, S., and Chuck, S. L. (2003)
Am. J. Physiol. Cell Physiol., 284, C1272-C1279.
17.Rubartelli, A., Bonifaci, N., and Sitia, R.
(1995) Cancer Res., 55, 675-680.
18.Nakamura, H., Herzenberg, L. A., Bai, J., Araya,
S., Kondo, N., Nishinaka, Y., Herzenberg, L. A., and Yodoi, J. (2001)
Proc. Natl. Acad. Sci. USA, 98, 15143-15148.
19.Jikimoto, T., Nishikubo, Y., Koshiba, M.,
Kanagawa, S., Morinobu, S., Morinobu, A., Saura, R., Mizuno, K., Kondo,
S., Toyokuni, S., Nakamura, H., Yodoi, J., and Kumagai, S. (2002)
Mol. Immunol., 38, 765-772.
20.Yamada, Y., Nakamura, H., Adachi, T., Sannohe,
S., Oyamada, H., Kayaba, H., Yodo, J., and Chihara, J. (2003)
Immunol. Lett., 86, 199-205.
21.Sumida, Y., Nakashima, T., Yoh, T., Nakajima, Y.,
Ishikawa, H., Mitsuyoshi, H., Sakamoto, Y., Okanoue, T., Kashima, K.,
Nakamura, H., and Yodoi, J. (2000) J. Hepatol., 33,
616-622.
22.Bertini, R., Howard, O. M., Dong, H. F.,
Oppenheim, J. J., Bizzarri, C., Sergi, R., Caselli, G., Pagliei, S.,
Romines, B., Wilshire, J. A., Mengozzi, M., Nakamura, H., Yodoi, J.,
Pekkari, K., Gurunath, R., Holmgren, A., Herzenberg, L. A., and Ghezzi,
P. (1999) J. Exp. Med., 189, 1783-1789.
23.Spyrou, G., Enmark, E., Miranda-Vizuete, A., and
Gustafsson, J. (1997) J. Biol. Chem., 272, 2936-2941.
24.Damdimopoulos, A. E., Miranda-Vizuete, A.,
Pelto-Huikko, M., Gustafsson, J.-E., and Spyrou, G. J. (2002) J.
Biol. Chem., 277, 33249-33257.
25.Chen, Y., Cai, J., Murphy, T. J., and Jones, D.
P. (2002) J. Biol. Chem., 277, 33242-33248.
26.Rybnikova, E., Damdimopoulos, A. E., Gustafsson,
J. E., Spyrou, G., and Pelto-Huikko, M. (2000) Eur. J.
Neurosci., 12, 1669-1678.
27.Makino, Y., Yoshikawa, N., Okamoto, K., Hirota,
K., Yodoi, J., Makino, I., and Tanaka, H. (1999) J. Biol. Chem.,
274, 3182-3188.
28.Ema, M., Hirota, K., Mimura, J., Abe, H., Yodoi,
J., Sogawa, K., Poellinger, L., and Fujii-Kuriyama, Y. (1999) EMBO
J., 18, 1905-1914.
29.Hirota, K., Matsui, M., Iwata, S., Nishiyama, A.,
Mori, K., and Yodoi, J. (1997) Proc. Natl. Acad. Sci. USA,
94, 3633-3638.
30.Hirota, K., Murata, M., Sachi, Y., Nakamura, H.,
Takeuchi, J., Mori, K., and Yodoi, J. (1999) J. Biol. Chem.,
274, 27891-27897.
31.Didier, C., Kerblat, I., Drouet, C., Favier, A.,
Beani, J. C., and Richard, M. J. (2001) Free Radic. Biol. Med.,
31, 585-598.
32.Wei, S. J., Botero, A., Hirota, K., Bradbury, C.
M., Markovina, S., Laszlo, A., Spitz, D. R., Goswami, P. C., Yodoi, J.,
and Gius, D. (2000) Cancer Res., 60, 6688-6695.
33.Wiesel, P., Foster, L. C., Pellacani, A., Layne,
M. D., Hsieh, C. M., Huggins, G. S., Strauss, P., Yet, S. F., and
Perrella, M. A. J. (2000) J. Biol. Chem., 275,
24840-24846.
34.Ueno, M., Masutani, H., Arai, R. J., Yamauchi,
A., Hirota, K., Sakai, T., Inamoto, T., Yamaoka, Y., Yodoi, J., and
Nikaido, T. (1999) J. Biol. Chem., 274, 35809-35815.
35.Takagi, Y., Horikawa, F., Nozaki, K., Sugino, T.,
Hashimoto, N., and Yodoi, J. (1998) Neurosci. Lett., 251,
25-28.
36.Powis, G., Gasdaska, J. R., and Baker, A. (1997)
Adv. Pharmacol., 38, 329-359.
37.Watson, W. H., Pohl, J., Montfort, W. R.,
Stuchlik, O., Reed, M. S., Powis, G., and Jones, D. P. (2003) J.
Biol. Chem., 278, 33408-33415.
38.Shibata, T., Yamada, T., Ishii, T., Kumazawa, S.,
Nakamura, H., Masutani, H., Yodoi, J., and Uchida, K. (2003) J.
Biol. Chem., 278, 26046-26054.
39.Ren, X., Bjornstedt, M., Shen, B., Ericson, M.
L., and Holmgren, A. (1993) Biochemistry, 32,
9701-9708.
40.Lee, K., Murakawa, M., Takahashi, S., Tsubuki,
S., Kawashima, S., Sakamaki, K., and Yonehara, S. (1998) J. Biol.
Chem., 273, 19160-19166.
41.Holmgren, A., and Fagerstedt, M. (1982) J.
Biol. Chem., 257, 6926-6930.
42.Fernando, M. R., Nanri, H., Yoshitake, S.,
Nagata-Kuno, K., and Minakami, S. (1992) Eur. J. Biochem.,
209, 917-922.
43.Das, K. C., Lewis-Molock, Y., and White, C. W.
(1997) Am. J. Respir. Cell. Mol. Biol., 17, 713-726.
44.Das, K. C., Guo, X. L., and White, C. W. (1999)
Am. J. Physiol., 276, L530-L539.
45.Nkabyo, Y. S., Ziegler, T. R., Gu, L. H., Watson,
W. H., and Jones, D. P. (2002) Am. J. Physiol. Gastrointest. Liver
Physiol., 283, G1352-G1359.
