REVIEW: TATA Box Polymorphisms in Human Gene Promoters and Associated Hereditary Pathologies
L. K. Savinkova1*, M. P. Ponomarenko1, P. M. Ponomarenko2, I. A. Drachkova1, M. V. Lysova1, T. V. Arshinova1, and N. A. Kolchanov1,2
1Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, ul. Akademika Lavrentieva 10, 630090 Novosibirsk, Russia; fax: (383) 333-1278; E-mail: icg-adm@bionet.nsc.ru2Novosibirsk State University, ul. Pirogova 2, 630090 Novosibirsk, Russia; fax: (383) 339-7101; E-mail: nauka@nsu.ru
* To whom correspondence should be addressed.
Received January 21, 2008; Revision received August 4, 2008
TATA-binding protein (TBP) is the first basal factor that recognizes and binds a TATA box on TATA-containing gene promoters transcribed by RNA polymerase II. Data available in the literature are indicative of admissible variability of the TATA box. The TATA box flanking sequences can influence TBP affinity as well as the level of basal and activated transcription. The possibility of mediated involvement in in vivo gene expression regulation of the TBP interactions with variant TATA boxes is supported by data on TATA box polymorphisms and associated human hereditary pathologies. A table containing data on TATA element polymorphisms in human gene promoters (about 40 mutations have been described), associated with particular pathologies, their short functional characteristics, and manifestation mechanisms of TATA-box SNPs is presented. Four classes of polymorphisms are considered: TATA box polymorphisms that weaken and enhance promoter, polymorphisms causing TATA box emergence and disappearance, and human virus TATA box polymorphisms. The described examples are indicative of the polymorphism-associated severe pathologies like thalassemia, the increased risk of hepatocellular carcinoma, sensitivity to H. pylori infection, oral cavity and lung cancers, arterial hypertension, etc.
KEY WORDS: TATA box, gene expression, SNP, hereditary pathologiesDOI: 10.1134/S0006297909020011
Abbreviations: SNP, single nucleotide polymorphisms (single nucleotide substitutions in genomic DNA); TBP, TATA-binding protein; TF, transcription factor; TPI, triosephosphate isomerase gene; UCP, uncoupling protein.
One of the basic distinctions between transcription processes in
prokaryotes and eukaryotes is that in prokaryotes the preinitiation
complex is formed and DNA is transcribed by a single DNA-dependent RNA
polymerase, while in eukaryotes these functions require three different
enzymes. Also, none of the eukaryotic RNA polymerases is capable of
independent transcription. It requires additional proteins
(transcription factors) the set of which, with the one exception of the
TATA-binding protein (TBP), is different for each enzyme. Each RNA
polymerase transcribes genes of a certain class.
Gene promoters transcribed by RNA polymerase II contain combinations of elements that are commonly supposed to be divided into the core promoter elements and regulatory elements [1-3]. Core promoter elements contain binding sites for general transcription factors (TF) and RNA polymerase II and define the precision in the transcription machinery arrangement relative to the transcription start nucleotide [1]. Core promoter elements of RNA polymerase II include the TATA box [1, 3] located at a distance of about 30 bp from the transcription initiating nucleotide and the initiatory element (Inr element) that occupies the transcription start region [1-7].
Numerous data available in the literature on TATA element variability suggest that promoters with the variant TATA box sequences and different stability of TBP–DNA complexes have a common mechanism of transcription regulation [8-13]. The interaction of the TBP with variant TATA boxes might be directly involved in regulation of gene expression in vivo. This is supported by data on TATA box polymorphisms and associated clinical pathologies in humans.
The goal of this review was to combine data available in the literature on single-nucleotide polymorphisms in and near TATA boxes that are associated with human hereditary diseases with their brief functional characterization. In addition, at the beginning of this review we characterize TATA boxes and the assembly of the basal transcription complex of RNA polymerase II.
CHARACTERISTICS OF TATA ELEMENTS OF RNA POLYMERASE II
PROMOTERS
It is assumed that the TATA-binding protein is the first basal factor that recognizes and binds TATA element within TATA-containing promoters by inducing helix bending by ~80° in the DNA promoter region [14, 15]. The TBP (the TFIID subunit) binding to TATA box induces assembly of the transcription preinitiation complex [14-16], and the TBP–TATA subcomplex is the recognition and binding site for other basal transcription factors and RNA polymerase II (Pol II) [17-19].
In vitro TBP binds a TATA box with rather high affinity (~1 nM), but unlike almost all transcription factors specifically interacting with DNA, it binds DNA via the small groove that, unlike the large one, contains very few specificity determinants [20]. Unlike different TF that are unnecessary for promoters free of their binding sites [21], TBP is necessary for transcription of almost all promoters including those without a TATA box. This is also confirmed by the fact that TBP is included in transcription complexes of all three eukaryotic RNA polymerases. Obviously, the specificity of TBP interactions on TATA-free promoters is achieved using mechanisms of direct and indirect interaction with activator proteins and TBP-associated factors (TAFs), whereas the DNA–protein interaction defines the interaction specificity on the TATA-containing promoters.
Data in the literature are indicative of permissible variability of the TATA box canonical sequence TATAAAA in natural promoters. Only about 30% of TATA-containing promoters of class II genes contain a canonical TATA box. Nevertheless, some TATA-containing promoters can be very sensitive to mutations in TATA elements. Moreover, in the context of different promoters, identical mutations in the TATA box differently influence promoter activity. Thus, it was shown that a nucleotide substitution in the TATA box of the hsp70 gene promoter (TATA→TGTA) results in about tenfold decrease in transcription level, while similar mutations in the his3 gene promoter (TATA→TGTA and TATA→TATG) decrease transcription level almost 100-fold [22, 23].
It was shown [24] that mutations in consensus T1A2T3A4A5A6A7, the functioning of which was analyzed in the context of 25 promoters, differently influence promoter activity. Thus, mutations in positions 1 and 6 and in positions 3 and 6 of the sequence (T1→C, T3→C, A6→G) decreased promoter activities to 30% of the wild type. Mutations resulting in sequences TAAAAAA and TATGAA decreased promoter activities to a non-registered level. Finer mutational effects were registered during comparison of the TATNTA sequence with TATNAA, where N is any nucleotide. When N = C, or G, or T, then T substitution for A5 resulted in double activity, but for N = A the same mutation decreased transcription to 30%. These experiments also show the role of the TATA box flanking sequences, namely, TATA boxes with identical mutations can be good or bad substrates depending on the context of the DNA fragment in which they are located. The TATA element flanking sequences can also influence the TBP affinity to TATA.
It was shown [25] that changes in flanking TATA sequences in MLP promoter lowers the binding constant of TBP compared to that for the natural promoter. The TATA element flanking sequences influence the level of basal transcription and response of activators. Two adenovirus TATA boxes, MLP and E4, were used in [26]. MLP is a strong basal promoter, while E4 is a weak one but highly inducible by activators. The replacement of flanking sequences of the MLP promoter by those of E4 produced MLP with enhanced response to activators and vice versa. Changes in the interaction of TBP with TATA elements depend on flanking sequences to almost the same extent as to the sequences on TATA boxes proper, and the flanking sequence motifs influencing the TBP/TATA interaction are unique for particular TATA boxes [27]. The architecture of a TATA box with flanking sequences is thought to have an important effect on the level of basal and activated transcription.
The exact mechanism of the activated transcription enhancement remains unclear. Besides, sequences flanking the TATA box of MLP promoter are of different significance. The sequence flanking the TATA box from the 5′ end is more conserved and has higher information content for TBP compared to the sequence flanking the TATA box from 3′ end [27].
We obtained similar results concerning the flanking sequence effects on the oligonucleotide affinities to TBP [28]. The recombinant human TBP was used for binding. Experimental determination of equilibrium Kd of TBP complexes with oligonucleotides has shown that the affinity of TBP to oligonucleotides with different AT content in the TATA box flanking sequences differs 25-30-fold. The data show that TATA box flank enrichment with low-melting AT pairs may have a negative effect on biological activity of TATA-containing promoters.
