Novel Family of Human Transposable Elements Formed Due to Fusion of the First Exon of Gene MAST2 with Retrotransposon SVA
O. B. Bantysh and A. A. Buzdin*
Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (495) 727-3863; E-mail: bu3din@mail.ru* To whom correspondence should be addressed.
Received July 22, 2009; Revision received September 21, 2009
We identified a novel human-specific family of transposable elements that consists of fused copies of the CpG-island containing the first exon of gene MAST2 and retrotransposon SVA. We propose a mechanism for the formation of this family termed CpG-SVA, comprising 5′-transduction by an SVA insert. After the divergence of human and chimpanzee ancestor lineages, retrotransposon SVA has inserted into the first intron of gene MAST2 in the sense orientation. Due to splicing of an aberrant RNA driven by MAST2 promoter, but terminally processed using SVA polyadenylation signal, the first exon of MAST2 has fused to a spliced 3′-terminal fragment of SVA retrotransposon. The above ancestor CpG-SVA element due to retrotranspositions of its own copies has formed a novel family represented in the human genome by 76 members. Recruitment of a MAST2 CpG island was most likely beneficial to the hybrid retrotransposons because it could significantly increase retrotransposition frequency. Also, we show that human L1 reverse transcriptase adds an extra cytosine residue to the 3′ terminus of the nascent first strand of cDNA.
KEY WORDS: molecular evolution, human DNA, retroelements, genomic transposable elements, regulation of transcriptionDOI: 10.1134/S0006297909120153
Abbreviations: REs, retroelements.
Retroelements (REs) are genomic transposable elements that proliferate
in the host DNA by using reverse transcription. This enzymatic activity
that results in synthesis of a cDNA copy on a RE RNA template is a
property of reverse transcriptase protein [1, 2]. Reverse transcriptases from different organisms
share significant sequence identity and are homologous to the catalytic
subunit of telomerase [3]. REs that utilize reverse
transcriptase encoded in their own “genome” are termed
“autonomous”, whereas those that lack a reverse
transcriptase gene are called “non-autonomous”.
Non-autonomous REs are “parasitizing” on autonomous ones by
recruiting their reverse transcriptase. REs occupy more than 40% of the
human genome and are represented by several millions of copies [4]. However, among dozens of human RE families,
members of only four of them were still retrotranspositionally active
until evolutionarily recent times [5]. For example,
only two groups of autonomous REs preserved their activities after
divergence of human and chimpanzee ancestors: endogenous retroviruses
HERV-K (HML-2) and L1 retrotransposons, and two groups of
non-autonomous REs – Alu and SVA retrotransposons. Members of all
these families had very different dynamics of retrotransposition, so
they are represented now in the genome by very diverse numbers of
human-specific (hs) representatives. There are approximately 150 hs
copies of HERV-K (HML-2) endogenous retroviruses [6], 1200 hs copies of L1 retrotransposons, and 5500
and 860 hs copies of Alu and SVA elements, respectively [5].
For retrotransposition Alu and SVA elements utilize L1-encoded reverse transcriptase [7]. Full-size L1 retrotransposon is about 6 kb long and encodes two open reading frames: reverse transcriptase/integrase and RNA binding protein [8]. The characteristic feature of L1 retroposition is generating of 10-18-bp-long direct repeats flanking the site of insertion [7]. The majority of L1 insertions have 5′-truncated termini as a result of abortive reverse transcription [9]. The non-autonomous family Alu appeared in the genome due to dimerization of an ancestral element derived from cellular 7SL RNA [10]. Alu elements (typically about 300-bp-long) are transcribed by RNA polymerase III [11]. In contrast to Alu, SVA retrotransposons are likely transcribed by RNA polymerase II, and their length is usually over 1500 bp.
SVA retrotransposons were formed less than 25 million years ago and are thought to be the youngest family among the primate REs. SVA are the complex REs formed by the fusion of so-called SINE-R sequence, variable number tandem repeat sequence (VNTR), and Alu [12]. The SVA family is still active as at least five cases of human inheritable diseases cased by de novo SVA insertion have been documented so far. Many SVA inserts are polymorphic in the human population [13].