46.Watson, W. H., and Jones, D. P. (2003) FEBS
Lett., 543, 144-147.
47.Sasada, T., Iwata, S., Sato, N., Kitaoka, Y.,
Hirota, K., Nakamura, K., Nishiyama, A., Taniguchi, Y., Takabayashi,
A., and Yodoi, J. (1996) J. Clin. Invest., 97,
2268-2276.
48.Andoh, T., Chock, P. B., and Chiueh, C. C. (2002)
J. Biol. Chem., 277, 9655-9660.
49.Gon, Y., Sasada, T., Matsui, M., Hashimoto, S.,
Takagi, Y., Iwata, S., Wada, H., Horie, T., and Yodoi, J. (2001)
Life Sci., 68, 1877-1888.
50.Shioji, K., Kishimoto, C., Nakamura, H.,
Masutani, H., Yuan, Z., Oka, S.-I., and Yodoi, J. (2002)
Circulation, 106, 1403-1409.
51.Tanaka, T., Nakamura, H., Nishiyama, A., Hosoi,
F., Masutani, H., Wada, H., and Yodoi, J. (2000) Free Radic.
Res., 33, 851-855.
52.Rhee, S. G., Kang, S. W., Chang, T.-S., Jeong,
W., and Kim, K. (2001) IUBMB Life, 52, 35-41.
53.Norberg, J., and Arner, E. S. J. (2001) Free
Radic. Biol. Med., 31, 1287-1312.
54.Das, K. C., and Das, C. K. (2000) Biochem.
Biophys. Res. Commun., 277, 443-447.
55.Arner, E. S. J., and Holmgren, A. (2000) Eur.
J. Biochem., 267, 6102-6109.
56.Matsui, M., Oshima, M., Oshima, H., Takaku, K.,
Maruyama, T., Yodoi, J., and Taketa, M. M. (1996) Develop.
Biol., 178, 179-185.
57.Nonn, L., Williams, R. R., Erickson, R. P., and
Powis, G. (2003) Mol. Cell. Biol., 23, 916-922.
58.Park, J., and Churchich, J. E. (1992) FEBS
Lett., 310, 1-4.
59.Bodenstein, J., and Follmann, H. (1991) Z.
Naturforsch. (C), 46, 270-279.
60.Pedrajas, J. R., Miranda-Vizuete, A., Javanmardy,
N., Gustafsson, J. A., and Spyrou, G. (2000) J. Biol. Chem.,
275, 16296-16301.
61.Reddy, P. G., Bhuyan, D. K., and Bhuyan, K. C.
(1999) Biochem. Biophys. Res. Commun., 265, 345-349.
62.Holmgren, A., and Bjornstedt, M. (1995) Meth.
Enzymol., 252, 199-208.
63.Nakamura, H., Masutani, H., and Yodoi, J. (2002)
Antioxid. Redox Signal., 4, 455-464.
64.Miyamoto, S., Kawano, H., Sakamoto, T., Soejima,
H., Kajiwara, I., Hokamaki, J., Hirai, N., Sugiyama, S., Yoshimura, M.,
Yasue, H., Nakamura, H., Yodoi, J., and Ogawa, H. (2004) Antioxid.
Redox Signal., 6, 75-80.
65.Miyamoto, S., Kawano, H., Takazoe, K., Soejima,
H., Sakamoto, T., Hokamaki, J., Yoshimura, M., Nakamura, H., Yodoi, J.,
and Ogawa, H. (2004) Thromb. Res., 113, 345-351.
66.Rundlof, A. K., and Arner, E. S. J. (2004)
Antioxid. Redox Signal., 6, 41-52.
67.Mitsui, A., Hamuro, J., Nakamura, H., Kondo, N.,
Hirabayashi, Y., Ishizaki-Koizumi, S., Hirakawa, T., Inoue, T., and
Yodoi, J. (2002) Antioxid. Redox Signal., 4, 693-696.
68.Takagi, Y., Mitsui, A., Nishiyama, A., Nozaki,
K., Sono, H., Gon, Y., Hashimoto, N., and Yodoi, J. (1999) Proc.
Natl. Acad. Sci. USA, 96, 4131-4136.
69.Hoshino, T., Nakamura, H., Okamoto, M., Kato, S.,
Araya, S., Nomiyama, K., Oizumi, K., Young, H. A., Aizawa, H., and
Yodoi, J. (2003) Am. J. Respir. Crit. Care Med., 168,
1075-1083.
70.Nakamura, H., Tamura, S., Watanabe, I., Iwasaki,
T., and Yodoi, J. (2002) Immunol. Lett., 82, 165-170.
71.Kasuno, K., Nakamura, H., Ono, T., Muso, E., and
Yodoi, J. (2003) Kidney Int., 64, 1273-1282.
72.Das, K. C. (2004) Antioxid. Redox Signal.,
6, 177-184.
73.Kondo, N., Ishii, Y., Kwon, Y. W., Tanito, M.,
Horita, H., Nishinaka, Y., Nakamura, H., and Yodoi, J. (2004) J.
Immunol., 172, 442-448.
74.Lowenstein, C. J. (2004) Circulation,
110, 1178-1179.
75.Hattori, I., Takagi, Y., Nakamura, H., Nozaki,
K., Bai, J., Kondo, N., Sugino, T., Nishimura, M., Hashimoto, N., and
Yodoi, J. (2004) Antioxid. Redox Signal., 6, 81-87.
76.Tanito, M., Nakamura, H., Kwon, Y. W., Teratani,
A., Masutani, H., Shioji, K., Kishimoto, C., Ohira, A., Horie, R., and
Yodoi, J. (2004) Antioxid. Redox Signal., 6, 89-97.
77.Ott, M., Robertson, J. D., Gogvadze, V.,
Zhivotovsky, B., and Orrenius, S. (2002) Proc. Natl. Acad. Sci.
USA, 99, 1259-1263.
78.Ly, J. D., Grubb, D. R., and Lawen, A. (2003)
Apoptosis, 8, 115-128.
79.Tanaka, T., Hosoi, F., Yamaguchi-Iwai, Y.,
Nakamura, H., Masutani, H., Ueda, S., Nishiyama, A., Takeda, S., Wada,
H., Spyrou, G., and Yodoi, J. (2002) EMBO J., 21,
1695-1703.