It should be emphasized that the experimental results agree with our previously developed algorithm for prediction of the affinity of TBP to DNA using base sequence of the latter [29]. Computer analysis of known eukaryotic gene promoters, using the algorithm developed on the basis of experimental data on the interaction of TBP with TATA-containing oligonucleotides [29], has shown that TATA boxes are surrounded by GC-enriched sites with lower affinity to TBP compared to that of random DNA sequences. The selection for flanking GC-enrichment during evolution can be explained by the fact that the high-melting GC-rich flanks make the TATA box more distinguishable for TBP during its search by linear diffusion, and they also localize the low-melting region of the TATA element as though they fix local DNA conformation formed upon TBP binding.
The considered single-nucleotide polymorphisms in regulatory gene regions, namely, in TATA elements of promoters, are indicative of their effect on in vivo regulation of human gene expression and on hereditary diseases usually emerging due to changes in this regulation.
CHARACTERISTICS OF FUNCTIONAL TATA BOX POLYMORHISMS IN HUMAN GENE
PROMOTERS
TATA Box Polymorphisms That Weaken Promoters
Globin genes. The highest content of TATA box damage was found in the cluster of human globin genes [30-38] associated with β- and δ-thalassemia caused by insufficient content of the normally structured globin chains or by the presence of defective globin chains.
Analysis of DNA sequence of cloned β-globin gene of Chinese β-thalassemia carriers revealed a single nucleotide substitution –28A>G inside the TATA box [30]. Expression from this gene in HeLa cells produced the 3-5-fold lower amount of β-globin mRNA compared to the normal β-gene. The β-globin RNA content in total RNA, isolated from erythroid cells of β-thalassemia patients, was 10 times lower than the norm. The authors noted that this means approaching of the results obtained on heterologous cells to those in vivo. Cloning of the β-globin gene of Kurdish Jewish β-thalassemia patients also revealed an SNP in the TATA box –28A>C [31] leading to the decrease or complete absence of globin mRNA transcripts (see table section 1).
TATA box single-nucleotide polymorphisms associated with hereditary
predisposition to human diseases
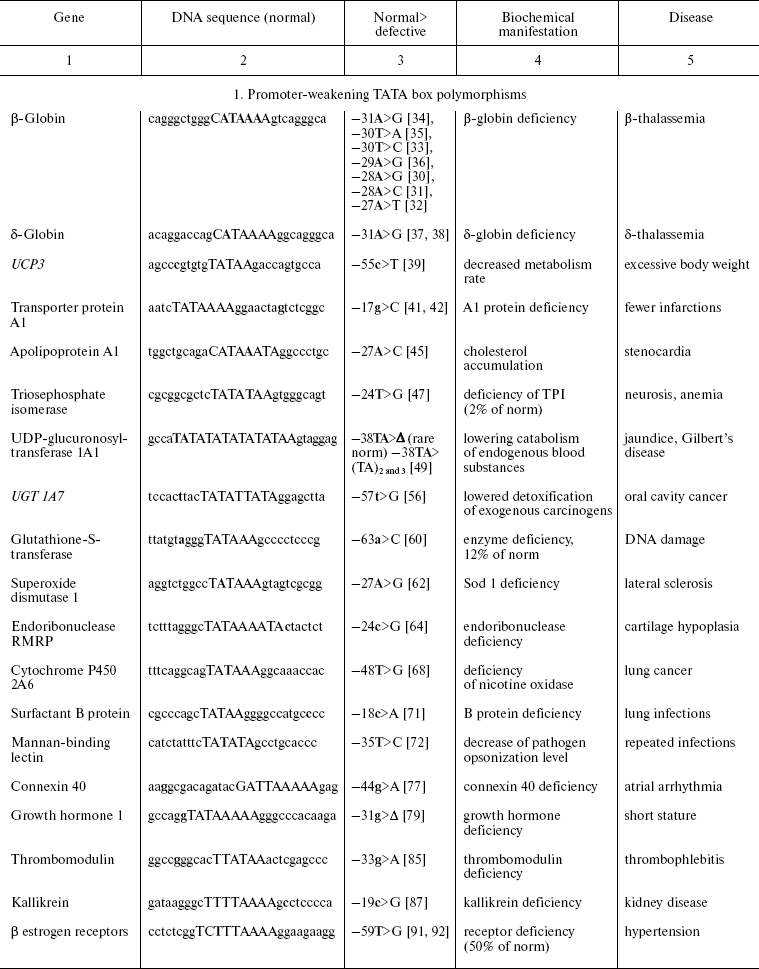
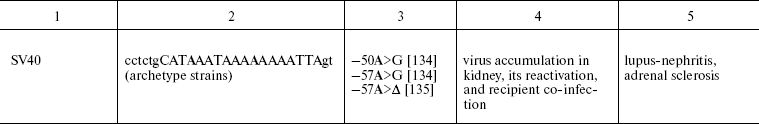
Note: TATA boxes are indicated in upper case; Δ, deletion;
(TA)2 and 3, TATA and TATATA, respectively; [X :
Z], fragment between positions X and Z.
* –82T>C, position numbers of promoter of the CYP
2B6 gene encoding cytochrome P450 2B6 as generally accepted from
translation start [114].
The SNP –27A>T causing β-thalassemia specific of endemics from Corsica Iceland was found [32]. Polymorphism –30T>C transforming the ATAAA sequence to ACAAA was found in β-thalassemia carriers from South China [33].
The SNP –31A>G was also found for the β globin gene [34]. The gene expression in heterologous COS cell line has shown that β-thalassemia was caused by gene transcription lowering by 45% compared to the norm.
Analysis of DNA sequence in β-thalassemia carriers from the Yugoslav population revealed the –30T>A mutation in the TATA box [35], and in this case the level of RNA synthesis was 13% of normal. Within the TATA box of Afro-American β-thalassemia patients a single G for A substitution in position 29 was found, which resulted in transcription of only 25% of the normal RNA level both in heterologous cells and in vivo [36]. Besides, the SNP deteriorated normal RNA splicing, which resulted in formation of RNA that did not encode β globin.
Mutation –31A>G was found in the TATA box of the δ-globin gene resulting in δ-thalassemia phenotype [37, 38]. Carriers of homozygous δ-thalassemia alleles exhibit an unusually low level of HbA2 synthesis.
Gene UCP3. Uncoupling proteins (UCP) are transporters of the inner mitochondrial membrane that decrease the efficiency of oxidative phosphorylation and ATP synthesis [39]. The polymorphism –55C>T at the distance 6 bp upstream from the supposed TATA box influences UCP3 gene expression and stability of its mRNA [39] and decreases the rate of metabolism and accordingly influences body mass index. Predisposition to obesity is more pronounced in carriers of homozygote TT than of CC or CT.
Gene ABCA1. Gene ABCA1 encodes membrane protein A1 transporter of ATP-binding cassette, which transfers various molecules including proteins, lipids, ions, and sugars and is important for reverse transport of cholesterol [40]. An SNP in the promoter region of the ABCA1 gene, –17G>C, located “downstream” from the TATA box, is not associated with change in plasma lipid level; it is able to change the ABCA1 activity only inside the vascular wall and to decrease 3.5-fold [41, 42] the risk of myocardial infarction and other vascular diseases. The exact mechanism of the functional effect of the SNP on ABCA1 expression is still not known.
Gene of apolipoprotein A1. Apolipoprotein A1 (APOA1) plays an important role in the reverse capture of cholesterol; therefore, its low level in the blood serum is a risk factor for stenocardia [43, 44]. Sequencing the APOA1 DNA of patients with the APOA1 protein deficiency revealed a mutation in the TATA box, –27A>C [45]. In carriers of APOA1 heterozygote a 45-55% level of apolipoprotein A1 in the blood plasma was observed, while in carriers of homozygotes the content of this protein did not exceed 10% of that in the wild type.
Gene of triosephosphate isomerase. The triosephosphate isomerase gene (TPI) is expressed in all cell types, being a member of the “housekeeping” genes [46]. Multiple electrophoretic and chromatographic TPI forms have been discovered in all human tissues, but all of them are encoded by a single gene and are the result of posttranslational protein modifications [46]. Molecular characteristics of alleles of Afro-Americans and Caucasians [47] revealed SNP –24T>G in the TATA box-like sequence located at a distance of 24 bp from transcription site. In people having the mutation in the TPI gene promoter, mRNA deficiency is observed, the enzyme activity being 2-10% of the norm, and neurological and muscular changes are characteristic of these patients.
Gene of UDP-glucuronosyltransferase 1A1. The UGT (UDP-glucuronosyltransferase) enzyme superfamily catalyzes catabolism of different endogenous substances including bilirubin and steroid hormones, as well as a number of xenobiotics [48].