Among the active human REs, SVA elements are probably the most poorly studied family. Evolution of this complex group is still going on, especially via quantitative and qualitative changes in tandem repeats, oligomerization, and acquisition of new sequences. This acquisition of genomic sequences by SVA elements can occur in the middle part of an SVA (e.g. due to pseudogene insertion into an SVA element), or on SVA termini. In the latter case new sequences can appear either on the 5′- or on the 3′-terminus of an SVA (so-called 5′ and 3′ SVA transduction). The 3′-transduction mechanism is based on slippage of an SVA polyadenylation signal by the transcriptional complex. In this case, transcriptional termination and RNA processing occur on any downstream polyadenylation signal, which results in forming of mature transcripts longer than the initial copy of an SVA. Like a normal SVA copy, it attaches to reverse transcriptase complex and serves as the template for the synthesis of cDNA, which in turn inserts into the genome. The size of a genomic sequence transferred in such a way can differ from several base pairs to more than 1500 bp. Probably the most striking example of this phenomenon is the transduction of the whole gene AMAC in the great ape genomes [14]. Due to SVA 3′-transduction, the human genome has three functional 1.2 kb-long copies of the AMAC gene. This type of transduction is also typical for L1 retrotransposons [15].
Another kind of transduction results in attaching of new sequences to the 5′ end of an SVA. There are two major mechanisms of this 5′-transduction. First, during the reverse transcription a template switching can occur that will result in fusion of different cellular RNA copies within one RE insert [16, 17]. Second, RE transcription initiation can proceed from any promoter located upstream in the genomic sequence. In this [/C:%5CDocuments%20and%20Settings%5CUSER%5CLocal%20Settings%5CTemp%5CWord_1 case] termination of transcription and RNA processing usually occur using a normal polyadenylation signal of a RE. This results in a mature RNA having on its 5′ end an additional copy of a flanking genomic sequence and a copy of the RE at its 3′ end. Subsequent reverse transcription and integration into the genome of a nascent cDNA results in a new RE genomic insert carrying a 5′ transduced part [18].
METHODS
DNA sequence analysis. Chimeric retrotransposons CpG-SVA were identified using consensus sequence of an SVA retrotransposon taken from the RepBase Update database (http://www.girinst.org/server/RepBase/) [19] and mRNA sequence of gene MAST2 (GeneBank accession number AB047005). For the mapping of chimeric elements, we used the BLAT program (http://genome.ucsc.edu/cgi-bin/hgBLAT) and UCSC Genome Browser. The February 2009 human genome assembly was used for in silico DNA analysis. Pseudogene flanking regions were investigated using the RepeatMasker program (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker). Direct repeats were identified by visual inspection of sequences flanking retrotransposon inserts. Homology searches against non-mammalian organisms were done using the BLAST server at NCBI (http://www.ncbi.nlm.nih.gov/BLAST) [20]. Transposable elements located close to CpG-SVA inserts were classified according to the RepBase Update database nomenclature (http://www.girinst.org/server/RepBase/). The data on MAST2 expression in human tissues was taken from the UCSC Genome Browser. The ClustalW program was used for multiple sequence alignment [21], and the PHYLIP software was utilized for phylogenetic tree construction [22]. Complete datasets including detailed information on CpG-SVA mapping, flanking direct repeats, and structural alignment are available upon request to the corresponding author’s E-mail: bu3din@mail.ru.