80.Wang, X. (2001) Genes Dev., 15,
2922-2933.
81.Ueda, S., Nakamura, H., Masutani, H., Sasada, T.,
Yonehara, S., Takabayashi, A., Yamaoka, Y., and Yodoi, J. (1998) J.
Immunol., 161, 6689-6695.
82.Grogan, T. M., Fenoglio-Prieser, C., Zeheb, R.,
Bellamy, W., Frutiger, Y., Vela, E., Stemmerman, G., Macdonald, J.,
Richter, L., Gallegos, A., and Powis, G. (2000) Hum. Pathol.,
31, 475-481.
83.Freemerman, A. J., and Powis, G. (2000)
Biochem. Biophys. Res. Commun., 274, 136-141.
84.Saitoh, M., Nishitoh, H., Fujii, H., Takeda, K.,
Tobiume, K., Sawada, Y., Kawabata, M., Miyazono, K., and Ichijo, H.
(1998) EMBO J., 17, 2596-2606.
85.Tobiume, K., Matsuzawa, A., Takahashi, T.,
Nishitoh, H., Morita, K.-I., Takeda, K., Minowa, O., Miyazono, K.,
Noda, T., and Ichijo, H. (2001) EMBO Rep., 2,
222-228.
86.Davis, R. J. (1994) Trends Biochem. Sci.,
19, 470-473.
87.Waskiewicz, A. J., and Cooper, J. A. (1995)
Curr. Opin. Cell. Biol., 7, 798-805.
88.Widmann, C., Gibson, S., Jarpe, M. B., and
Johnson, G. L. (1999) Physiol. Rev., 79, 143-180.
89.Ichijo, H., Nishida, E., Irie, K., Dijke, P. T.,
Saitoh, M., Moriguchi, T., Takagi, M., Matsumoto, K., Miyazono, K., and
Gotoh, Y. (1997) Science, 275, 90-94.
90.Tobiume, K., Inage, T., Takeda, K., Enomoto, S.,
Miyazono, K., and Ichijo, H. (1997) Biochem. Biophys. Res.
Commun., 239, 905-910.
91.Geleziunas, R., Xu, W., Takeda, K., Ichijo, H.,
and Greene, W. C. (2001) Nature, 410, 834-838.
92.Sumbayev, V. V. (2003) Arch. Biochem.
Biophys., 415, 133-136.
93.Liu, Y., and Min, W. (2002) Circ. Res.,
90, 1259-1266.
94.Qin, J., Clore, G. M., Kennedy, W. M., Huth, J.
R., and Gronenborn, A. M. (1995) Structure, 3,
289-297.
95.Wang, P. F., Veine, D. M., Ahn, S. H., and
Williams, C. H., Jr. (1996) Biochemistry, 35,
4812-4819.
96.Nishiyama, A., Masutani, H., Nakamura, H.,
Nishinaka, Y., and Yodoi, J. (2001) IUBMB Life, 52,
29-33.
97.Allen, R. G. (1998) Age, 21,
47-76.
98.Gabbita, S. P., Robinson, K. A., Stewart, C. A.,
Foyd, R. A., and Hensley, K. (2000) Arch. Biochem. Biophys.,
376, 1-13.
99.Forman, H. J., Torres, M., and Fukuto, J. (2002)
Mol. Cell. Biochem., 234/235, 49-62.
100.Haddad, J. J. (2002) Cell. Signal.,
14, 879-897.
101.Aggarwal, B. B. (2000) Biochem.
Pharmacol., 60, 1033-1039.
102.Grippo, J. F., Holmgren, A., and Pratt, W. B.
(1985) J. Biol. Chem., 260, 93-97.
103.Hayashi, S., Hajiro-Nakanishi, K., Makino, Y.,
Eguchi, H., Yodoi, J., and Tanaka, H. (1997) Nucleic Acids Res.,
25, 4035-4040.
104.Kim, Y.-C., Yamaguchi, Y., Kondo, N., Masutani,
H., and Yodoi, J. (2003) Oncogene, 22, 1860-1865.
105.Shaulian, E., and Karin, M. (2002) Nature
Cell Biol., 4, E131-E136.
106.Sheikh, M. S., and Fornace, A. J. (2000) J.
Cell. Physiol., 182, 171-181.
107.Floher, L., Brigelius-Floher, R., Saliou, C.,
Traber, M. G., and Packer, L. (1997) Free Radic. Biol. Med.,
22, 1115-1126.
108.Rupec, R. A., and Baeurle, P. A. (1995) Eur.
J. Biochem., 234, 632-640.
109.Das, K. C. (2001) J. Biol. Chem.,
276, 4662-4670.
110.Hirano, M., Osada, S., Aoki, T., Hirai, S.,
Hosaka, M., Inoue, J., and Ohno, S. (1996) J. Biol. Chem.,
271, 13234-13238.
111.Stewart, Z. A., and Pietenpol, J. A. (2001)
Chem. Res. Toxicol., 14, 243-263.
112.Hainaut, P., and Mann, K. (2001) Antiox.
Redox Signal., 3, 611-623.
113.Nguyen, T., Huang, H. C., and Pickett, C. B.
(2000) J. Biol. Chem., 275, 15466-15473.
114.Dinkova-Kostova, A. T., Holtzclaw, W. D., Cole,
R. N., Itoh, K., Wakabayashi, N., Katoh, Y., Yamamoto, M., and Talalay,
P. (2002) Proc. Natl. Acad. Sci. USA, 99,
11908-11913.
115.Lyakhovich, V. V., Vavilin, V. A., Zenkov, N.
K., and Menshchikova, E. B. (2006) Biochemistry (Moscow),
71, 962-974.
116.Tanito, M., Masutani, H., Nakamura, H., Oka,
S., Ohira, A., and Yodoi, J. (2002) Neurosci. Lett., 326,
142-146.
117.Srivastava, S., Watowich, S. J., Petrash, J.
M., Srivastava, S. K., and Bhatnagar, A. (1999) Biochemistry,
38, 42-54.
118.Uchida, K., Kanematsu, M., Sakai, K., Matsuda,
T., Hattori, N., Mizuno, Y., Suzuki, D., Miyata, T., Noguchi, N., Niki,
E., and Osawa, T. (1998) Proc. Natl. Acad. Sci. USA, 95,
4882-4887.