A variable number of TA repeats was found in the human gene UGT1A1 promoter, which is inversely proportional to the level of gene expression. It was shown in some works [49-54] that additional TA pairs, (TA)6→(TA)7, in the promoter TATA boxes cause lowering the UGT1A1 gene expression and reduced bilirubin conjugation with serum albumin, resulting in bilirubinemia, or Gilbert’s disease. In this case, the UGT activity in the liver, important for bilirubin excretion, decreases to 30% of the norm.
Investigations among Afro-Americans, Caucasians, and Japanese revealed ethnic difference in UGT1A1 genetic polymorphisms [49]. The rare case with five repeats (TA)5 is mainly characteristic of Afro-Americans, the case with six repeats (TA)6 is most widespread in the Japanese population, and in the case of hyperbilirubinemia the haplotype with (TA)8 is frequent in the latter. The differences in the haplotype distribution among populations suggest the possibility of ethnic distinctions in the extent of enzymatic detoxification of drugs.
Gene UGT1A7. Since UGT1A7, a member of UDP-glucuronosyltransferase superfamily, provides for detoxification of some tobacco carcinogens, the authors of [55] tried to determine whether allele 1A7 variants are associated with the risk of oral cavity cancer. They analyzed UGT1A7 gene expression in different tissues and genotypes of individuals with oral cavity cancer and without it. It was shown that the risk of oral cavity cancer increased 8-10-fold in smokers who are carriers of allele –57T>G.
It was shown during investigation of the gene UGT1A7 promoter variants, important for irinotecan detoxification, that SNP –57T>G located upstream from the TATA box decreases transcription activity of promoter by 70%, which provides for genetic and biochemical grounds for individual drug metabolism including irinotecan [56]. Irinotecan is a cytostatic specifically inhibiting topoisomerase I by preventing intracellular DNA replication [57, 58].
Gene GSTM3. Proteins of the glutathione-S-transferase (GST) superfamily play an important role in cell protection against damaging effects of the environment and influence cell sensitivity to drugs [59]; therefore, low levels of the glutathione-S-transferase M3 gene expression can result in multiple damages to cellular DNA, differences in drug metabolism, and sensitivity to different diseases. In children with low level of gene expression, SNP –63A>C adjacent to the TATA box was revealed [60]. Gene expression in homozygotes –63CC was eight times lower than in –63AA.
Gene of superoxide dismutase 1. Amyotrophic lateral sclerosis is a progressive neurodegenerative disease manifested by gradual development and spreading of muscular atrophy due to damage to corresponding parts of the nervous system [61]. An amyotrophic lateral sclerosis type associated with single-nucleotide polymorphism –27A>G of the TATA box of the Cu/Zn superoxide dismutase 1 gene (SOD1 gene) has been described [62]. This SNP disturbs the canonical sequence of the TATA box of the superoxide dismutase gene promoter and reduces gene expression.
Gene RMRP. Cells of vertebrates contain a site-specific endoribonuclease (RNase MRP; RMRP), a ribonucleoprotein cleaving mitochondrial RNA [63]. The –24C>G polymorphism located next to the TATA box of the RMRP gene promoter, together with other SNP of the gene, leads to lowering the transcript levels and is associated with disturbed development of cartilaginous tissue [64].
Gene of cytochrome P450 2A6. Human cytochrome P450 2A6 (CYP 2A6) is the main nicotine oxidase and plays an important role in activation of some pre-carcinogens and detoxification of many drugs [65]. Gene polymorphisms responsible for individual distinctions in nicotine metabolism in smokers have been described [66, 67]. The SNP –48T>G that in experiment on “pulse” transfection of human B16A2 cells halved the reporter gene expression compared to that from the wild-type promoter was described [68]. The authors found experimentally for the first time the fact of polymorphous destruction of the natural TATA box of the cytochrome P450 superfamily genes. It is significant that the fixed population frequencies of the defective TATA box appeared to be very high: 5.2% in Sweden, 7.2% in Turkey, and 15.7% in China, which is a strong argument in favor this allele being a tumor risk factor in smokers.
Gene SP-B. Surfactant proteins (SP) A, B, C, and D and phospholipids form a surface-active complex preventing collapse of lung alveoli upon expiration [69]. Surfactant protein B (SP-B) is one of two small hydrophobic proteins influencing surface-active properties of phospholipids [70]. Analysis of potential regulatory function of SNP –18C>A in the SP-B gene promoter revealed enhanced promoter activity in the case of the C allele and almost threefold increase in the protein amount [71]. Production of a lower amount of protein is characteristic of carriers of homozygotes with AA allele, which may be the cause of lung diseases.
Gene of mannan-binding lectin 2. Gene MBL2 of mannan-binding lectin 2 encodes C-type lectin serving as a soluble receptor of inborn immune response upon initiation, regulation, and amplification [72, 73]. MBL2 deficiency and its low level in blood serum are the basis for general deficiency of opsonin (a blood plasma protein that binds bacteria and macrophages and thus stimulates their “agglutination”) and are associated with the risk of repeated infections [74-76]. The SNP –35T>C (TATATA→TACATA) in the MBL2 gene promoter TATA box [72], not previously described, leads to disturbance in TFIID binding to the TATA box, lowering the gene expression level, and pathogen opsonization in the organism.
Connexin-40 gene. Two tightly bound SNP, –44G>A and +71A>G, are found in regulatory regions of the connexin-40 gene (Cx-40) of a specific auricle membrane protein [77]. The use of a plasmid carrying a reporter construct with SNP –44A has shown the reduction of gene activity by 65% compared to the wild-type carrying G in this position, which can be responsible for hereditary atrial arrhythmia.
Gene of growth hormone 1. In humans, height depends on a complex interaction of genetic and environmental factors and is also associated with hereditary mutations in the growth hormone gene GH1 [78, 79]. It was shown that deletion of G adjacent to the TATA box lowering gene expression and human stature.
Thrombomodulin gene. Thrombomodulin (TM), a receptor of endothelial cell surface glycoprotein, plays an important role in procoagulant conversion into physiological anticoagulation factor thrombin [80-84]. Examination of 205 patients with thrombophlebitis revealed SNP –33G>A [85] upstream from the TATA box. The use of constructs carrying fragments of the normal promoter and promoter with SNP for transfection of EAhy926 cell line showed lowering transcription activity of the TM gene promoter by more than 60%.
Kallikrein gene. The human kallikrein gene (KLK1) encodes the inactive form prekallikrein that is activated during intracellular proteolysis and is transformed to kinins involved in regulation of renal vascular tonus [86]. SNP –19C>G located 2 bp downstream from the TATA box of the kallikrein gene promoter is rather frequent but is not associated with hypertension, but it leads to alteration of in vitro transcription activity and significantly reduces the enzyme amount, which increases the risk of kidney and vascular system diseases [87].
Gene of β estrogen receptors. Protein of estrogen receptors ER is represented by two isoforms, α and β, which are expressed from promoters ERα and ERβ, respectively. Experiments on ERα- and ERβ-knockout mice have shown that the receptor plays an important role in development of reproductive and non-reproductive sex distinctions [88, 89]. Multiple alignment of 1600 bp fragments of human, chimpanzee, mouse, and rat genomic DNA around known ERβ transcription starts revealed a strict homology region immediately before the ERβ transcription start, and its in vivo methylation was registered [90]. Sequencing of this region in 50 Afro-Americans and 50 Caucasians and comparison of the DNA revealed the –59T>G polymorphism inside the unique TATA-like fragment of this conserved region. Human prostate cancer cells LNCaP were transfected by plasmid pGL3 containing the promoter allele variants –59T or –59G and inserted reporter luciferase gene [91]. Since promoter activity of the rare (1%) allele –59G was two times lower than that of the frequent (99%) –59T, the authors concluded [90, 91] that SNP –59T>G damages the normal TATA box of ER promoter and causes the deficiency of β estrogen receptor. It was shown [92] that SNP –59T>G is also involved in creation of variability of phenotypic reactions to drug therapy, in particular to tamoxifen.