RESULTS AND DISCUSSION
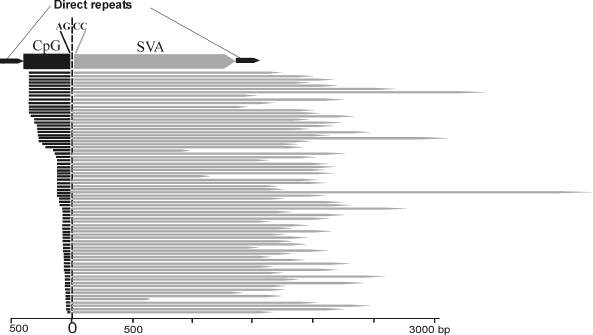
Identification of a novel chimeric family of retrotransposons. Detailed structural analysis of the human-specific SVA retrotransposons revealed 76 elements of an unusual structure. At the 5′ termini these elements carried copies of the first exon of the MAST2 gene, whereas at the 3′ end – SVA retrotransposon sequences. The border between exonic and SVA parts was located exactly between canonical acceptor splice site AG from the exonic part and a non-canonical donor splice-site CC from the SVA part (396 position in the SVA consensus sequence). Lengths of both parts of the chimeric elements significantly varied: from 35 to 383 bp for the 5′-terminal part and from 662 to 4255 bp for the 3′ terminal part. The border between the two parts was constant in all the chimeras (Fig. 1). On the 3′ terminus, the chimeras harbored a poly(A) sequence of variable length. These bipartite elements were flanked by 12-18 bp long direct repeats. The presence of the direct repeats surrounding chimeric inserts suggests implication of L1 retrotranspositional machinery in their formation, whereas the poly(A) sequence indicates that retrotransposed RNA was transcribed by RNA polymerase II. The identified family of chimeric REs was called “CpG-SVA” because its 5′ terminal part complementary to the first exon of the MAST2 gene included a CpG island sequence. CpG-SVA elements were found only in human genomic DNA, whereas separately both SVA retrotransposons and MAST2 exon sequence exist in the genomes of all great apes. Therefore, CpG-SVA can be regarded as a new human-specific family of retrotransposons.
While the present manuscript was under review, two other papers describing the same family of hybrid retrotransposons (CpG-SVA) were simultaneously published, where this family was termed either “MAST2-SVA” [23] or “SVA-F1” [24].Fig. 1. Structure of chimeric CpG-SVA retrotransposons. CpG-SVA inserts are flanked by direct repeats. Lengths of 5′ terminal (exonic) part vary from 35 to 383 bp, lengths of 3′ (SVA-derived) part vary from 662 to 4255 bp. The 5′-terminal parts are homologous to the first exon of the MAST2 gene, 3′-terminal parts – to SVA retrotransposon. The junction point between the two parts is identical in all CpG-SVA elements (canonical splice acceptor site AG from the side of the exonic part and non-canonical splice donor site CC from the side of SVA). All SVA fragments start from position 396 of the SVA consensus sequence.
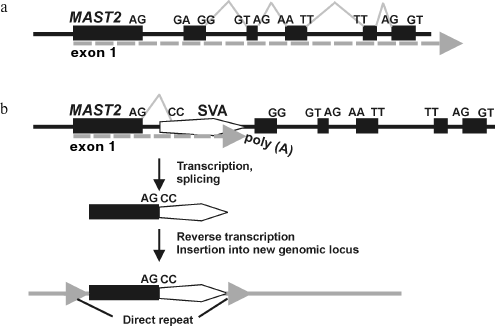
Mechanism of CpG-SVA family formation. Based on the structural features of the identified CpG-SVA family members, we purposed a mechanism for their formation (Fig. 2). At the first stage, the SVA retrotransposon most probably has inserted into the first intron of the MSAT2 gene in the sense orientation. After that there was formed an aberrant RNA driven by the MAST2 promoter and terminally processed using the SVA polyadenylation signal. This RNA was further spliced, which resulted in a fusion of the first exon of MAST2 with a 3′-terminal fragment of an SVA (starting from 393 nucleotide of the SVA consensus sequence). This spliced chimeric RNA was then reverse transcribed by the L1 retrotranspositional machinery followed by integration of a nascent cDNA into the genome. This resulted in emerging of the master copy of CpG-SVA inserted into human DNA and flanked by direct repeats. The newly inserted CpG-SVA element appeared to be transcriptionally active, possible due to the enclosed CpG-islet, and gave rise to a new family of REs.
This hypothesis is supported by the observation that there is the canonical MAST2 gene acceptor splice site AG of on the border between the MAST2- and SVA-derived fragments. The putative donor splice site CC within an SVA is not canonical, which might be explained by the peculiarities of MAST2 exon–intronic structure where non-canonical splice sites form the majority (Fig. 2a).Fig. 2. Proposed mechanism of CpG-SVA family formation. a) Schematic representation of genomic locus comprising human gene MAST2. Dotted arrow designates transcriptional direction, exons and splice sites are shown. b) Insert of an SVA retrotransposon in the sense orientation has changed gene exon–intronic structure and gave rise to aberrantly spliced mRNA polyadenylated at the SVA sequence. A copy of this mRNA has inserted into a new locus of the human genome and gave rise to the CpG-SVA family that continued proliferation in human DNA. However, the ancestral allele of the MAST2 gene comprising the SVA insert was lost due to the negative selection.