119.Anderson, M. M., Hazen, S. L., Hsu, F. F., and
Heinecke, J. W. (1997) J. Clin. Invest., 99, 424-432.
120.Ghilarducci, D. P., and Tjeerdema, R. (1995)
Rev. Environ. Contam. Toxicol., 144, 95-146.
121.Horton, N. D., Mamiya, B. M., and Kehrer, J. P.
(1997) Toxicology, 122, 111-122.
122.Ramu, K., Perry, C. S., Ahme, T., Pakenham, G.,
and Kehrer, J. P. (1996) Toxicol. Appl. Pharmacol., 140,
487-498.
123.Kehrer, J. P., and Biswal, S. S. (2000)
Toxicol. Sci., 57, 6-15.
124.Horton, N. D., Biswal, S. S., Corrigan, L. L.,
Bratta, J., and Kehrer, J. P. (1999) J. Biol. Chem., 274,
9200-9206.
125.Watson, W. H., Yang, X., Choi, Y. E., Jones, D.
P., and Kehrer, J. P. (2004) Toxicol. Sci., 78, 3-14.
126.Li, L., Hamilton, R. F., and Holian, A. (1999)
Am. J. Physiol., 277, L550-L557.
127.Kumar, S., Rabson, A. B., and Gelinas, C.
(1992) Mol. Cell. Biol., 12, 3094-3106.
128.Biswal, S., Acquaah-Mensah, G., Datta, K., Wu,
X., and Kehrer, J. P. (2002) Chem. Res. Toxicol., 15,
180-186.
129.Biswal, S., Maxwell, T., Rangasamy, T., and
Kehrer, J. (2003) Carcinogenesis, 24, 1401-1406.
130.Holmgren, A. (1989) J. Biol. Chem.,
264, 13963-13966.
131.Gromer, S., Schirmer, R. H., and Becker, K.
(1999) Redox Rep., 4, 221-228.
132.Holmgren, A. (2000) Antioxid. Redox
Signal., 2, 811-820.
133.Miranda-Vizuete, A., Damdimopoulos, A. E., and
Spyrou, G. (1999) Biochim. Biophys. Acta, 1447,
113-118.
134.Sun, Q. A., Wu, Y., Zappacosta, F., Jeang, K.
T., Lee, B. J., Hatfield, D. L., and Gladyshev, V. N. (1999) J.
Biol. Chem., 274, 24522-24530.
135.Sun, Q. A., Kirnarsky, L., Sherman, S., and
Gladyshev, V. N. (2001) Proc. Natl. Acad. Sci. USA, 98,
3673-3678.
136.Rundlof, A. K., Carlsten, M., Giacobini, M. M.,
and Arner, E. S. (2000) Biochem. J., 347, 661-668.
137.Sun, Q. A., Zappacosta, F., Factor, V. M.,
Wirth, P. J., Hatfield, D. L., and Gladyshev, V. N. (2001) J. Biol.
Chem., 276, 3106-3114.
138.Kanzok, S. M., Fechner, A., Bauer, H.,
Ulschmid, J. K., Muller, H. M., Botella-Munoz, J., Schneuwly, S.,
Schirmer, R., and Becker, K. (2001) Science, 291,
643-646.
139.Holmgren, A. (1977) J. Biol. Chem.,
252, 4600-4606.
140.Gladyshev, V. N., Jeang, K. T., and Stadtman,
T. C. (1996) Proc. Natl. Acad. Sci. USA, 93,
6146-6151.
141.Zhong, L., Arner, E. S. J., Ljung, J., Aslund,
F., and Holmgren, A. (1998) J. Biol. Chem., 273,
8581-8591.
142.Lee, S. R., Bar-Noy, S., Kwon, J., Levine, R.
L., Stadtman, T. C., and Rhee, S. G. (2000) Proc. Natl. Acad. Sci.
USA, 97, 2521-2526.
143.Zhong, L., Arner, E. S. J., and Holmgren, A.
(2000) Proc. Natl. Acad. Sci. USA, 97, 5854-5859.
144.Menshchikova, E. B., Lankin, V. Z., Zenkov, N.
K., Bondar', I. A., Krugovykh, N. F., and Trufakin, V. A. (2006)
Oxidative Stress. Prooxidants and Antioxidants [in Russian],
Slovo, Moscow.
145.Hill, K. E., McCollum, G. W., Boeglin, M. E.,
and Burk, R. F. (1997) Biochem. Biophys. Res. Commun.,
234, 293-295.
146.Marcocci, L., Flohe, L., and Packer, L. (1997)
Biofactors, 6, 351-358.
147.Berggren, M., Gallegos, A., Gasdaska, J., and
Powis, G. (1999) Anticancer Res., 17, 3377-3380.
148.Sandalova, T., Zhong, L., Lindqvist, Y.,
Holmgren, A., and Schneider, G. (2001) Proc. Natl. Acad. Sci.
USA, 98, 9533-9538.
149.Arscott, L. D., Gromer, S., Schirmer, R. H.,
Becker, K., and Williams, C. H., Jr. (1997) Proc. Natl. Acad. Sci.
USA, 94, 3621-3626.
150.Williams, C. H. (1992) Chemistry and
Biochemistry of Flavoenzymes (Muller, F., ed.) Vol. 3, CRC Inc.,
Boca Raton, FL, pp. 121-211.
151.Gromer, S., Wissing, J., Behne, D., Ashman, K.,
Schirmer, R. H., Flohe, L., and Becker, K. (1998) Biochem. J.,
332, 591-592.
152.Nordberg, J., Zhong, L., Holmgren, A., and
Arner, E. S. J. (1998) J. Biol. Chem., 273,
10835-10842.
153.Kanzok, S. M., Schirmer, R. H., Turbachova, I.,
Iozef, R., and Becker, K. (2000) J. Biol. Chem., 275,
40180-40186.
154.Ferrari, D. M., and Soling, H. D. (1999)
Biochem. J., 339, 1-10.
155.Lundstrom, J., and Holmgren, A. (1990) J.
Biol. Chem., 265, 9114-9120.
156.Lundstrom-Ljung, J., Birnbach, U., Rupp, K.,
Soling, H. D., and Holmgren, A. (1995) FEBS Lett., 357,
305-308.
157.Andersson, M., Holmgren, A., and Spyrou, G.
(1996) J. Biol. Chem., 271, 10116-10120.