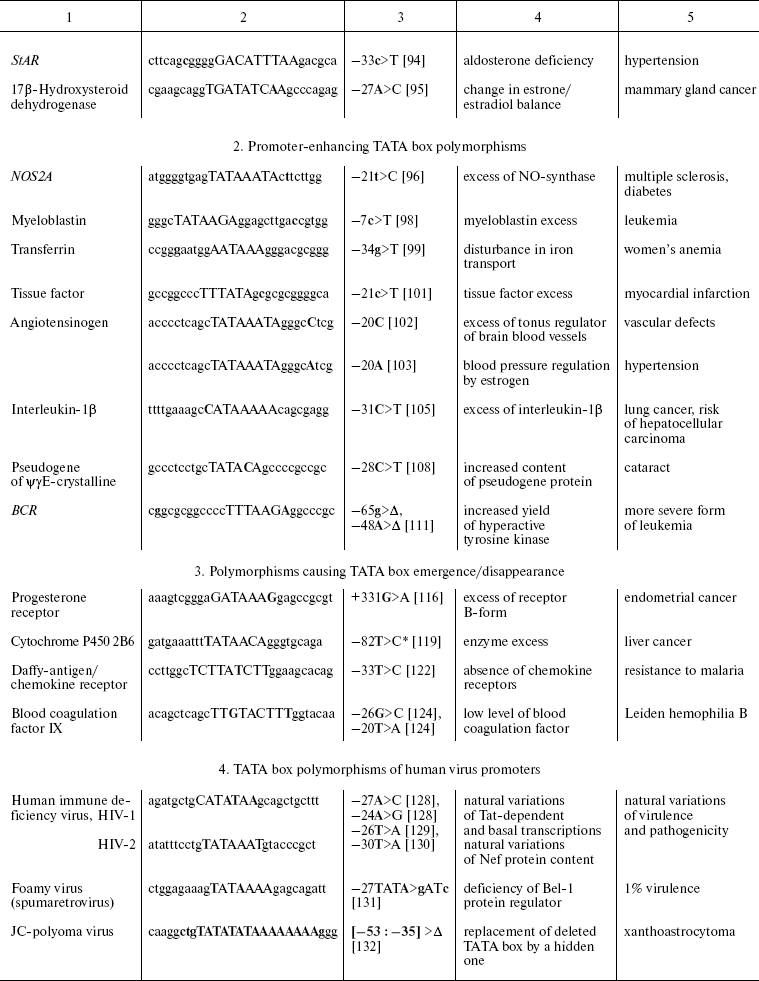
Gene StAR. Cholesterol transfer from the external to internal mitochondrial membrane, where it is cleaved to pregnenolone, is mediated by StAR protein (steroidogenic acute regulatory protein) whose gene has been identified and cloned [93]. Polymorphism –33C>T, found in this gene promoter, influences StAR gene expression regulation and is associated with lowered promoter response to activation by cAMP and aldosterone synthesis activators [94]. These SNP carriers are characterized by reduced response to physiological stimulators (35-40% of the norm), which is associated with hypertension emerging due to change in aldosterone synthesis regulation and predisposition to diabetes.
Gene of hydroxysteroid dehydrogenase. Gene EDH17B2 encodes type 1 17β-hydroxysteroid dehydrogenase (17HSD) that catalyzes the reversible reaction between estrone and estradiol. The involvement of the EDH17B2 gene in the regulation of estrogen balance makes it a likely candidate gene of familial mammary gland and ovary cancers [95]. Examination of patients has shown that point mutation –27A>C in the TATA box of the gene decreases promoter activity by 45% [95] and is responsible for a variant of hereditary mammary gland cancer.
Promoter-Enhancing TATA Box Polymorphisms
Gene NOS2A. It has been shown that a proximal promoter of the nitrogen oxide synthase gene (NO-synthase) NOS2A contains several SNP, including SNP –21T>C adjacent to the TATA box [96] (table section 2). This polymorphism is associated with enhanced sensitivity to various pathologies like multiple sclerosis, cerebral malaria, diabetes, acute respiratory and lung diseases, etc. Examination of such patients revealed enhanced expression of the NO-synthase gene.
Myeloblastin (Wegener’s autoantigen) gene. Determination of base sequence of 5′-terminal region of the proteinase 3 gene (PR3) has shown that its mRNA is synthesized in HL-60 cells and in cells of myeloid leukemia patients, and that myeloblastin and proteinase 3 (also identified as Wegener’s autoantigen) are encoded by the same mRNA [97]. Analysis of the PR3 gene promoter sequence revealed T substitution for C in position –7 resulting in enhanced expression of the PR3 gene whose protein product is considered as a pathogenic factor in degenerative and inflammatory diseases [98].
Transferrin gene. Transferrin (Tf) is the main iron-transporting protein in blood plasma. Polymorphism –34G>T in the transferrin gene promoter between the Sp1 binding site and the TATA box was found, which leads to changes in cellular iron homeostasis causing anemia in women [99].
Tissue factor gene. Tissue factor (TF) is a 47 kDa transmembrane protein expressed in a broad set of tissues including outer blood vessel membrane, where it quickly activates coagulation in response to deterioration of integrity [100]. Studying the effect of –21C>T polymorphism, located next to the TATA box, on the susceptibility to thrombophlebitis and myocardial infarction has shown that this SNP enhances TF gene expression, which can result in increased risk of these diseases [101].
Gene of angiotensinogen. Angiotensinogen is a precursor of one of the most powerful vasoactive hormones, angiotensin II. The human angiotensinogen gene (ANG) contains SNP –20A>C located between the TATA box and the transcription origin. Reporter constructs containing the angiotensinogen gene promoter with C in position –20 have basal activity 2-3-fold exceeding that in the case of allele –20A. Correspondingly, allele –20C with enhanced expression of cerebral angiotensinogen (regulator of blood vessel tonus and lumen) was associated with developmental pathologies of small cerebral blood vessels [102].
When A is in position –20, a binding site for estrogen receptor (ERα) is formed and the expression of the angiotensinogen gene is regulated in a gender-specific way, increasing the risk of hypertension and stroke in women [103].
Gene of interleukin 1β. IL 1β is an antiinflammatory cytokine launching the cascade of inflammatory reactions via induction of genes associated with inflammation and production of reactive free radicals [104]. The gene contains SNP –31C>T at a distance of 31 bp upstream from the transcription origin. The rare (1%) allele –31T contains a canonical TATA box TATAAAAA, unlike the widespread allele with C in position –31, exhibiting weak DNA–protein interactions in vitro [105] that are indicative of the absence of the TATA box. The association between allele –31T and risk of non-small-cell lung carcinoma was found in patients of the Norwegian population [106] (table section 3). A high risk of hepatocellular carcinoma and high sensitivity to H. pylori infection are associated with the same TATA-box-containing allele (–31T) [106, 107].
Gene of ψγE crystalline. Hereditary cataract is a genotypically and phenotypically heterogeneous disease. The γ-crystalline gene cluster (CRYG) is a family of genes encoding the main structural proteins of the crystalline lens [108], but only two genes encode the full-sized protein and four genes encode only its fragments. Human Coppock-like cataract is caused by a mutation within the TATA element of the ψγE gene promoter—TATACA is replaced by TATATA and a more perfect TATA box is formed (SNP –28C>T). This stimulates 10-fold increase in the promoter activity and increase by 30% in the mutant ψγE gene expression over the wild-type pseudogene [108]. The 6 kDa protein product of the pseudogene is an N-terminal fragment of γ-crystalline, the increased content of which causes the lens opacification or cataract.
BCR gene. Chromosomal translocation leading to emergence of Philadelphia chromosome characteristic of chronic myeloid leukemia (CML) fuses the breakpoint cluster region (BCR) of chromosome 22 with protooncogene c-ABL of chromosome 9 [109, 110]. The translocation results in a fused BCR/ABL gene encoding chimeric protein p210, a hyperactive tyrosine kinase [111-113]. Analysis of the human BCR gene encoding chimeric BCR/ABL mRNA revealed a promoter containing the TATA-box-like sequence TTTAA with SNP and deletions –48A and –65G before and after the TATA box [111]; these increase promoter activity, causing enhanced gene expression and more severe chronic myeloid leukemia.