Interestingly, at present there is no fixed SVA insert into the MAST2 gene intron in the human genome. Apparently, an ancestral allele containing the above SVA element in a gene intron was eliminated by negative selection as it could not provide functional MAST2 mRNA formation because of the aberrant splicing of transcripts and/or preliminary polyadenylation on the SVA sequence.
Further evolution of the CpG-SVA family. We have found among the CpG-SVA elements several cases of 5′ and 3′ transduction of unrelated genomic DNA, proven by the mapping of the enclosing direct repeats. As in the classical 3′ transduction mechanism, it is likely that the downstream genomic fragments were captured due to “getting through” of SVA polyadenylation signals by the RNA polymerase II complex with the subsequent termination on any downstream sequence. In case of 5′ CpG-SVA transduction, there was apparently transcription of CpG-SVA elements initiated from upstream genomic promoters. Overall, we identified 18 and 11 cases of the 5′ and 3′ CpG-SVA transductions, respectively. In reality there may be some additional cases of CpG-SVA transductions not identified in this study because we have not been able to identify flanking direct repeats for five CpG-SVA elements. The size of the transferred genomic sequence differed from 8 to 854 bp for 5′- and from 141 to 734 bp for 3′-transduction events. Remarkably, four CpG-SVA elements contained both 5′ and 3′ transduced sequences (table, elements No. 4, 6, 7, 8). These four elements were highly similar and consisted of 364 bp long MAST2 exon and 2143-3361 bp long SVA sequences. SVA length variations were caused by the instability its internal satellite repeat modules. The double transducer CpG-SVAs were flanked by Alu sequence (member of evolutionally ancient AluSc family) at the 5′-termini and by the 400-bp-long sequence including evolutionarily ancient AluSp element at the 3′ ends. These structure similarities suggest common ancestry of these four elements from a single progenitor CpG-SVA element.
Genomic coordinates of identified CpG-SVA elements
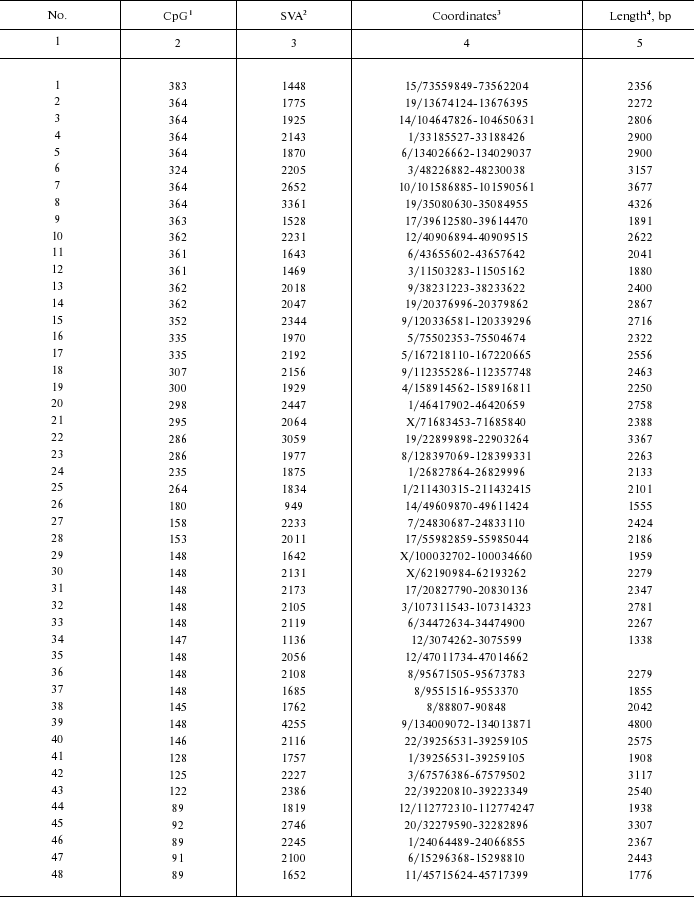

1 Length of exonic part.
2 Length of SVA part.
3 Genomic coordinates (chromosome number/coordinates in
chromosome); genome assembly from February 2009.