158.Holmgren, A., and Lyckeborg, C. (1980) Proc.
Natl. Acad. Sci. USA, 77, 5149-5152.
159.Luthman, M., and Holmgren, A. (1982)
Biochemistry, 21, 6628-6633.
160.Buettner, G. R. (1993) Arch. Biochem.
Biophys., 300, 535-543.
161.Chopra, M., and Thurnham, D. I. (1999) Proc.
Nutr. Soc., 58, 663-671.
162.May, J. M., Mendiratta, S., Qu, Z. C., and
Loggins, E. (1999) Free Radic. Biol. Med., 26,
1513-1523.
163.Tamura, T., Gladyshev, V., Liu, S., and
Stadtman, T. C. (1995/1996) Biofactors, 5, 99-102.
164.Packer, L., Witt, E. H., and Tritschler, H. J.
(1995) Free Radic. Biol. Med., 19, 227-250.
165.Kagan, V. E., Shvedova, A., Serbinova, E.,
Khan, S., Swanson, C., Powell, R., and Packer, L. (1992) Biochem.
Pharmacol., 44, 1637-1649.
166.Teichert, J., and Preiss, R. (1992) Int. J.
Clin. Pharmacol. Ther. Toxicol., 30, 511-512.
167.Constantinescu, A., Pick, U., Handelman, G. J.,
Haramaki, N., Han, D., Podda, M., Tritschler, H. J., and Packer, L.
(1995) Biochem. Pharmacol., 50, 253-261.
168.Pick, U., Haramaki, N., Constantinescu, A.,
Handelman, G. J., Tritschler, H. J., and Packer, L. (1995) Biochem.
Biophys. Res. Commun., 206, 724-730.
169.Kumar, S., Bjornstedt, M., and Holmgren, A.
(1992) Eur. J. Biochem., 207, 435-439.
170.Bjornstedt, M., Xue, J., Huang, W., Akesson,
B., and Holmgren, A. (1994) J. Biol. Chem., 269,
29382-29384.
171.Ganther, H. E. (1971) Biochemistry,
10, 4089-4098.
172.Bjornstedt, M., Kumar, S., and Holmgren, A.
(1992) J. Biol. Chem., 267, 8030-8034.
173.Bjornstedt, M., Kumar, S., Bjorkhem, L.,
Spyrou, G., and Holmgren, A. (1997) Biomed. Environ. Sci.,
10, 271-279.
174.Manevich, Y., and Fisher, A. B. (2005) Free
Radic. Biol. Med., 38, 1422-1432.
175.Fujii, J., and Ikeda, Y. (2002) Redox
Rep., 7, 123-130.
176.Manevich, Y., Feinstein, S., and Fisher, A. B.
(2004) Proc. Natl. Acad. Sci. USA, 101, 3780-3785.
177.Chance, B., Sies, H., and Boveris, A. (1979)
Physiol. Rev., 59, 527-605.
178.Scandalios, J. G., Guan, L., and Polidoros, A.
N. (1997) Oxidative Stress and the Molecular Biology of Antioxidant
Defenses (Scandalios, J. G., ed.) Cold Spring Harbor Laboratory
Press, Cold Spring Harbor, New York, pp. 343-406.
179.Moore, R. B., Mankad, M. V., Shriver, S. K.,
Mankad, V. N., and Plishker, G. A. (1991) J. Biol. Chem.,
266, 18964-18968.
180.Neumann, C. A., Krause, D. S., Carman, C. V.,
Das, S., Dubey, D. P., Abraham, J. L., Bronson, R. T., Fujiwara, Y.,
Orkin, S. H., and VanEtten, R. A. (2003) Nature, 424,
561-565.
181.Lee, T. H., Kim, S. U., Yu, S. L., Kim, S. H.,
Park, D. S., Moon, H. B., Dho, S. H., Kwon, K. S., Kwon, H. J., Han, Y.
H., Jeong, S., Kang, S. W., Shin, H. S., Lee, K. K., Rhee, S. G., and
Yu, D. Y. (2003) Blood, 101, 5033-5038.
182.Wang, X., Phelan, S. A., Forsman-Semb, K.,
Taylor, E. F., Petros, C., Brown, A., Lerner, C. P., and Paigen, B.
(2003) J. Biol. Chem., 278, 25179-25190.
183.Rhee, S. G., Chang, T. S., Bae, Y. S., Lee, S.
R., and Kang, S. W. (2003) J. Am. Soc. Nephrol., 14,
S211-S215.
184.Thannickal, V. J., and Fanburg, B. L. (2000)
Am. J. Physiol. Lung Cell. Mol. Physiol., 279,
L1005-L1028.
185.Kang, S. W., Chae, H. Z., Seo, M. S., Kim, K.,
Baines, I. C., and Rhee, S. G. (1998) J. Biol. Chem.,
273, 6297-6302.
186.Kang, S. W., Chang, T. S., Lee, T. H., Kim, E.
S., Yu, D. Y., and Rhee, S. G. (2004) J. Biol. Chem.,
279, 2535-2543.
187.Shau, H., Huang, A. C., Faris, M., Nazarian,
R., DeVellis, J., and Chen, W. (1998) Biochem. Biophys. Res.
Commun., 249, 683-686.
188.Kim, H., Lee, T. H., Park, E. S., Suh, J. M.,
Park, S. J., Chung, H. K., Kwon, O. Y., Kim, Y. K., Ro, H. K., and
Shong, M. (2000) J. Biol. Chem., 275, 18266-18270.
189.Wood, Z. A., Poole, L. B., and Karplus, P. A.
(2003) Science, 300, 650-653.
190.Jin, D. Y., Chae, H. Z., Rhee, S. G., and
Jeang, K. T. (1997) J. Biol. Chem., 272, 30952-30961.
191.Li, B., Ishii, T., Tan, C. P., Soh, J. W., and
Goff, S. P. (2002) J. Biol. Chem., 277, 12418-12422.
192.Chang, J. W., Jeon, H. B., Lee, J. H., Yoo, J.
S., Chun, J. S., Kim, J. H., and Yoo, Y. J. (2001) Biochem. Biophys.
Res. Commun., 289, 507-512.
193.Mizusawa, H., Ishii, T., and Bannai, S. (2000)
Neurosci. Lett., 283, 57-60.