Polymorphisms Causing TATA Box Emergence or Disappearance
Progesterone receptor gene. It is known that in addition to hereditary factors, development of endometrial cancer is stimulated by exogenous and endogenous estrogens [114] not equilibrated by progestins [115]. The antiproliferative effect of progesterone requires progesterone receptor (PR) existing in two isoforms, PR-A and PR-B, and antiproliferative effects depend on stoichiometric balance of these isoforms [114]. Sequencing progesterone gene receptors of endometrial cancer patients revealed six variable regions including SNP +331G>A in the promoter region of the PR-B gene. Using computer modeling, the authors predicted that polymorphism +331G>A creates a potential TATA box in the TATA-free PR-B gene promoter, which also results in emergence of a new transcription initiation site [116]. Biochemical analysis confirmed that this polymorphism enhances gene transcription by stimulation of increased production of PR-B isoform [117]. The disturbance of balance between isoforms doubles the risk of endometrial cancer, especially in women with increased body mass.
Gene of cytochrome P450 2B6. In vitro studies of the liver cytochrome P450 2B6 gene (CYP 2B6) showed a high extent of its individual variability in expression at the level of mRNA and protein syntheses, as well as high catalytic activity [118]. Experimental comparison of known transcription and translation starts of a broad set of the CYP 2B family genes from different organisms and multiple alignment of their genomic sequences have shown that all their unique ATG translation starts coincided at the conserved alignment site, while transcription starts were distributed over a variable 40 bp site at a distance of 12 bp from the translation start [119]. Therefore, it appeared most reasonable in the case of the CYP 2B gene family to introduce a unified position numbering from the ATG translation start (position +1) both for coding (+) and regulatory (–) regions.
Detailed analysis of the CYP 2B6 gene promoter revealed SNP –82T>C that decreases TBP binding to the TATA box and simultaneously increases C/EBP affinity to this newly-formed binding site. This means that the natural TATA box was transformed to a potential binding site for transcription factor C/EBP [119] that threefold increased transcription activity and almost twofold the content of mature mRNA. Due to the CYP 2B6 precarcinogen-activating properties, SNP –82T>C is associated with a protein increasing the probability of liver cancer [119].
Gene DARC. Gene DARC encodes Duffy antigen receptor for chemokines in endothelial cells and erythrocytes [120]. A pathogenic feature of the protein is due to its usage by Plasmodium vivax for penetration into erythrocytes, thus causing malaria [121]. As a rule, at a distance of 30 bp before the erythroid-specific gene transcription site, there is a TATA/GATA box owing to which GATA-1, the main protein regulator of the erythroid system, is involved in the preinitiation complex [122]. SNP –33T>C destroys the TATA/GATA box of the DARC gene, thus preventing the emergence on the erythrocyte surface of a chemokine receptor, a target for Plasmodium vivax, and so causing resistance to malaria in West African aborigines.
Gene of blood coagulation factor IX. Leiden’s hemophilia B is characterized by a low content of blood coagulation factor IX in patients, especially in children, the maximum being 60% of the norm [123].
It was shown [124] that hemophilia in children is associated with two SNP in promoter of the blood coagulation factor gene fIX, –26G>C and –20T>A, destroying the overlapping binding site TATA/HNF-4. HNF-4 is the nuclear hepatocyte factor that regulates expression of factor IX. The inability of HNF-4 to efficiently bind to the altered site results in hemophilia. Experiments with plasmid constructs containing in the promoter SNP –26G>C in one case and SNP –20T>A in the other have shown manifold decrease in basal and HNF-4-induced expression of the reporter gene [124].
TATA Box Polymorphisms of Human Virus Promoters
It is known that RNA- and DNA-containing retroviruses and polyoma viruses integrate their genomes into the host cell DNA upon infection and use the cell apparatus for their own multiplication. This becomes possible due to powerful viral promoters containing a standard TATA box and binding sites for other TF of eukaryotic cells, which are recognized by cell proteins as their own.
Human immune deficiency virus. At present two types of human immune deficiency virus (HIV), HIV-1 and HIV-2, are known; they differ in structure, antigen composition, and epidemiological characteristics [125]. HIV-1 exhibits higher pathogenicity and more pronounced genetic variability compared to HIV-2, and unlike HIV-2, it is widespread over the world, mainly in a number of districts in West Africa [125]. The HIV-1 genome consists of RNA whose DNA copy contains on the 5′ and 3′ termini long terminal repeats (LTR), the prototype of the promoter–enhancer block containing standard eukaryotic TATA box and enhancers, with which cellular DNA-binding proteins (Sp1 and NF-κB, NFAT-1, etc.) interact [126, 127].
It was shown [128] that polymorphisms found in TATA boxes of the promoter subtypes HIV-1 A-D, F-J, –27A>C and –24A>G (table section 4), cause disturbance of basal and Tat-dependent transcription (Tat is the viral gene expression activator) and correspondingly, alteration of the pathogenic potential of the virus.
The TATA box of HIV-1 subtype E contains the TAAAA sequence instead of TATAA (SNP –26T>A) and undergoes inactivation upon its sequence replacement by the canonical one [129].
The role of nef protein (a highly conserved protein of primate lentiviruses involved in replication regulation and immunomodulation) in HIV-2 pathogenesis was studied [130]. The SNP revealed in the promoter the TATA box region (–32T>A) is still not associated with any disease outside of its involvement in change in pathogenicity.
Foamy virus or human spumaretrovirus. Foamy virus (spumaretrovirus) is an exogenous retrovirus (HSRV) that contains a functionally active promoter upstream from the bel-genes. The bel-1 gene product is a trans-activator of the LTR-directed transcription and is absolutely necessary for replication of the virus [131]. It was shown that mutations of the TATA box in the bel-1 gene promoter transform it into the GATC sequence used as transcription start, while the region upstream by 33 bp is used, respectively, as the TATA box, which results in production of proviral clones with infectivity decreased by 100-fold.
JC virus. JC virus is a DNA-containing polyoma viruses (like BK and SV40) inducing brain tumors in experimental animals and in young people [132]. Genomic regions of the JC virus were analyzed in a nine-year-old boy with pleomorphic xanthoastrocytoma (a slowly growing tumor with good prognosis), and it was found that they mutated (a hidden TATA box replaced the deleted one and a GG deletion at –35) relative to the archetypal form oncogenic for animals; just this explains the benign character of these tumors [133].
Virus SV40. After primary infection, SV40, like other polyoma viruses, settles in kidneys and can be reactivated upon lowered immunity and infect the kidney recipient [134, 135]. Sequence analysis of the regulatory regions of the virus has shown that not archetypal SV40 strains are prevalent, but rather strains carrying several mutations in the 5′ regulatory region (–50A>G, –57A>G) which are associated with lupus-nephritis, while deletion –47A, found in the region of T-antigen binding, is associated with adrenal sclerosis [134].
About 106 SNP are registered in the NCBI dbSNP database [136]. Most of them are functionally silent, but a significant number of these SNP, especially in coding and regulatory gene regions, will be of biological and/or medical significance [137]. At the present time SNP of regulatory gene regions, influencing their expression level, are an important but relatively poorly studied class of genetic variations, unlike polymorphisms of coding gene regions. Two main mechanisms of SNP exhibition in both gene regions are known: the change in the pattern and/or level of gene transcription and direct change in biochemical properties of the gene product [138]. The SNP manifestations in coding and regulatory gene regions are indistinguishable by their severity for human health. Thus, it was shown that SNP in coding regions of DNA metabolism and repair genes, causing codon change and single amino acid mutation, can influence individual susceptibility to oncological diseases and their therapy [139]. SNP in the coding region of the human LIMD1 gene are associated with mammary gland and lung cancers [140], SNP in the coding region of apolipoprotein E gene are associated with human chronic non-infectious diseases and predisposition to a number of cardiovascular and neurodegenerative diseases [45], while SNP in the coding region of the interleukin-1β gene is associated with cancer of the pancreas and is hardly compatible with life [105], etc. It is known that SNP of introns also cause human phenotypic variability and enhance the burden of diseases. For example, two single-nucleotide substitutions in intron 6 of the tryptophan oxygenase gene are associated with human mental diseases (Turrett’s syndrome, hyperactive-child syndrome, predisposition to alcoholism and narcomania) [143]. An SNP of intron 2 of the IL-1Rα gene (the interleukin-1 antagonist receptor gene) is associated with a variant of stomach cancer [144], an SNP in an AU-rich region of the glucocorticoid β receptor gene is associated with rheumatoid arthritis [145], etc.