4 Overall length of CpG-SVA element, including direct repeats
and transduced sequences.
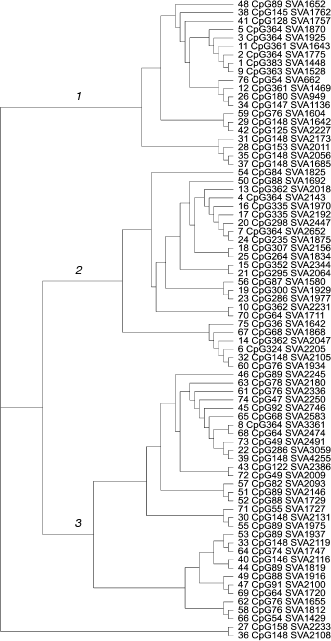
Evolutionary dynamics of the CpG-SVA family. Once the exonic parts of the chimeras varied in length, but not in their primary structure, the SVA-derived parts had very different lengths and primary structure. In the SVA parts there were different genetic changes like insertions, deletions, duplications, quantitative changes in tandem repeat composition, and even insertions of retrotransposons. Together with the presence of transduced genomic sequences, this enabled us to construct a phylogenetic tree for the members of the CpG-SVA family to trace their reciprocal neighborhood. According to the primary structure similarity, CpG-SVA elements were grouped into three major branches (Fig. 3). Interestingly, although there was a kind of correlation between the size of the “exonic” part and sequence localization on the tree, all three above branches contained elements having exonic parts of very different lengths. There was also no connection between the position on a tree and lengths of the SVA parts. In several cases different tree branches included elements with exactly the same lengths of exonic part. For example, branch 2 contained one CpG-SVA element with 364 bp long exonic part, whereas branch 1 had five such elements. Exonic parts of seven elements from branch 2 and of one element from branch 3 were 148-bp-long. There were also similar coincidences for the lengths 64, 76, 88, and 361 bp. These coincidences of exonic part sizes suggest that there were multiple independent events when CpG-SVA elements with identical exonic parts were formed.
The observed peculiarities of distribution of lengths of CpG-SVA exonic parts can be explained by the following factors: (i) there could be multiple functional transcription start sites within CpG-SVA, or (ii) in some cases reverse transcription of the CpG-SVA RNA could terminate before the complete copying of the template has finished. The resulting shortened CpG-SVA inserts could, in turn, generate new elements having even shorter exonic parts, etc.Fig. 3. Phylogenetic tree of the CpG-SVA elements. For each element, its number (according to the table) and lengths of its exonic and SVA parts are shown.
What are the functions of the exonic part of CpG-SVA? Considering that (i) the first exon of the MAST2 gene includes a CpG island, (ii) CpG islands usually play major roles in gene transcriptional regulation, and (iii) MAST2 is strongly upregulated in testis, it can be hypothesized that the exonic part provides increased transcription of CpG-SVA family members in testis. This may be beneficial for the CpG-SVA family as it facilitates fixation of new inserts in the genome. To be fixed, an RE insertion must occur into germ line cells, e.g. those localized in testis. Indeed, in terms of proliferation in the genome, the evolutionarily young family CpG-SVA should be considered as a very successful one: offspring of only one among more than 1000 SVA copies that resided in human DNA at that time (i.e. <0.1%) have generated 76 new fixed inserts (~9% of all 860 human-specific SVA elements) [5]. Experimental investigation of this hypothesis will be a matter of our further studies.