194.Lee, S. C., Chae, H. Z., Lee, J. E., Kwon, B.
D., Lee, J. B., Won, Y. H., Ahn, K. Y., and Kim, Y. P. (2000) J.
Invest. Dermatol., 115, 1108-1114.
195.Bast, A., Wolf, G., Oberbaumer, I., and
Walther, R. (2002) Diabetologia, 45, 867-876.
196.Kinnula, V. L., Lehtonen, S., Kaarteenaho-Wiik,
R., Lakari, E., Paakko, P., Kang, S. W., Rhee, S. G., and Soini, Y.
(2002) Thorax, 57, 157-164.
197.Novoselov, S. V., Peshenko, I. V., Popov, V.
I., Novoselov, V. I., Bystrova, M. F., Evdokimov, V. J., Kamzalov, S.
S., Merkulova, M. I., Shuvaeva, T. M., Lipkin, V. M., and Fesenko, E.
E. (1999) Cell Tissue Res., 298, 471-480.
198.Chuchalin, A. G., Novoselov, V. I., Shifrina,
O. N., Soodaeva, S. K., Yanin, V. A., and Barishnikova, L. M. (2003)
Respir. Med., 97, 147-151.
199.Immenschuh, S., Baumgart-Vogt, E., Tan, M.,
Iwahara, S., Ramadori, G., and Fahimi, H. D. (2003) J. Histochem.
Cytochem., 51, 1621-1631.
200.Banmeyer, I., Marchand, C., Verhaeghe, C.,
Vucic, B., Rees, J. F., and Knoops, B. (2004) Free Radic. Biol.
Med., 36, 65-77.
201.Seo, M. S., Kang, S. W., Kim, K., Baines, I.
C., Lee, T. H., and Rhee, S. G. (2000) J. Biol. Chem.,
275, 20346-20354.
202.Ishii, T., Yamada, M., Sato, H., Matsue, M.,
Taketani, S., Nakayama, K., Sugita, Y., and Bannai, S. (1993) J.
Biol. Chem., 268, 18633-18636.
203.Immenschuh, S., and Baumgart-Vogy, E. (2005)
Antioxid. Redox Signal., 7, 768-777.
204.Immenschuh, S., Iwahara, S.-I., Satoh, H.,
Nell, C., Katz, N., and Muller-Eberhard, U. (1995) Biochemistry,
34, 13407-13411.
205.Maines, M. D. (1997) Annu. Rev. Pharmacol.
Toxicol., 37, 517-554.
206.Otterbein, L. E., and Choi, A. M. (2000) Am.
J. Physiol. Lung Cell. Mol. Physiol., 279, L1029-L1037.
207.Siow, R. C. M., Ishii, T., Sato, H., Taketani,
S., Leake, D. S., Sweiry, J. H., Pearson, J. D., Bannai, S., and Mann,
G. E. (1995) FEBS Lett., 368, 239-242.
208.Nakaso, K., Kitayama, M., Mizuta, E., Fukuda,
H., Ishii, T., Nakashima, K., and Yamada, K. (2000) Neurosci.
Lett., 293, 49-52.
209.Kim, H. S., Manevich, Y., Feinstein, S. I.,
Pak, J. H., Ho, Y. S., and Fisher, A. B. (2003) Am. J. Physiol. Lung
Cell. Mol. Physiol., 285, L363-L369.
210.Nguyen, T., Sherratt, P. J., and Pickett, C. B.
(2003) Annu. Rev. Pharmacol. Toxicol., 43, 233-260.
211.Ishii, T., Itoh, K., Takahashi, S., Sato, H.,
Yanagawa, T., Katoh, Y., Bannai, S., and Yamamoto, M. (2000) J.
Biol. Chem., 275, 16023-16029.
212.Hess, A., Wijayanti, N., Neuschafer-Rube, A.
P., Katz, N., Kietzmann, T., and Immenschuh, S. (2003) J. Biol.
Chem., 278, 45419-45434.
213.Gopalakrishna, R., and Jaken, S. (2000) Free
Radic. Biol. Med., 28, 1349-1361.
214.Nishizuka, Y. (1992) Science,
258, 607-614.
215.Immenschuh, S., Stritzke, J., Iwahara, S.-I.,
and Ramadori, G. (1999) Hepatology, 30, 118-127.
216.Chang, T. S., Jeong, W., Choi, S. Y., Yu, S.,
Kang, S. W., and Rhee, S. G. (2002) J. Biol. Chem., 277,
25370-25376.
217.Reynolds, C. M., Meyer, J., and Poole, L. B.
(2002) Biochemistry, 41, 1990-2001.
218.Koo, K. H., Lee, S., Jeong, S. Y., Kim, E. T.,
Kim, H. J., Kim, K., Song, K., and Chae, H. Z. (2002) Arch. Biochem.
Biophys., 397, 312-318.
219.Iwahara, S., Satoh, H., Song, D.-X., Webb, J.,
Burlingame, A. L., Nagae, Y., and Muller-Eberhard, U. (1995)
Biochemistry, 34, 13398-13406.
220.Ishii, T., Kawane, T., Taketani, S., and
Bannai, S. (1995) Biochem. Biophys. Res. Commun., 216,
970-975.
221.Lee, S. P., Hwang, Y. S., Kim, Y. J., Kwon, K.
S., Kim, H. J., Kim, K., and Chae, H. Z. (2001) J. Biol. Chem.,
276, 29826-29832.
222.Sauer, H., Wartenberg, M., and Hescheler, J.
(2001) Cell. Physiol. Biochem., 11, 173-186.
223.Shackelford, R. E., Kaufmann, W. K., and
Paules, R. S. (2000) Free Radic. Biol. Med., 28,
1387-1404.
224.Oberley, T. D. (2002) Am. J. Pathol.,
160, 403-408.
225.Irani, K., Xia, Y., Zweier, J. L., Sollott, S.
J., Der, C. J., Fearon, E. R., Sundaresan, M., Finkel, T., and
Goldschmidt-Clermont, P. J. (1997) Science, 275,
1649-1652.
226.Li, N., and Oberley, T. D. (1998) J. Cell.
Physiol., 177, 148-160.
227.Prosperi, M. T., Ferbus, D., Karczinski, I.,
and Goubin, G. (1993) J. Biol. Chem., 268,
11050-11056.