This review shows that at the present time about 40 polymorphisms in the promoter TATA elements are associated with different human diseases and are confirmed by molecular-biological studies. In the above-described examples certain SNP result in lowering transcription activity of promoters, which increases 8-10-fold the risk of oral cavity cancer in smokers [55], stenocardia [43, 44], neurodegenerative diseases [62], etc. Other SNP, on the contrary, increase promoter transcription activity, which results in increased sensitivity to hypertension, stenocardia, lung diseases, and disseminated sclerosis [96] and are associated with pathologies in the development of small brain blood vessels [102]. The emergence of a latent TATA box as a result of mutation may be responsible for Coppock-like cataract [108] or endometrial cancer [116]. There are also cases of destruction of TBP binding sites overlapping with factors GATA-1 and HNF-4 [122, 124]. Then TATA box polymorphisms affect binding of these transcription factors, which is also of clinical importance.
Thus, it is difficult to overestimate the consequences of phenotypic manifestation of a variety of SNP located in TATA boxes and flanking their sequences. The significance of these SNP also concerns the possibility of disturbance of coordinated gene expression, the observance of which is especially necessary in the process of organogenesis accompanied by proliferation and transport of particular cell groups that are strictly ordered in time and space.
Obviously, the need for experimental and bioinformatic investigation of TATA element SNP has appeared, which will stimulate identification of genes involved in emergence of complex human diseases like hypertension, arthritis, and oncological diseases [146, 147] and the elucidation of the role of SNP in the development of these diseases and predisposition to them, in different sensitivity to environmental factors and drug preparations, etc. [148]. During these experimental and bioinformatic investigations, up-to-date computer approaches should be elaborated for detection of SNP not yet functionally annotated that are potentially able to influence important processes and systems of the human body. Results of such investigations will be widely used in studies of gene expression regulation and genome analysis, in pharmacology upon elaboration of new multipurpose drugs, as well as in medical-genetic investigations.
The authors are grateful to T. I. Merkulova for reading and providing valuable advice on the manuscript and to K. V. Gunbina for help in material collection and systematization.
This work was supported by the Russian Foundation for Basic Research (grants 09-04-00653 and 08-04-01048).
REFERENCES
1.Aso, T., Conaway, J. W., and Conaway, R. C. (1994)
J. Biol. Chem., 269, 26575-26583.
2.Colgan, J., and Manley, J. L. (1995) Proc. Natl.
Acad. Sci. USA, 92, 1955-1959.
3.Butler, J. E., and Kadonaga, J. T. (2002) Genes
Dev., 16, 2583-2592.
4.Struh, K. (1987) Cell, 49,
295-297.
5.Smale, S. T., and Kadonaga, J. T. (2003) Annu.
Rev. Biochem., 72, 449-479.
6.Breathnach, R., and Chambon, P. (1981) Annu.
Rev. Biochem., 50, 349-383.
7.Conaway, R. C., and Conaway, J. W. (1997) Progr.
Nucleic Acids Res. Mol. Biol., 56, 327-346.
8.Basehoar, A. D., Zanton, S. J., and Pugh, B. F.
(2004) Cell, 116, 699-709.
9.Gershenzon, N. I., and Ioshikhes, I. P. (2005)
Bioinformatics, 21, 1295-1300.
10.Patikoglou, G. A., Kim, J. L., Sun, L., Yang, S.
H., Kodadek, T., and Burley, S. K. (1999) Genes Dev., 13,
3217-3230.
11.Perier, R. C., Praz, V., Junier, T., Bonnard, C.,
and Bucher, P. (2000) Nucleic Acids Res., 28,
302-303.
12.Stewart, J. J., Fischbeck, J. A., Chen, X., and
Stargell, L. A. (2006) J. Biol. Chem., 281,
22665-22673.
13.Bucher, P. (1990) J. Mol. Biol.,
212, 563-578.
14.Kim, J. L., Nikolov, D. B., and Burley, S. K.
(1993) Nature, 365, 520-527.
15.Kim, Y., Geige, J. H., Hahn, S., and Sigler, P.
B. (1993) Nature, 365, 512-520.
16.Buratowski, S., Hahn, S., Guarente, L., and
Sharp, P. A. (1989) Cell, 56, 549-561.
17.Buratowski, S. (1994) Cell, 77,
1-3.
18.Reinberg, D., and Roeder, R. G. (1987) J.
Biol. Chem., 262, 3310-3321.
19.Sawadogo, M., and Sentenac, A. (1990) Annu.
Rev. Biochem., 59, 711-754.
20.Maldonado, E., Ha, I., Cortes, P., Weis, L., and
Reinberg, D. (1990) Mol. Cell Biol., 10, 6335-6347.
21.Pugh, B. F. (2000) Gene, 255,
1-14.
22.Caron, C., Rousset, R., Beraud, C., Moncollin,
V., et al. (1993) EMBO J., 12, 4269-4278.
23.Gill, G., and Tjian, R. (1991) Cell,
65, 333-340.
24.Wobbe, C. R., and Struhl, K. (1990) Mol. Cell.
Biol., 10, 3859-3867.
25.Librizzi, M. D., Brenowitz, M., and Willis, I. M.
(1998) J. Biol. Chem., 273, 4563-4568.
26.Wolner, B. S., and Gralla, J. D. (2001) J.
Biol. Chem., 276, 6260-6266.
27.Faiger, H., Ivanchenko, M., Cohen, I., and Haran,
T. E. (2006) Nucleic Acids Res., 34, 104-119.
28.Savinkova, L. K., Drachkova, I. A., Ponomarenko,
M. P., Lysova, M. V., Arshinova, T. V., and Kolchanov, N. A. (2007)
Ecol. Genet., V, 44-49.
29.Ponomarenko, M. P., Ponomarenko, Y. V., Frolov,
A. S., Podkolodny, N. L., Savinkova, L. K., Kolchanov, N. A., and
Overton, G. C. (1999) Bioinformatics, 15, 687-703.
30.Orkin, S. H., Sexton, J. P., Cheng, T. C., Goff,
S. C., Giardina, P. J., Lee, J. I., and Kazazian, H. H., Jr. (1983)
Nucleic Acids Res., 11, 4727-4734.
31.Poncz, M., Ballantine, M., Solowiejczyk, D.,
Barak, I., Schwartz, E., and Surrey, S. (1982) J. Biol.
Chem., 257, 5994-5996.
32.Badens, C., Jassim, N., Martini, N., Mattei, J.
F., Elion, J., and Lena-Russo, D. (1999) Hemoglobin, 23,
339-344.
33.Cai, S. P., Zhang, J. Z., Doherty, M., and Kan,
Y. W. (1989) Am. J. Hum. Genet., 45, 112-114.
34.Takihara, Y., Nakamura, T., Yamada, H., Takagi,
Y., and Fukumaki, Y. (1986) Blood, 67, 547-550.
35.Fei, Y. J., Stoming, T. A., Efremov, G. D.,
Efremov, D. G., Battacharia, R., et al. (1988) Biochem. Biophys.
Res. Commun., 153, 741-747.
36.Antonarakis, S. E., Irkin, S. H., Cheng, T. C.,
et al. (1984) Proc. Natl. Acad. Sci. USA, 81,
1154-1158.
37.Frischknecht, H., and Dutly, F. (2005)
Hemoglobin, 29, 151-154.
38.Frishknecht, H., Troxler, H., Forstar, C., and
Dutly, F. (2006) Hemoglobin, 30, 23-27.
39.Otabe, S., Clement, K., Dina, C., Pelloux, V.,
Guy-Grand, B., Froguel, P., and Vasseur, F. (2000) Diabetol.,
43, 245-249.
40.Dean, M., and Allikmets, R. (1995) Curr. Opin.
Genet. Dev., 5, 779-785.
41.Santamarina-Fojo, S., Peterson, K., Knapper, C.,
et al. (2000) Proc. Natl. Acad. Sci. USA, 97,
7987-7992.
42.Zwarts, K. Y., Clee, S. M., Zwinderman, A. H., et
al. (2002) Clin. Genet., 61, 115-125.
43.Gordon, D., and Rifkind, B. M. (1998) N. Engl.
J. Med., 321, 1311-1315.
44.Rubin, E. M., Krauss, R. M., Spangler, E. A.,
Verstuyft, J. G., and Gift, S. M. (1991) Nature, 353,
265-267.
45.Matsunaga, A., Sasaki, J., et al. (1999)
Arterioscler. Thromb. Vasc. Biol., 19, 348-355.
46.Brown, J. R., Daar, I. O., Krug, J. R., and
Maquat, L. E. (1985) Mol. Cell. Biol., 5, 1694-1706.