L1 reverse transcriptase adds an extra cytosine residue. In 58 of 73 CpG-SVA elements (~80%), the element insert started with the guanine residue that was absent either in genomic pre-integration site or in the sequence of the first MAST2 exon. Among the other 15 cases, in 12 the insert started with a guanine residue that was included in the exonic sequence. Therefore, in 96% of the cases the insert started with a guanine residue, regardless of its presence in the retrotransposon consensus sequence. This phenomenon can be explained by the template-independent addition of the extra cytosine residue to the very 3′ end of the nascent first strand of cDNA by the L1 reverse transcriptase. As far as we know, this property of L1 reverse transcriptase has not been described before in the literature.
We thank Prof. Jiri Hejnar (Institute of Molecular Genetics, Prague) for valuable comments to the manuscript and fruitful discussion.
This study was supported by a “Molecular and Cellular Biology” grant from the Presidium of the Russian Academy of Sciences and by the Russian Foundation for Basic Research grants 08-04-00720-a and 09-04-12302.
REFERENCES
1.Baltimore, D. (1970) Nature, 226,
1209-1211.
2.Temin, H. M., and Mizutani, S. (1970)
Nature, 226, 1211-1213.
3.Eickbush, T. H. (1997) Science, 277,
911-912.
4.Buzdin, A. A. (2004) Cell. Mol. Life Sci.,
61, 2046-2059.
5.Mills, R. E., Bennett, E. A., Iskow, R. C., Luttig,
C. T., Tsui, C., Pittard, W. S., and Devine, S. E. (2006) Am. J.
Hum. Genet., 78, 671-679.
6.Buzdin, A. (2007) Sci. World J., 7,
1848-1868.
7.Jurka, J. (1997) Proc. Natl. Acad. Sci. USA,
94, 1872-1877.
8.Kazazian, H. H., Jr., and Goodier, J. L. (2002)
Cell, 110, 277-280.
9.Furano, A. V. (2000) Progr. Nucleic Acid Res.
Mol. Biol., 64, 255-294.
10.Ullu, E., and Tschudi, C. (1984) Nature,
312, 171-172.
11.Kramerov, D. A., and Vassetzky, N. S. (2005)
Int. Rev. Cytol., 247, 165-221.
12.Wang, H., Xing, J., Grover, D., Hedges, D. J.,
Han, K., Walker, J. A., and Batzer, M. A. (2005) J. Mol. Biol.,
354, 994-1007.
13.Goodier, J. L., and Kazazian, H. H. (2008)
Cell, 135, 23-35.
14.Xing, J., Wang, H., Belancio, V. P., Cordaux, R.,
Deininger, P. L., and Batzer, M. A. (2006) Proc. Natl. Acad. Sci.
USA, 103, 17608-17613.
15.Babushok, D. V., Ostertag, E. M., and Kazazian,
H. H. (2007) Cell. Mol. Life Sci., 64,
542-554.
16.Buzdin, A., Gogvadze, E., Kovalskaya, E.,
Volchkov, P., Ustyugova, S., Illarionova, A., Fushan, A., Vinogradova,
T., and Sverdlov, E. (2003) Nucleic Acids Res., 31,
4385-4390.
17.Gogvadze, E., Barbisan, C., Lebrun, M. H., and
Buzdin, A. (2007) BMC Genom., 8, 360.
18.Brosius, J. (1999) Genetica, 107,
209-238.
19.Jurka, J., Kapitonov, V. V., Pavlicek, A.,
Klonowski, P., Kohany, O., and Walichiewicz, J. (2005) Cytogenet.
Genome Res., 110, 462-467.
20.Altschul, S. F., Gish, W., Miller, W., Myers, E.
W., and Lipman, D. J. (1990) J. Mol. Biol., 215,
403-410.
21.Thompson, J. D., Higgins, D. G., and Gibson, T.
J. (1994) Nucleic Acids Res., 22, 4673-4680.
22.Felsenstein, J. (1993) Distributed by the author,
Department of Genetics, University of Washington, Seattle.
23.Hancks, D., Ewing, A., Chen, J. E., Tokunaga, K.,
and Kazazian, H. (2009) Genome Res., August 3 (E-pub ahead of
print).
24.Damert, A., Raiz, J., Horn, A., Lower, J., Wang,
H., Xing, J., Batzer, M., Lower, R., and Schumann, G. (2009) Genome
Res., July 27 (E-pub ahead of print).