228.Wen, S.-T., and VanEtten, R. A. (1997) Genes
Dev., 11, 2456-2467.
229.Mu, Z. M., Yin, X. Y., and Prochownik, E. V.
(2002) J. Biol. Chem., 277, 43175-43184.
230.Gu, L., Zheng, H., Murray, S. A., Ying, H., and
Xiao, Z. X. J. (2003) Biochem. Biophys. Res. Commun.,
302, 384-391.
231.Chen, G., and Goeddel, D. V. (2002)
Science, 296, 1634-1635.
232.Schreck, R., Rieber, P., and Baeuerle, P. A.
(1991) EMBO J., 10, 2247-2258.
233.Zhang, P., Liu, B., Kang, S. W., Seo, M. S.,
Rhee, S. G., and Obeid, L. M. (1997) J. Biol. Chem., 272,
30615-30618.
234.Chu, S. H., Lee-Kang, J., Lee, K. H., and Lee,
K. (2003) Pharmacology, 69, 12-19.
235.Kim, Y. J., Lee, W. S., Ip, G., Chae, H. Z.,
Park, E. M., and Park, Y. M. (2006) Cancer Res., 66,
7136-7142.
236.Bushweller, J. H., Billeter, M., Holmgren, A.,
and Wuthrich, K. (1994) J. Mol. Biol., 235,
1585-1597.
237.Nordstrand, K., Aslund, F., Holmgren, A.,
Otting, G., and Berndt, K. D. (1999) J. Mol. Biol., 286,
541-552.
238.Schafer, F. Q., and Buettner, G. R. (2001)
Free Radic. Biol. Med., 30, 238-239.
239.Daily, D., Vlamis-Gardikas, A., Offen, D.,
Mittelmann, L., Melamed, E., Holmgren, A., and Barzilai, A. (2001)
J. Biol. Chem., 276, 21618-21626.
240.Klatt, P., Pineda-Molina, E., DeLacoba, M. G.,
Padilla, A. C., Martinez-Galisteo, E., Barzena, J. A., and Lamas, S.
(1999) FASEB J., 13, 1481-1490.
241.Tew, K. D. (2007) Biochem. Pharmacol.,
73, 1257-1269.
242.Kallis, G. B., and Holmgren, A. (1980) J.
Biol. Chem., 255, 10261-10265.
243.Holmgren, A., and Luthman, M. (1978)
Biochemistry, 17, 4071-4077.
244.Martin, J. L. (1995) Structure,
3, 245-250.
245.Barrett, W. C., DeGnore, J. P., Konig, S.,
Fales, H. M., Keng, Y. F., Zhang, Z. Y., Yim, M. B., and Chock, P. B.
(1999) Biochemistry, 38, 6699-6705.
246.Borges, C. R., Geddes, T. J., Watson, J. T.,
and Kuhn, D. M. (2002) J. Biol. Chem., 277,
48295-48302.
247.Nulton-Persson, A. C., Starke, D. W., Mieyal,
J. J., and Szweda, L. I. (2003) Biochemistry, 42,
4235-4242.
248.Rao, R. K., and Clayton, L. W. (2002)
Biochem. Biophys. Res. Commun., 293, 610-616.
249.Wang, J., Boja, E. S., Tan, W., Tekle, E.,
Fales, H. M., English, S., Mieyal, J. J., and Chock, P. B. (2001) J.
Biol. Chem., 276, 47763-47766.
250.Wang, J., Tekle, E., Oubrahim, H., Mieyal, J.
J., Stadtman, E. R., and Chock, P. B. (2003) Proc. Natl. Acad. Sci.
USA, 100, 5103-5106.
251.Gravina, S. A., and Mieyal, J. J. (1993)
Biochemistry, 32, 3368-3376.
252.Yoshitake, S., Nanri, H., Fernando, M. R., and
Minakami, S. (1994) J. Biochem., 116, 42-46.
253.Ruoppolo, M., Lundstrom-Ljung, J., Talamo, F.,
Pucci, P., and Marino, G. (1997) Biochemistry, 36,
12259-12267.
254.Lundberg, M., Johansson, C., Chandra, J.,
Enoksson, M., Jacobsson, G., Ljung, J., Johansson, M., and Holmgren, A.
(2001) J. Biol. Chem., 276, 26269-26275.
255.Wingert, R. A., Galloway, J. L., Barut, B.,
Foott, H., Fraenkel, P., Axe, J. L., Weber, G. J., Dooley, K.,
Davidson, A. J., Schmid, B., Paw, B. H., Shaw, G. C., Kingsley, P.,
Palis, J., Schubert, H., Chen, O., Kaplan, J., and Zon, L. I. (2005)
Nature, 436, 1035-1039.
256.Lysell, J., Vladic, Y. S., Ciarlo, N.,
Holmgren, A., and Sahlin, L. (2003) Gynecol. Endocrinol.,
17, 303-310.
257.Holmgren, A. (1976) Proc. Natl. Acad. Sci.
USA, 73, 2275-2279.
258.Holmgren, A. (1979) J. Biol. Chem.,
254, 3664-3671.
259.Holmgren, A., Ohlsson, I., and Grankvist, M. L.
(1978) J. Biol. Chem., 253, 430-436.
260.Pineda-Molina, E., Klatt, P., Vazquez, J.,
Marina, A., de Lacoba, M. G., Perez-Sala, D., and Lamas, S. (2001)
Biochemistry, 40, 14134-14142.
261.Takashima, Y., Hirota, K., Nakamura, H.,
Nakamura, T., Akiyama, K., Cheng, F. S., Maeda, M., and Yodoi, J.
(1999) Immunol. Lett., 68, 397-401.
262.Bandyopadhyay, S., Starke, D. W., Mieyal, J.
J., and Gronostajski, R. M. (1998) J. Biol. Chem., 273,
392-397.
263.Hirota, K., Matsui, M., Murata, M., Takashima,
Y., Cheng, F. S., Itoh, T., Fukuda, K., and Yodoi, J. (2000)
Biochem. Biophys. Res. Commun., 274, 177-182.
264.Nakamura, T., Ohno, T., Hirota, K., Nishiyama,
A., Nakamura, H., Wada, H., and Yodoi, J. (1999) Free Radic.
Res., 31, 357-365.