47.Watanabe, M., Zingg, B. C., and Mohrenweiser, H.
W. (1996) Am. J. Hum. Genet., 58, 308-316.
48.Bosma, P. J., Chowdhury, J. R., Bakker, C., et
al. (1995) N. Engl. J. Med., 333, 1171-1175.
49.Kaniwa, N., Kurose, K., Jinno, H., Tanaka-Kagawa,
T., Saito, Y., Saeki, M., Sawada, J., Tohkin, M., and Hasegawa, R.
(2005) Drug Metab. Dispos., 33, 458-465.
50.Mackenzie, P. I., Owens, I. S., Burchell, B., et
al. (1997) Pharmacogenetics, 7, 255-269.
51.Beutler, E., Gelbart, T., and Demina, A. (1998)
Proc. Natl. Acad. Sci. USA, 95, 8170-8174.
52.Rauchschwalbe, S. K., Zuhlsdorf, M. T., Schuhly,
U., and Kuhlman, J. (2002) Int. J. Clin. Pharmacol.
Ther., 40, 233-240.
53.Sugatani, J., Yamakawa, K., Yoshinari, K., et al.
(2002) Biochem. Biophys. Res. Commun., 292, 492-497.
54.Burchel, B., and Hume, R. (1999) J.
Gastroenterol. Hepatol., 14, 960-966.
55.Zheng, Z., Park, J. Y., Guillemette, C., Schantz,
S. P., and Lazarus, P. (2001) J. Natl. Cancer Inst., 93,
1411-1418.
56.Lankisch, T. O., Vogel, A., Eilermann, S.,
Fiebeler, A., Krone, B., Barut, A., Manns, M. P., and Strassburg, C. P.
(2005) Mol. Pharmacol., 67, 1732-1739.
57.Haaz, M. C., Rivory, L., Jantet, S.,
Ratanasavanh, D., and Robert, J. (1997) Pharmacol. Toxicol.,
80, 91-96.
58.Catimel, G., Chabot, G. G., Guastalla, J. P., et
al. (1995) Ann. Oncol., 6, 133-140.
59.Smith, M. T., Evans, C. G., Doane-Setzer, P.,
Castro, V. M., Tahir, M. K., and Mannervik, B. (1989) Cancer
Res., 49, 2621-2625.
60.Liu, X., Campbell, M. R., Pittman, G. S.,
Faulkner, E. C., Watson, M. A., and Bell, D. A. (2005) Cancer
Res., 65, 99-104.
61.Marden, J. J., Harraz, M. M., Williams, A. J.,
Nelson, K., Luo, M., Paulson, H., and Engelhardt, J. F. (2007) J.
Clin. Invest., 117, 2913-2919.
62.Niemann, S., Broom, W. J., and Brown, R. H., Jr.
(2007) Muscle Nerve, 36, 704-707.
63.Topper, J. N., and Clayton, D. A. (1990)
Nucleic Acids Res., 18, 793-799.
64.Bonafe, L., Schmitt, K., Eich, G., Giedion, A.,
and Superti-Furga, A. (2002) Clin. Genet., 61,
146-151.
65.Pelkonen, O., Rautio, A., Raunio, H., and
Pasanen, M. (2000) Toxicology, 144, 139-147.
66.Oscarson, M. (2001) Drug Metab. Dispos.,
29, 91-95.
67.Pianezza, M. L., Sellers, E. M., and Tyndale, R.
F. (1998) Nature, 393, 750.
68.Pitarque, M., von Richter, O., Oke, B., Berkkan,
H., Oscarson, M., and Ingelman-Sundberg, M. (2001) Biochem. Biophys.
Res. Commun., 292, 455-460.
69.Goerke, J. (1998) Biochim. Biophys. Acta,
1408, 79-89.
70.Pilot-Matias, T. J., Kister, S. E., Fox, J. L.,
Kropp, K., Glasser, S. W., and Whitsett, J. A. (1989) DNA,
8, 75-86.
71.Steagall, W. K., Lin, J. P., and Moss, J. (2007)
Am. J. Physiol. Lung Cell. Mol. Physiol., 292,
L448-L453.
72.Boldt, A. B., Culpi, L., Tsuneto, L. T., de
Souza, I. R., Kun, J. F., and Petzl-Erler, M. L. (2006) Hum.
Immunol., 67, 722-734.
73.Madsen, H. O., Satz, M. L., Hogh, B., Svejgaard,
A., and Garred, P. (1998) J. Immunol., 161,
3169-3175.
74.Super, M., Levinsky, R. J., and Turner, M. W.
(1990) Clin. Exp. Immunol., 79, 144-150.
75.Garred, P., Madsen, H. O., Hofmann, B., and
Svejgaard, A. (1995) Lancet, 346, 941-943.
76.Garred, P., Madsen, H. O., Balslev, U., Hofmann,
B., Pedersen, C., Gerstoft, J., and Svejgaard, A. (1997) Lancet,
349, 236-240.
77.Groenewegen, W. A., Firouzi, M., Bezzina, C. R.,
et al. (2003) Circ. Res., 92, 14-22.
78.Chen, E. Y., Liao, Y. C., Smith, D. H., et al.
(1989) Genomics, 4, 479-497.
79.Horan, M., Millar, D. S., Hedderich, J., Lewis,
G., Newsway, V., et al. (2003) Hum. Mutat., 21,
408-423.
80.Conway, E. M., and Rosenberg, R. D. (1988)
Mol. Cell Biol., 8, 5588-5592.
81.Hirokawa, K., and Aoki, N. (1991) J. Cell.
Physiol., 147, 157-165.
82.Hirokawa, K., and Aoki, N. (1991) Biochem.
J., 276, 739-743.
83.Bartha, K., Brisson, C., Archipoff, G., de la
Salle, C., Lanza, F., Cazenave, J. P., and Beretz, A. (1993) J.
Biol. Chem., 268, 421-429.
84.Ye, J., Esmon, C. T., and Johnson, A. E. (1993)
J. Biol. Chem., 268, 2373-2379.
85.Le Flem, L., Picard, V., et al. (1999)
Arterioscler. Thromb. Vasc. Biol., 19, 1098-1104.
86.Evans, B. A., Yun, Z. Y., Close, J. A., et al.
(1988) Biochemistry, 27, 3124-3129.
87.Slim, R., Torremocha, F., Moreau, T., et al.
(2002) J. Am. Soc. Nephrol., 13, 968-976.
88.Ogawa, S., Eng, V., Taylor, J., Lubahn, D. B.,
Korach, K. S., and Pfaff, D. W. (1998) Endocrinology,
139, 5070-5081.
89.Ogawa, S., Chan, J., Chester, A. E., Gustafsson,
J. A., Korach, K. S., and Pfaff, D. W. (1999) Proc. Natl. Acad. Sci.
USA, 96, 12887-12892.
90.Schubert, C., Pencalla, A., Fliegner, D.,
Kintscher, U., et al. (2006) in Berliner Symposium
Geschlechterforschung in der Medizin. Dokumentation, p. 36.
91.Philips, S., Bermes, A., Nguyen, A. T., Luzcando,
R., Narayanan, P. B., Flockhart, D. A., and Skaar, T. C. (2005)
Clin. Pharmacol. Ther., 77, P63.
92.Philips, S., Richter, A., Oesterreich, S., Rae,
J., Perumal, N., Flockhart, D., and Skaar, T. (2007) Clin.
Pharmacol. Ther., 81, S87.
93.Clark, B. J., Wells, J., King, S. R., and Stocco,
D. M. (1994) J. Biol. Chem., 269, 28314-28322.
94.Casal, A. J., Sinclair, V. J., Capponi, A. M.,
Nicod, J., Huynh-Do, U., and Ferrari, P. (2006) J. Mol.
Endocrinol., 37, 71-80.
95.Peltoketo, H., Piao, Y., Mannermaa, A., Ponder,
B. A., Isomaa, V., Poutanen, M., Winqvist, R., and Vihko, R. (1994)
Genomics, 23, 250-252.
96.Burgner, D., Rockett, K., Ackerman, H., Hull, J.,
Usen, S., Pinder, M., and Kwiatkowski, D. P. (2003) Genes and
Immunity, 4, 506-514.
97.Labbaye, C., Musette, P., and Cayre, Y. E. (1991)
Proc. Natl. Acad. Sci. USA, 88, 9253-9256.