265.Chrestensen, C. A., Starke, D. W., and Mieyal,
J. J. (2000) J. Biol. Chem., 275, 26556-26565.
266.Daily, D., Vlamis-Gardikas, A., Offen, D.,
Mittelmann, L., Melamed, E., Holmgren, A., and Barzilai, A. (2001)
J. Biol. Chem., 276, 1335-1344.
267.Davis, D. A., Newcomb, F. M., Starke, D. W.,
Ott, D. E., Mieyal, J. J., and Yarchoan, R. (1997) J. Biol.
Chem., 272, 25935-25940.
268.Lind, C., Gerdes, R., Schuppe-Koistinen, I.,
and Cotgreave, I. A. (1998) Biochem. Biophys. Res. Commun.,
247, 481-486.
269.Nakamura, H., Bai, J., Nishinaka, Y., Ueda, S.,
Sasada, T., Ohshio, G., Imamura, M., Takabayashi, A., Yamaoka, Y., and
Yodoi, J. (2000) Cancer Detect. Prev., 24, 53-60.
270.Gladyshev, V. N., Liu, A., Novoselov, S. V.,
Krysan, K., Sun, Q. A., Kryukov, V. M., Kryukov, G. V., and Lou, M. F.
(2001) J. Biol. Chem., 276, 30374-30380.
271.Johansson, C., Lillig, C. H., and Holmgren, A.
(2004) J. Biol. Chem., 279, 7537-7543.
272.Beer, S. M., Taylor, E. R., Brown, S. E., Dahm,
C. C., Costa, N. J., Runswick, M. J., and Murphy, M. P. (2004) J.
Biol. Chem., 279, 47939-47951.
273.Taylor, E. R., Hurrell, F., Shannon, R. J.,
Lin, T. K., Hirst, J., and Murphy, M. P. (2003) J. Biol. Chem.,
278, 19603-19611.
274.Holmgren, A. (1979) J. Biol. Chem.,
254, 3672-3678.
275.Lillig, C. H., Berndt, C., Vergnolle, O., Lonn,
M. E., Hudemann, C., Bill, E., and Holmgren, A. (2005) Proc. Natl.
Acad. Sci. USA, 102, 8168-8173.
276.Berndt, C., Hudemann, C., Hanschmann, E. M.,
Axelsson, R., Holmgren, A., and Lillig, C. H. (2007) Antioxid. Redox
Signal., 9, 151-157.
277.Soderdahl, T., Enoksson, M., Lundberg, M.,
Holmgren, A., Ottersen, O. P., Orrenius, S., Bolcsfoldi, G., and
Cotgreave, I. A. (2003) FASEB J., 17, 124-126.
278.Lillig, C. H., Lonn, M. E., Enoksson, M.,
Fernandes, A. P., and Holmgren, A. (2004) Proc. Natl. Acad. Sci.
USA, 101, 13227-13232.
279.Enoksson, M., Fernandes, A. P., Prast, S.,
Lillig, C. H., Holmgren, A., and Orrenius, S. (2005) Biochem.
Biophys. Res. Commun., 327, 774-779.
280.Holmgren, A., Johansson, C., Berndt, C., Lonn,
M. E., Hudemann, C., and Lillig, C. H. (2005) Biochem. Soc.
Trans., 33, 1375-1377.
281.Rodriguez-Manzaneque, M. T., Tamarit, J.,
Belli, G., Ros, J., and Herrero, E. (2002) Mol. Biol. Cell,
13, 1109-1121.
282.Fernandes, A. P., Fladvad, M., Berndt, C.,
Andresen, C., Lillig, C. H., Neubauer, P., Sunnerhagen, M., Holmgren,
A., and Vlamis-Gardikas, A. (2005) J. Biol. Chem., 280,
24544-24552.
283.Muhlenhoff, U., Gerber, J., Richhardt, N., and
Lill, R. (2003) EMBO J., 22, 4815-4825.
284.Molina-Navarro, M. M., Casas, C., Piedrafita,
L., Belli, G., and Herrero, E. (2006) FEBS Lett., 580,
2273-2280.
285.Tamarit, J., Belli, G., Cabiscol, E., Herrero,
E., and Ros, J. (2003) J. Biol. Chem., 278,
25745-25751.
286.Xia, B., Vlamis-Gardikas, A., Holmgren, A.,
Wright, P. E., and Dyson, H. J. (2001) J. Mol. Biol.,
310, 907-918.
287.Berndt, C., Lillig, C. H., and Holmgren, A.
(2007) Am. J. Physiol. Heart Circ. Physiol., 292,
H1227-H1236.
288.Okuda, M., Inoue, N., Azumi, H., Seno, T.,
Sumi, Y., Hirata, K. I., Kawashima, S., Hayashi, Y., Itoh, H., Yodoi,
J., and Yokoyama, M. (2001) Arterioscler. Thromb. Vasc. Biol.,
21, 1483-1487.
289.Nakamura, H., Vaage, J., Valen, G., Padilla, C.
A., Bjornstedt, M., and Holmgren, A. (1998) Free Radic. Biol.
Med., 24, 1176-1186.
290.Lundberg, M., Fernandes, A. P., Kumar, S., and
Holmgren, A. (2004) Biochem. Biophys. Res. Commun., 319,
801-809.
291.Adachi, T., Pimentel, D. R., Heibeck, T., Hou,
X., Lee, Y. J., Jiang, B., Ido, Y., and Cohen, R. A. (2004) J. Biol.
Chem., 279, 29857-29862.
292.Berry-Lane, P. A., Patterson, C., VanDerMerwe,
M., Hu, Z., Holland, S. M., Yeh, E. T. H., and Runge, M. S. (2001)
J. Clin. Invest., 108, 1513-1522.
293.Dong, M., Feng, F. Y., Lin, C., Zhang, X.
Y., Fu, M., Liang, X., Zha, Y. Y., Lu, H. Y., and Wu, M. (2004)
Zhonghua Yi Xie Za Zhi, 84, 323-328.
294.Kalinina, E. V., Chernov, N. N., Saprin, A. N.,
Kotova, Ya. N., Gavrilova, Yu. A., Chermnykh, N. S., and Shcherbak, N.
P. (2007) Byul. Eksp. Biol. Med., 144, 301-303.
295.Kalinina, E. V., Novichkova, M. D., Scherbak,
N. P., Solomka, V. S., and Saprin, A. N. (2001) Adv. Exp. Med.
Biol., 500, 241-244.