98.Zimmer, M., Medcalf, R. L., Fink, T. M.,
Mattmann, C., Lichter, P., and Jenne, D. E. (1992) Proc. Natl. Acad.
Sci. USA, 89, 8215-8219.
99.Lee, P. L., Halloran, C., and Beutler, E. (2001)
Blood Cells Mol. Dis., 27, 539-548.
100.Ruf, W., and Edgington, T. S. (1994)
FASEB, 8, 385-390.
101.Arnaud, E., Barbalat, V., Nicaud, V., et al.
(2000) Arterioscler. Thromb. Vasc. Biol., 20,
892-898.
102.Zhao, Y. Y., Zhou, J., Narayanan, C. S., Cui,
Y., and Kumar, A. (1999) Hypertension, 33, 108-115.
103.Schmidt, H., Aulchenko, Y. S., Schweignofer,
N., et al. (2004) Stroke, 35, 2592-2597.
104.Fornari, M. C., Pedreira, S., Niveloni, S., et
al. (1998) Am. J. Gastroenterol., 93, 413-418.
105.Fosang, A. J., Last, K., Knauper, V., Murphy,
G., and Neame, P. J. (1996) FEBS Lett., 380, 17-20.
106.Zienolddiny, S., Ryberg, D., Maggini, V.,
Skaug, V., Canzian, F., and Haugen, A. (2004) Int. J. Cancer,
109, 353-356.
107.Wang, Y., Kato, N., Hoshida, Y., Taniguchi, H.,
et al. (2003) Hepatology, 37, 65-71.
108.Brakenhoff, R. H., Henskens, H. A., van Rossum,
M. W., Lubsen, N. H., and Schoenmakers, J. G. (1994) Hum. Mol.
Genet., 3, 279-283.
109.Shah, N. P., Witte, O. N., and Denny, C. T.
(1991) Mol. Cell. Biol., 11, 1854-1860.
110.De Klein, A., van Kessel, A. G., Grosveld, G.,
et al. (1982) Nature, 300, 765-767.
111.Tian, H., Zheng, W. Y., Fu, Y. G., Lin, J. H.,
Lu, F. J., and Zhou, S. Y. (2004) Ai Zheng, 23,
812-815.
112.Lifshitz, B., Fainstein, E., Marcelle, C.,
Shtivelman, E., Amson, R., Gale, R. P., and Canaani, E. (1988)
Oncogene, 2, 113-117.
113.Zhu, Q. S., Heisterkamp, N., and Groffen, J.
(1990) Nucleic Acids Res., 18, 7119-7125.
114.Kumar, N. S., Richer, J., Owen, G., Litman, E.,
Horwitz, K. B., and Leslie, K. K. (1998) Cancer Res., 58,
1860-1865.
115.Parazzini, F., la Vecchia, C., Moroni, S.,
Chatenoud, L., and Ricci, E. (1994) Int. J. Cancer, 59,
460-462.
116.De Vivo, I., Huggins, G. S., Hankinson, S. E.,
Lescault, P. J., Boezen, M., Colditz, G. A., and Hunter, D. J. (2002)
Proc. Natl. Acad. Sci. USA, 99, 12263-12268.
117.Liu, Z., Wong, J., et al. (2001) Proc. Natl.
Acad. Sci. USA, 98, 12426-12431.
118.Lang, T., Klein, K., Fischer, J., Nussler, A.
K., Neuhaus, P., Hofmann, U., Eichelbaum, M., Schwab, M., and Zanger,
U. M. (2001) Pharmacogenetics, 11, 399-415.
119.Zukunft, J., Lang, T., Richter, T.,
Hirsch-Ernst, K. I., Nussler, A. K., Klein, K., Schwab, M., Eichelbaum,
M., and Zanger, U. M. (2005) Mol. Pharmacol., 67,
1772-1782.
120.Iwamoto, S., Li, J., Sugimoto, N., Okuda, H.,
and Kajii, E. (1996) Biochem. Biophys. Res. Commun., 222,
852-859.
121.Tournamille, C., Colin, Y., Cartron, J. P., and
Le Van Kim, C. (1995) Nature Genet., 10, 224-228.
122.Penner, C. G., and Davie, J. R. (1994) FEBS
Lett., 342, 273-277.
123.Yoshitake, S., Schach, B. G., Foster, D. C.,
Davie, E. W., and Kurachi, K. (1985) Biochemistry, 24,
3736-3750.
124.Reijnen, M. J., Sladek, F. M., Bertina, R. M.,
and Reitsma, P. H. (1992) Proc. Natl. Acad. Sci. USA,
89, 6300-6303.
125.Turner, B. G., and Summers, M. F. (1999) J.
Mol. Biol., 285, 1-32.
126.Berkhout, B., and Jeang, K. T. (1992) J.
Virol., 66, 139-149.
127.Sakaguchi, M., Zenzie-Gregory, B., Groopman, J.
E., Smale, S. T., and Kim, S. Y. (1991) J. Virol., 65,
5448-5456.
128.Estable, M. C., Bell, B., Merzouki, A.,
Montaner, J. S., et al. (1996) J. Virol., 70,
4053-4062.
129.Jeeninga, R. E., Hoogenkamp, M., Armand-Ugon,
M., et al. (2000) J. Virol., 74, 3740-3751.
130.Padua, E., Jenkins, A., Brown, S., Bootman, J.,
Paixao, M. T., Almond, N., and Berry, N. (2003) J. Gen. Virol.,
84, 1287-1299.
131.Lochelt, M., Yu, S. F., Linial, M. L., and
Flugel, R. M. (1995) Virology, 206, 601-610.
132.Rollison, D. E., Utaipat, U., Ryschkewitsch,
C., et al. (2005) Int. J. Cancer, 113, 769-774.
133.Boldorini, R., Caldarelli-Stefano, R., Monga,
G., Zocchi, M., Mediati, M., Tosoni, A., and Ferrante, P. (1998) J.
Neurovirol., 4, 242-245.
134.Li, R. M., Mannon, R. B., Kleiner, D., Tsokos,
M., Bynum, M., Kirk, A. D., and Kopp, J. B. (2002)
Transplantation, 74, 1497-1504.
135.Li, R. M., Branton, M. H., Tanawattanacharoen,
S., Falk, R. A., Jennette, J. C., and Kopp, J. B. (2002) J.
Am. Soc. Nephrol., 13, 2320-2330.
136.Sherry, S. T., Ward, M. H., Kholodov, M.,
Baker, L., Phan, L., Smigielski, E. M., and Sirotkin, K. (2001)
Nucleic Acids Res., 29, 308-311.
137.Teufel, A., Krupp, M., Weinmann, A., and Galle,
P. R. (2006) Int. J. Mol. Med., 17, 967-973.
138.Simoni, M., Nieschlag, E., and Gromoll, J.
(2002) Hum. Reprod. Update, 8, 413-421.
139.Bartsch, H., Dally, H., Popanda, O., Risch, A.,
and Schmezer, P. (2007) Recent Res. Cancer Res., 174,
19-36.
140.Huggins, C. J., Gill, M., and Andrulis, I. L.
(2007) Cancer Genet. Cytogenet., 178, 36-41.
141.Hubacek, J. A., Pitha, J., Adamkova, V.,
Skodova, Z., Lanska, V., and Poledne, R. (2003) Physiol. Res.,
52, 195-200.
142.Barber, M. D., Powell, J. J., Lynch, S. F.,
Fearon, K. C., and Ross, J. A. (2000) Br. J. Cancer, 83,
1443-1447.
143.Comings, D. E., Gade, R., Muchleman, D., and
Chue, C. (1996) Pharmacogenetics, 6, 307-318.
144.Machado, J. C., Pharoon, P., Sousa, S., et al.
(2001) Gastroentherology, 121, 823-829.
145.Derijk, R. H., Schaaf, M. J., Turner, G., et
al. (2001)'' [javascript:AL_get(this,%20'jour',%20'J%20Rheumatol.'); J.
Rheumatol.], 28, 2383-2388.
146.Lander, E. S. (1996) Science,
274, 536-539.
147.Chakravarti, A. (1999) Nature Genet.,
21, 56-60.
148.Halushka, M. K., Fan, J. B., Bentley, K., Hsie,
L., Shen, N., Weder, A., Cooper, R., Lipshutz, R., and Chakravarti, A.
(1999) Nature Genet., 22, 239-247.